
Combining Human Genetics of Multiple Sclerosis with Oxidative Stress Phenotype for Drug Repositioning

 , , , ,
, , , , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Collection
2.2. g:Profiler Analysis
2.3. Drug Searching
2.4. In Silico Prediction of Physicochemical Properties of Drugs
3. Results
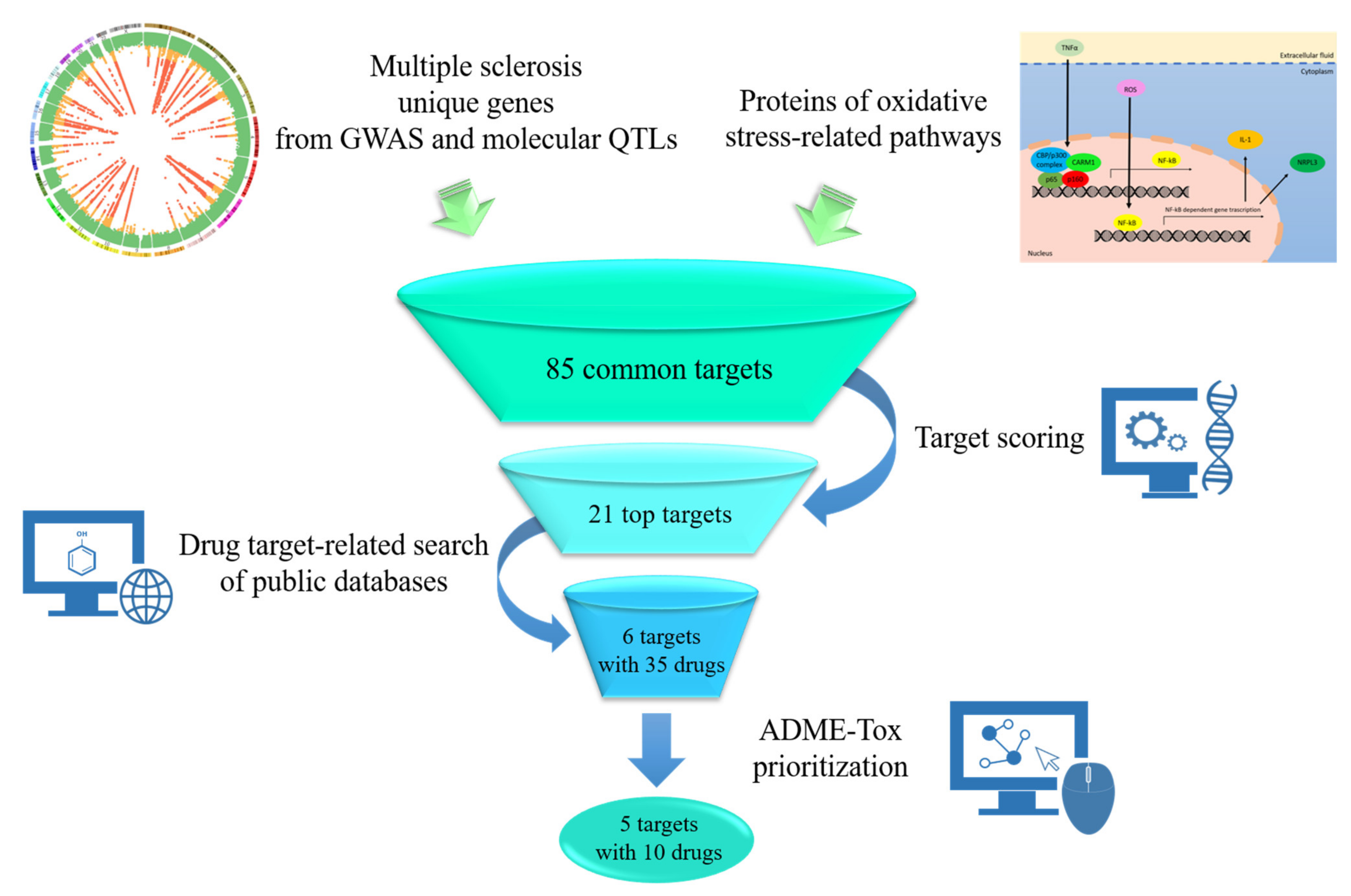
3.1. Genetic-Driven Identification of Targets Linked to Oxidative Pathways in MS
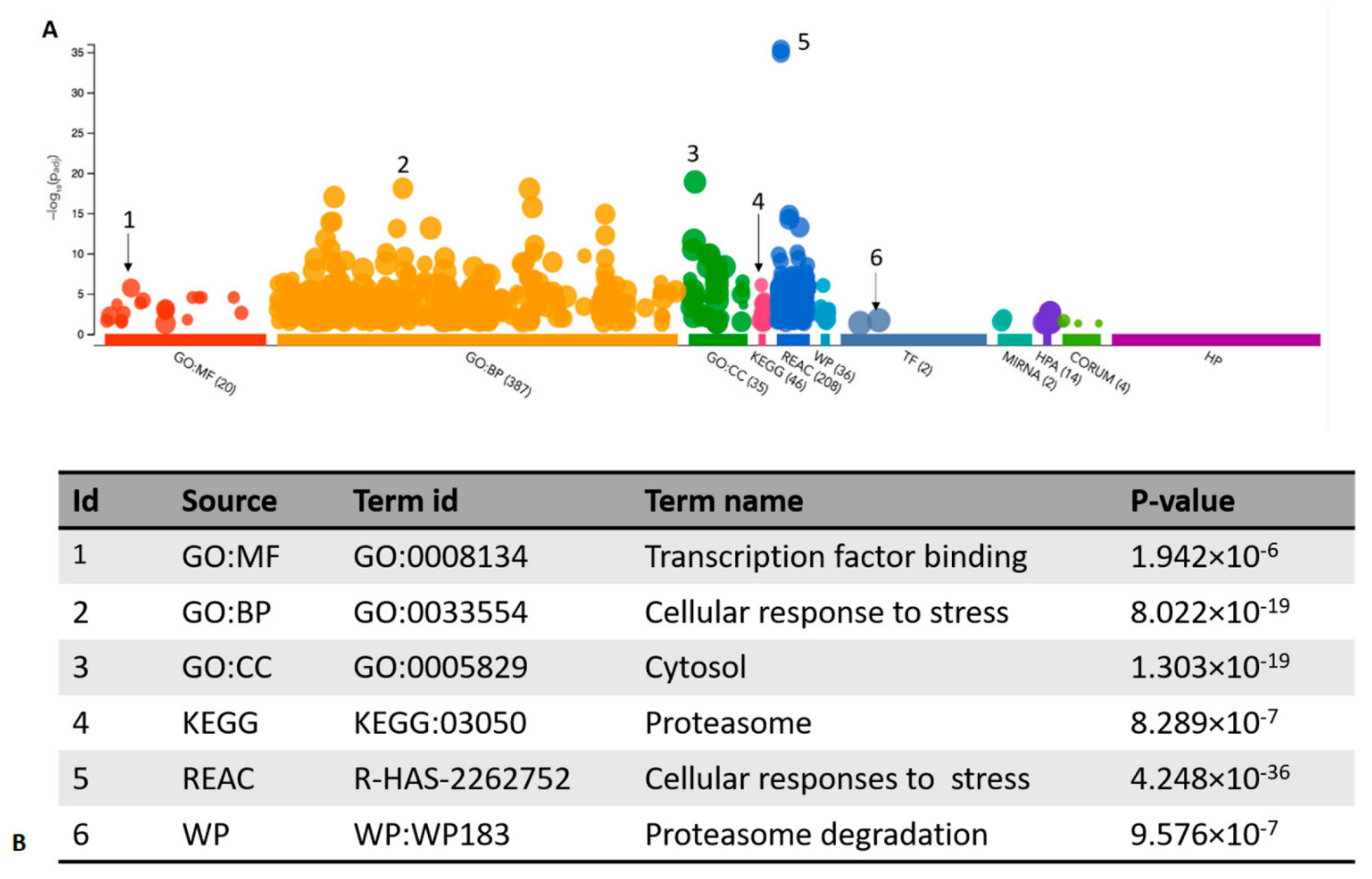
3.2. Functional Enrichment Analysis of the Identified Targets
3.3. Target Prioritization and Drug Search
3.4. Pharmacokinetic Prioritization of the Selected Drugs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Reich, D.S.; Lucchinetti, C.F.; Calabresi, P.A. Multiple Sclerosis. N. Engl. J. Med. 2018, 378, 169–180. [Google Scholar] [CrossRef] [PubMed]
- Hauser, S.L.; Cree, B.A.C. Treatment of Multiple Sclerosis: A Review. Am. J. Med. 2020, 133, 1380–1390. [Google Scholar] [CrossRef] [PubMed]
- Lassmann, H. Pathogenic Mechanisms Associated with Different Clinical Courses of Multiple Sclerosis. Front. Immunol. 2019, 9, 3116. [Google Scholar] [CrossRef] [PubMed]
- Pegoretti, V.; Swanson, K.A.; Bethea, J.R.; Probert, L.; Eisel, U.L.M.; Fischer, R. Inflammation and Oxidative Stress in Multiple Sclerosis: Consequences for Therapy Development. Oxidative Med. Cell. Longev. 2020, 2020, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.-Y.; Gui, L.-N.; Liu, Y.-Y.; Shi, S.; Cheng, Y. Oxidative Stress Marker Aberrations in Multiple Sclerosis: A Meta-Analysis Study. Front. Neurosci. 2020, 14, 823. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.; Nioi, P.; Pickett, C.B. The Nrf2-Antioxidant Response Element Signaling Pathway and Its Activation by Oxidative Stress. J. Biol. Chem. 2009, 284, 13291–13295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgan, M.J.; Liu, Z.G. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res. 2011, 21, 103–115. [Google Scholar] [CrossRef] [Green Version]
- Takata, T.; Araki, S.; Tsuchiya, Y.; Watanabe, Y. Oxidative Stress Orchestrates MAPK and Nitric-Oxide Synthase Signal. Int. J. Mol. Sci. 2020, 21, 8750. [Google Scholar] [CrossRef]
- Veroni, C.; Serafini, B.; Rosicarelli, B.; Fagnani, C.; Aloisi, F.; Agresti, C. Connecting Immune Cell Infiltration to the Multitasking Microglia Response and TNF Receptor 2 Induction in the Multiple Sclerosis Brain. Front. Cell. Neurosci. 2020, 14, 190. [Google Scholar] [CrossRef]
- Gopal, S.; Mikulskis, A.; Gold, R.; Fox, R.J.; Dawson, K.T.; Amaravadi, L. Evidence of activation of the Nrf2 pathway in multiple sclerosis patients treated with delayed-release dimethyl fumarate in the Phase 3 DEFINE and CONFIRM studies. Mult. Scler. J. 2017, 23, 1875–1883. [Google Scholar] [CrossRef]
- Yevgi, R.; Demir, R. Oxidative stress activity of fingolimod in multiple sclerosis. Clin. Neurol. Neurosurg. 2021, 202, 106500. [Google Scholar] [CrossRef]
- Adamczyk, B.; Wawrzyniak, S.; Kasperczyk, S.; Adamczyk-Sowa, M. The Evaluation of Oxidative Stress Parameters in Serum Patients with Relapsing-Remitting Multiple Sclerosis Treated with II-Line Immunomodulatory Therapy. Oxidative Med. Cell. Longev. 2017, 2017, 1–12. [Google Scholar] [CrossRef]
- Miller, E.D.; Dziedzic, A.; Saluk-Bijak, J.; Bijak, M. A Review of Various Antioxidant Compounds and their Potential Utility as Complementary Therapy in Multiple Sclerosis. Nutrients 2019, 11, 1528. [Google Scholar] [CrossRef] [Green Version]
- Forman, H.J.; Zhang, H. Targeting oxidative stress in disease: Promise and limitations of antioxidant therapy. Nat. Rev. Drug Discov. 2021, 20, 689–709, Erratum in 2021, 20, 652. [Google Scholar] [CrossRef]
- Cook, D.; Brown, D.; Alexander, R.; March, R.; Morgan, P.; Satterthwaite, G.; Pangalos, M.N. Lessons learned from the fate of AstraZeneca’s drug pipeline: A five-dimensional framework. Nat. Rev. Drug Discov. 2014, 13, 419–431. [Google Scholar] [CrossRef]
- Nelson, M.R.; Tipney, H.; Painter, J.L.; Shen, J.; Nicoletti, P.; Shen, Y.; Floratos, A.; Sham, P.C.; Li, M.J.; Wang, J.; et al. The support of human genetic evidence for approved drug indications. Nat. Genet. 2015, 47, 856–860. [Google Scholar] [CrossRef]
- Plenge, R.M.; Scolnick, E.M.; Altshuler, D. Validating therapeutic targets through human genetics. Nat. Rev. Drug Discov. 2013, 12, 581–594. [Google Scholar] [CrossRef]
- Floris, M.; Olla, S.; Schlessinger, D.; Cucca, F. Genetic-Driven Druggable Target Identification and Validation. Trends Genet. 2018, 34, 558–570. [Google Scholar] [CrossRef]
- International Multiple Sclerosis Genetics Consortium. Multiple sclerosis genomic map implicates peripheral immune cells and microglia in susceptibility. Science 2019, 365, eaav7188. [Google Scholar] [CrossRef] [Green Version]
- Cano-Gamez, E.; Trynka, G. From GWAS to Function: Using Functional Genomics to Identify the Mechanisms Underlying Complex Diseases. Front. Genet. 2020, 11, 424. [Google Scholar] [CrossRef]
- Eddershaw, P.J.; Beresford, A.P.; Bayliss, M.K. ADME/PK as part of a rational approach to drug discovery. Drug Discov. Today 2000, 5, 409–414. [Google Scholar] [CrossRef]
- GWAS Catalog (downloaded file: Gwas_catalog_v1.0.2-associations_e98_r2021-03-01.tsv). Available online: https://www.ebi.ac.uk/gwas/home (accessed on 1 March 2021).
- Buniello, A.; MacArthur, J.A.L.; Cerezo, M.; Harris, L.W.; Hayhurst, J.; Malangone, C.; McMahon, A.; Morales, J.; Mountjoy, E.; Sollis, E.; et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 2019, 47, D1005–D1012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LinDA. Available online: http://linda.irgb.cnr.it/ (accessed on 16 March 2021).
- Onano, S.; Cucca, F.; Pala, M. P18.25A The eQTLs Catalog and LinDA browser: A platform for prioritising target genes of GWAS variants. Abstracts from the 51st European Society of Human Genetics Conference: Oral Presentations. Eur. J. Hum. Genet. 2019, 27, 748–869. [Google Scholar] [CrossRef]
- Lift Genome Annotations. Available online: https://www.genome.ucsc.edu/cgi-bin/hgLiftOver (accessed on 15 March 2021).
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, D. The Human Genome Browser at UCSC. Genome Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef] [Green Version]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.; Daly, M.J.; et al. PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [Green Version]
- Plink v.1.9 Software. Available online: http://pngu.mgh.harvard.edu/purcell/plink/ (accessed on 18 March 2021).
- Genomes Project Consortium; Auton, A.; Brooks, L.D.; Durbin, R.M.; Garrison, E.P.; Kang, H.M.; Korbel, J.O.; Marchini, J.L.; McCarthy, S.; McVean, G.A.; et al. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef] [Green Version]
- VEP. Available online: https://grch37.ensembl.org/Tools/VEP (accessed on 16 March 2021).
- Kircher, M.; Witten, D.M.; Jain, P.; O’Roak, B.J.; Cooper, G.M.; Shendure, J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef] [Green Version]
- Reactome. Available online: https://reactome.org/ (accessed on 1 March 2021).
- Fabregat, A.; Sidiropoulos, K.; Viteri, G.; Marin-Garcia, P.; Ping, P.; Stein, L.; D’Eustachio, P.; Hermjakob, H. Reactome diagram viewer: Data structures and strategies to boost performance. Bioinformatics 2018, 34, 1208–1214. [Google Scholar] [CrossRef]
- g:Profiler (version e94_eg41_p11_9f195a1). Available online: https://biit.cs.ut.ee/gprofiler/gost (accessed on 24 March 2021).
- Raudvere, U.; Kolberg, L.; Kuzmin, I.; Arak, T.; Adler, P.; Peterson, H.; Vilo, J. g:Profiler: A web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 2019, 47, W191–W198. [Google Scholar] [CrossRef] [Green Version]
- Open Targets Platform. Available online: https://platform.opentargets.org/ (accessed on 29 March 2021).
- Ochoa, D.; Hercules, A.; Carmona, M.; Suveges, D.; Gonzalez-Uriarte, A.; Malangone, C.; Miranda, A.; Fumis, L.; Carvalho-Silva, D.; Spitzer, M.; et al. Open Targets Platform: Supporting systematic drug–target identification and prioritisation. Nucleic Acids Res. 2021, 49, D1302–D1310. [Google Scholar] [CrossRef]
- Hecker, N.; Ahmed, J.; Von Eichborn, J.; Dunkel, M.; Macha, K.; Eckert, A.; Gilson, M.K.; Bourne, P.E.; Preissner, R. SuperTarget goes quantitative: Update on drug-target interactions. Nucleic Acids Res. 2012, 40, D1113–D1117. [Google Scholar] [CrossRef]
- DrugBank. Available online: https://go.drugbank.com/ (accessed on 29 March 2021).
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A Major Update to the DrugBank Database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef]
- DGIdb. Available online: https://www.dgidb.org/ (accessed on 5 April 2021).
- Freshour, S.L.; Kiwala, S.; Cotto, K.C.; Coffman, A.C.; McMichael, J.F.; Song, J.J.; Griffith, M.; Griffith, O.L.; Wagner, A.H. Integration of the Drug-Gene Interaction Database (DGIdb 4.0) with open crowdsource efforts. Nucleic Acids Res. 2021, 49, D1144–D1151. [Google Scholar] [CrossRef]
- Jassal, B.; Matthews, L.; Viteri, G.; Gong, C.; Lorente, P.; Fabregat, A.; Gillespie, M.; Stephan, R.; Milacic, M.; Rothfels, K.; et al. The reactome pathway knowledgebase. Nucleic Acids Res. 2020, 4, D498–D503. [Google Scholar] [CrossRef]
- Pharos. Available online: https://pharos.nih.gov/ (accessed on 14 June 2021).
- Sheils, T.K.; Mathias, S.L.; Kelleher, K.J.; Siramshetty, V.B.; Nguyen, D.-T.; Bologa, C.G.; Jensen, L.J.; Vidović, D.; Koleti, A.; Schürer, S.C.; et al. TCRD and Pharos 2021: Mining the human proteome for disease biology. Nucleic Acids Res. 2020, 49, D1334–D1346. [Google Scholar] [CrossRef]
- International Multiple Sclerosis Genetics Consortium (IMSGC); Beecham, A.H.; Patsopoulos, N.A.; Xifara, D.K.; Davis, M.F.; Kemppinen, A.; Cotsapas, C.; Shah, T.S.; Spencer, C.; Booth, D.; et al. Analysis of immune-related loci identifies 48 new susceptibility variants for multiple sclerosis. Nat. Genet. 2013, 45, 1353–1360. [Google Scholar] [CrossRef]
- International Multiple Sclerosis Genetics Consortium; Wellcome Trust Case Control Consortium 2; Sawcer, S.; Hellenthal, G.; Pirinen, M.; Spencer, C.C.; Patsopoulos, N.A.; Moutsianas, L.; Dilthey, A.; Su, Z.; et al. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature 2011, 476, 214–219. [Google Scholar] [CrossRef]
- Manuel, A.M.; Dai, Y.; Freeman, L.A.; Jia, P.; Zhao, Z. An integrative study of genetic variants with brain tissue expression identifies viral etiology and potential drug targets of multiple sclerosis. Mol. Cell. Neurosci. 2021, 115, 103656. [Google Scholar] [CrossRef]
- Nabirotchkin, S.; Peluffo, A.E.; Rinaudo, P.; Yu, J.; Hajj, R.; Cohen, D. Next-generation drug repurposing using human genetics and network biology. Curr. Opin. Pharmacol. 2020, 51, 78–92. [Google Scholar] [CrossRef]
- Adamczyk, B.; Adamczyk-Sowa, M. New Insights into the Role of Oxidative Stress Mechanisms in the Pathophysiology and Treatment of Multiple Sclerosis. Oxidative Med. Cell. Longev. 2016, 2016, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Kawai, T.; Okochi, Y.; Ozaki, T.; Imura, Y.; Koizumi, S.; Yamazaki, M.; Abe, M.; Sakimura, K.; Yamashita, T.; Okamura, Y. Unconventional role of voltage-gated proton channels (VSOP/Hv1) in regulation of microglial ROS production. J. Neurochem. 2017, 142, 686–699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.H.; Chang, L.S. Arachidonic acid induces Fas and FasL upregulation in human leukemia U937 cells via Ca2+/ROS-mediated suppression of ERK/c-Fos pathway and activation of p38 MAPK/ATF-2 pathway. Toxicol Lett. 2009, 191, 140–148. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Xu, J.; Xiong, X.; Deng, Y. Salidroside inhibits MAPK, NF-κB, and STAT3 pathways in psoriasis-associated oxidative stress via SIRT1 activation. Redox Rep. 2019, 24, 70–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Covic, M.; Hassa, P.O.; Saccani, S.; Buerki, C.; Meier, N.I.; Lombardi, C.; Imhof, R.; Bedford, M.T.; Natoli, G.; Hottiger, M.O. Arginine methyltransferase CARM1 is a promoter-specific regulator of NF-kappaB-dependent gene expression. EMBO J. 2005, 24, 85–96. [Google Scholar] [CrossRef] [Green Version]
- Suresh, S.; Huard, S.; Dubois, T. CARM1/PRMT4: Making Its Mark beyond Its Function as a Transcriptional Coactivator. Trends Cell Biol. 2021, 31, 402–417. [Google Scholar] [CrossRef]
- Schmitz, M.L.; Kracht, M. Cyclin-Dependent Kinases as Coregulators of Inflammatory Gene Expression. Trends Pharmacol. Sci. 2016, 37, 101–113. [Google Scholar] [CrossRef]
- Yang, G.; Wright, C.J.; Hinson, M.D.; Fernando, A.P.; Sengupta, S.; Biswas, C.; La, P.; Dennery, P.A. Oxidative stress and inflammation modulate Rev-erbα signaling in the neonatal lung and affect circadian rhythmicity. Antioxid. Redox Signal. 2014, 21, 17–32. [Google Scholar] [CrossRef] [Green Version]
- Griffin, P.; Dimitry, J.M.; Sheehan, P.W.; Lananna, B.V.; Guo, C.; Robinette, M.L.; Hayes, M.E.; Cedeño, M.R.; Nadarajah, C.; Ezerskiy, L.A.; et al. Circadian clock protein Rev-erbα regulates neuroinflammation. Proc. Natl. Acad. Sci. USA 2019, 116, 5102–5107. [Google Scholar] [CrossRef] [Green Version]
- Makhoba, X.H.; Viegas, C., Jr.; Mosa, R.A.; Viegas, F.P.D.; Pooe, O.J. Potential Impact of the Multi-Target Drug Approach in the Treatment of Some Complex Diseases. Drug Des. Devel. Ther. 2020, 14, 3235–3249. [Google Scholar] [CrossRef]
- Telenti, A.; di Iulio, J. Regulatory genome variants in human susceptibility to infection. Hum Genet. 2020, 139, 759–768. [Google Scholar] [CrossRef]
- Drew, A.E.; Moradei, O.; Jacques, S.L.; Rioux, N.; Boriack-Sjodin, A.P.; Allain, C.; Scott, M.P.; Jin, L.; Raimondi, A.; Handler, J.L.; et al. Identification of a CARM1 Inhibitor with Potent In Vitro and In Vivo Activity in Preclinical Models of Multiple Myeloma. Sci. Rep. 2017, 7, 17993. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; de Boer, M.; van der Wel, E.J.; Van Eck, M.; Hoekstra, M. PRMT4 inhibitor TP-064 inhibits the pro-inflammatory macrophage lipopolysaccharide response in vitro and ex vivo and induces peritonitis-associated neutrophilia in vivo. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2021, 1867, 166212. [Google Scholar] [CrossRef]
- Hwang, J.W.; Cho, Y.; Bae, G.-U.; Kim, S.-N.; Kim, Y.K. Protein arginine methyltransferases: Promising targets for cancer therapy. Exp. Mol. Med. 2021, 53, 788–808. [Google Scholar] [CrossRef]
- Rose, P.; Won, Y.K.; Ong, C.N.; Whiteman, M. β-Phenylethyl and 8-methylsulphinyloctyl isothiocyanates, constituents of watercress, suppress LPS induced production of nitric oxide and prostaglandin E2 in RAW 264.7 macrophages. Nitric Oxide 2005, 12, 237–243. [Google Scholar] [CrossRef]
- Dey, M.; Ribnicky, D.; Kurmukov, A.G.; Raskin, I. In Vitro and in Vivo Anti-Inflammatory Activity of a Seed Preparation Containing Phenethylisothiocyanate. J. Pharmacol. Exp. Ther. 2006, 317, 326–333. [Google Scholar] [CrossRef] [Green Version]
- Gupta, P.; Adkins, C.; Lockman, P.; Srivastava, S.K. Metastasis of Breast Tumor Cells to Brain Is Suppressed by Phenethyl Isothiocyanate in a Novel In Vivo Metastasis Model. PLoS ONE 2013, 8, e67278. [Google Scholar] [CrossRef]
- Vitali, F.; Berghout, J.; Fan, J.; Li, J.; Li, Q.; Li, H.; Lussier, Y.A. Precision drug repurposing via convergent eQTL-based molecules and pathway targeting independent disease-associated polymorphisms. Pac. Symp. Biocomput. 2019, 24, 308–319. [Google Scholar]
- Cho, H.; Zhao, X.; Hatori, M.; Yu, R.T.; Barish, G.D.; Lam, M.T.; Chong, L.W.; Di Tacchio, L.; Atkins, A.R.; Glass, C.K.; et al. Regulation of circadian behaviour and metabolism by REV-ERB-α and REV-ERB-β. Nature 2012, 485, 123–127. [Google Scholar] [CrossRef] [Green Version]
- Finan, C.; Gaulton, A.; Kruger, F.A.; Lumbers, R.T.; Shah, T.; Engmann, J.; Galver, L.; Kelley, R.; Karlsson, A.; Santos, R.; et al. The druggable genome and support for target identification and validation in drug development. Sci. Transl. Med. 2017, 9, eaag1166. [Google Scholar] [CrossRef]
- Lau, A.; So, H.-C. Turning genome-wide association study findings into opportunities for drug repositioning. Comput. Struct. Biotechnol. J. 2020, 18, 1639–1650. [Google Scholar] [CrossRef]
- Webb, L.M.; Guerau-De-Arellano, M. Emerging Role for Methylation in Multiple Sclerosis: Beyond DNA. Trends Mol. Med. 2017, 23, 546–562. [Google Scholar] [CrossRef] [PubMed]
- Blanc, R.S.; Richard, S. Arginine Methylation: The Coming of Age. Mol. Cell 2017, 65, 8–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandran, U.; Mehendale, N.; Patil, S.; Chaguturu, R.; Patwardhan, B. Network pharmacology. In Innovative Approaches in Drug Discovery; Elsevier: Amsterdam, The Netherlands, 2017; pp. 1127–1164. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Target | Drug | Mechanism of Action | Status * | Database |
|---|---|---|---|---|
| CARM1 | BIIB021 | HSP90 and CARM1 inhibitor | Phase II for breast cancer and gastrointestinal stromal tumors | DGIdb |
| MAPK1 | PHENETHYL ISOTHIOCYANATE, (PEITC) | Bioactive compound activates ERK signal | Phase II lung cancer, tobacco use disorder and oral cancer | Super Target |
| CDK4 | ABEMACICLIB | CDK4/6 inhibitor | Approved for breast cancer | DGIdb, DrugBank, OpenTarget |
| CDK4 | ALVOCIDIB | CDKs inhibitor | Phase II for chronic lymphocytic leukemia; relapsed or refractory multiple myeloma; B-cell lymphoma; sarcoma; acute myeloid leukemia; prostate cancer; advanced ovarian epithelial cancer or primary peritoneal cancer; adenocarcinoma; kidney cancer; melanoma; endometrial cancer | DGIdb; DrugBank; OpenTarget |
| CDK4 | MILCICLIB | CDKs inhibitor | Phase II for malignant thymoma and hepatocellular carcinoma | DGIdb; OpenTarget |
| CDK4 | PHA-793887 | CDKs inhibitor | Phase I for advanced- metastatic solid tumors | DGIdb |
| STAT3 | ATIPRIMOD | Blocks STAT3 activation | Phase II for neuroendocrine cancer and multiple myeloma | DGIdb |
| STAT3 | ENMD 1198 | Mitosis inhibitors; tubulin modulators; STAT3 inhibitor | Phase I for advanced cancer | DrugBank |
| STAT3 | ERLOTINIB | EGFR inhibitor; stimulated phosphorylation and activation of STAT3 | Approved for lung and pancreatic cancer | SuperTarget; DGIdb |
| FOS | PILOCARPINE | Muscarinic receptor agonist- induced c-fos expression | Approved for the treatment of presbyopia | DGIdb |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Olla, S.; Steri, M.; Formato, A.; Whalen, M.B.; Corbisiero, S.; Agresti, C. Combining Human Genetics of Multiple Sclerosis with Oxidative Stress Phenotype for Drug Repositioning. Pharmaceutics 2021, 13, 2064. https://doi.org/10.3390/pharmaceutics13122064
Olla S, Steri M, Formato A, Whalen MB, Corbisiero S, Agresti C. Combining Human Genetics of Multiple Sclerosis with Oxidative Stress Phenotype for Drug Repositioning. Pharmaceutics. 2021; 13(12):2064. https://doi.org/10.3390/pharmaceutics13122064
Chicago/Turabian StyleOlla, Stefania, Maristella Steri, Alessia Formato, Michael B. Whalen, Silvia Corbisiero, and Cristina Agresti. 2021. "Combining Human Genetics of Multiple Sclerosis with Oxidative Stress Phenotype for Drug Repositioning" Pharmaceutics 13, no. 12: 2064. https://doi.org/10.3390/pharmaceutics13122064