
Novel N7-Arylmethyl Substituted Dinucleotide mRNA 5′ cap Analogs: Synthesis and Evaluation as Modulators of Translation †

 ,
,  , , , , and
, , , , and
Abstract
:
1. Introduction
2. Results and Discussion
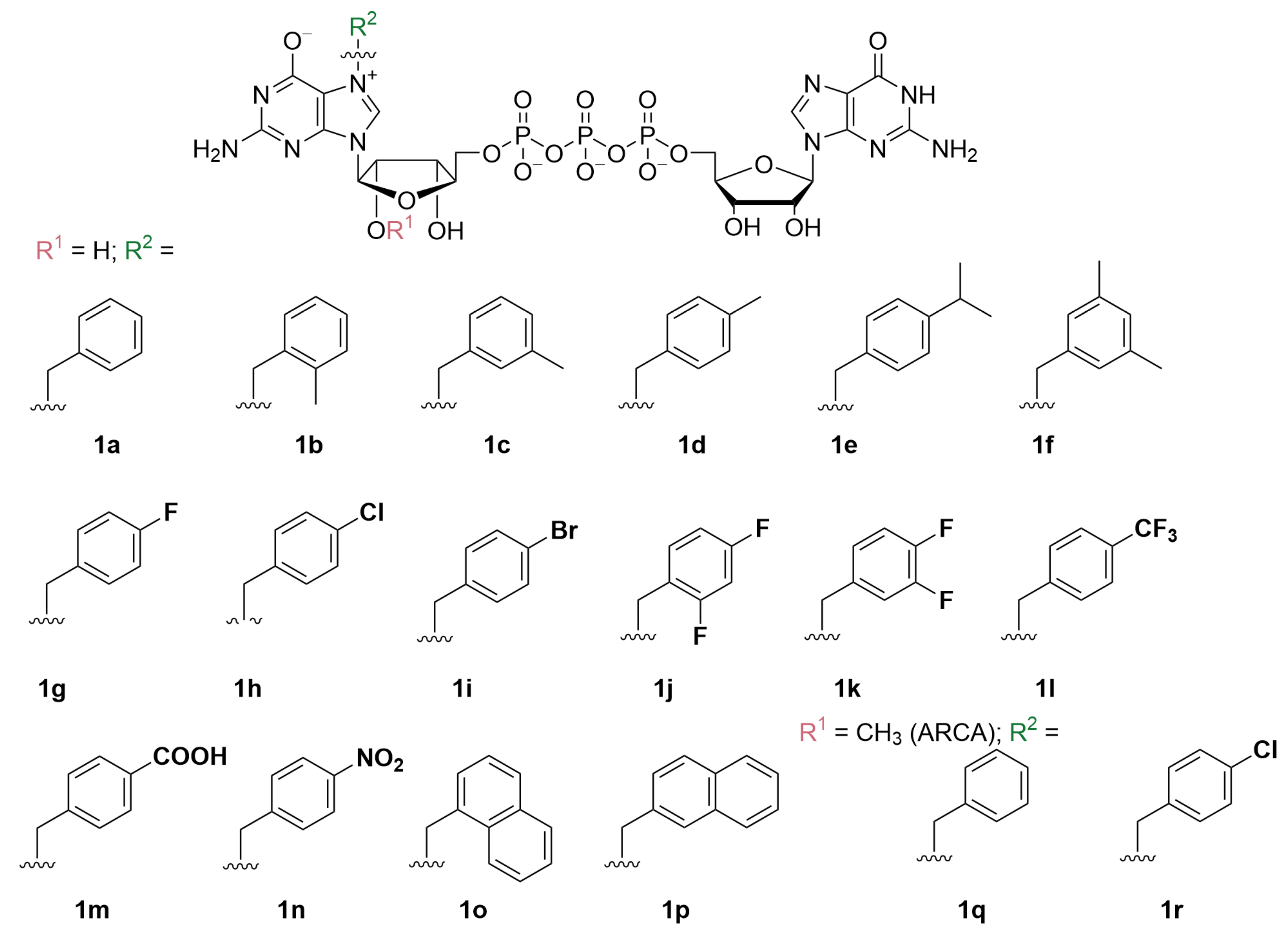
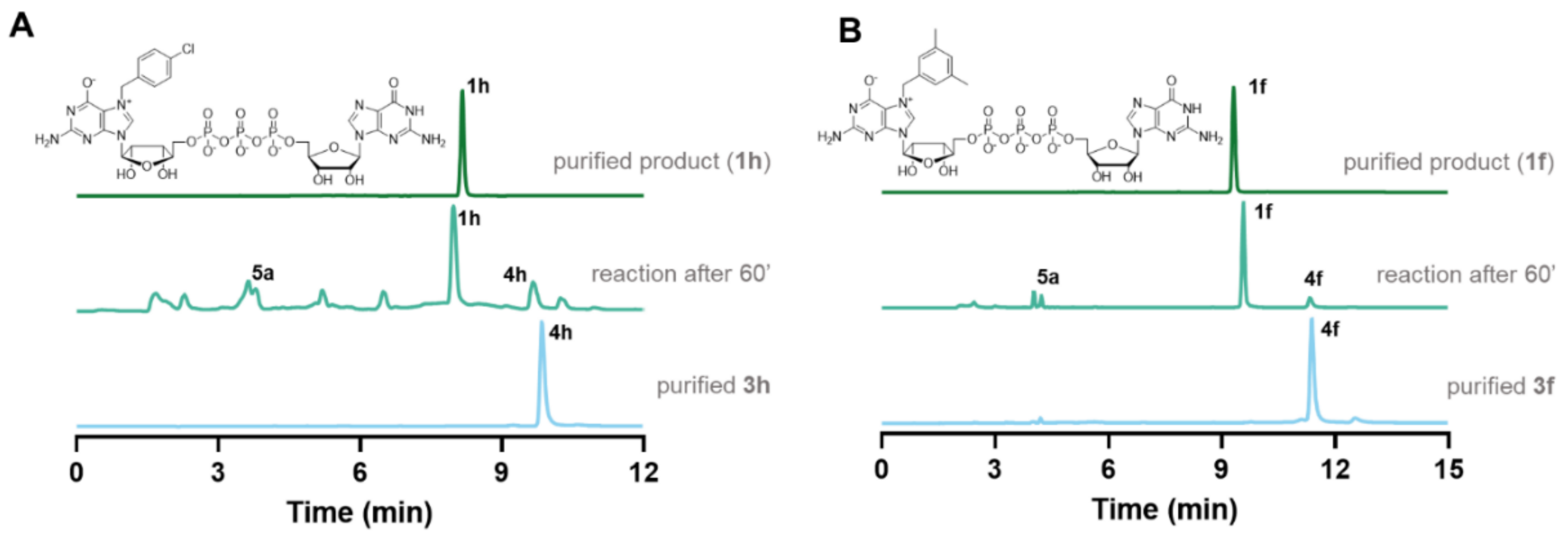
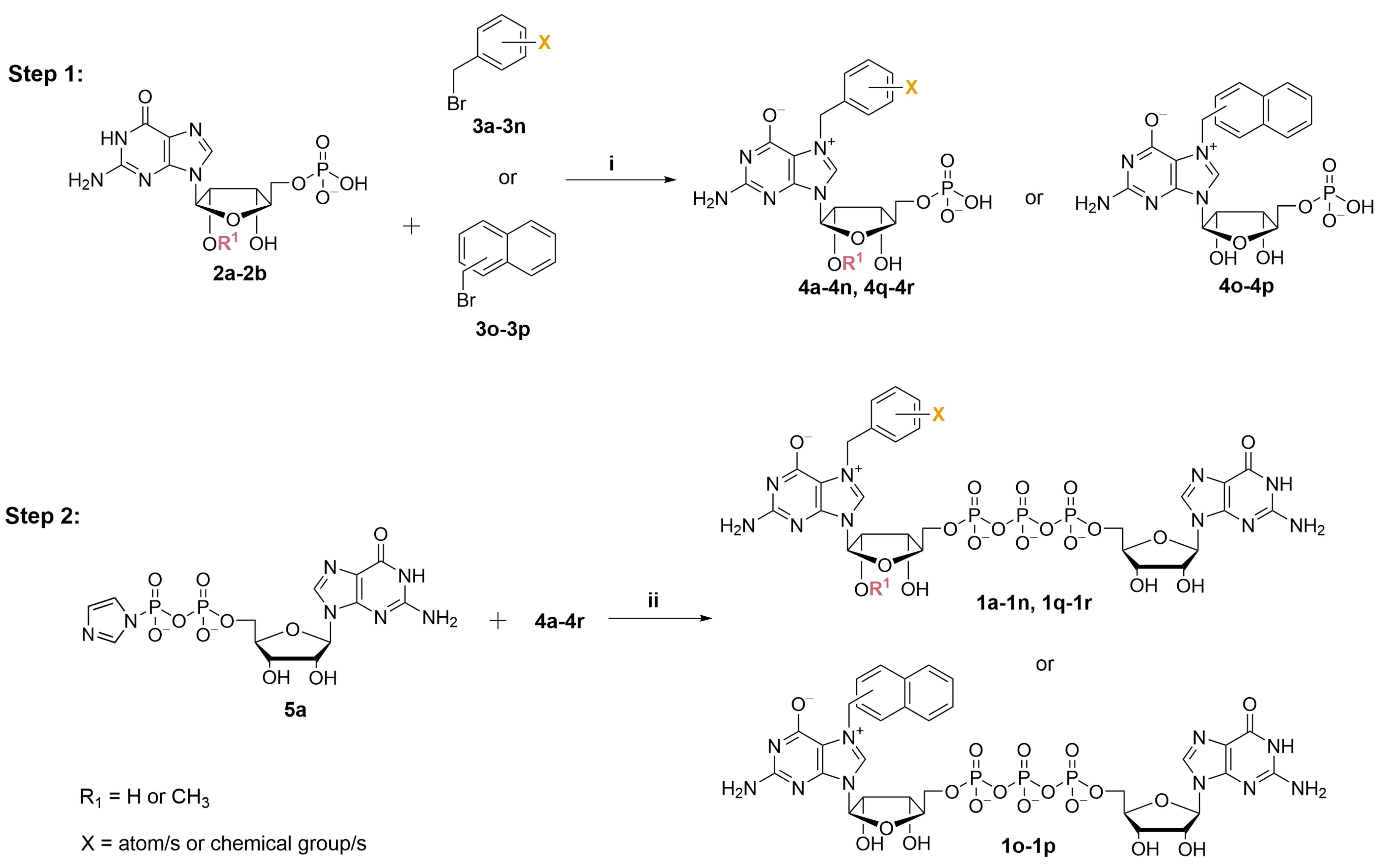
2.1. Synthesis of N7-Arylmethyl Cap Analogs
2.2. Interaction with eIF4E and Susceptibility to Hydrolysis by DcpS
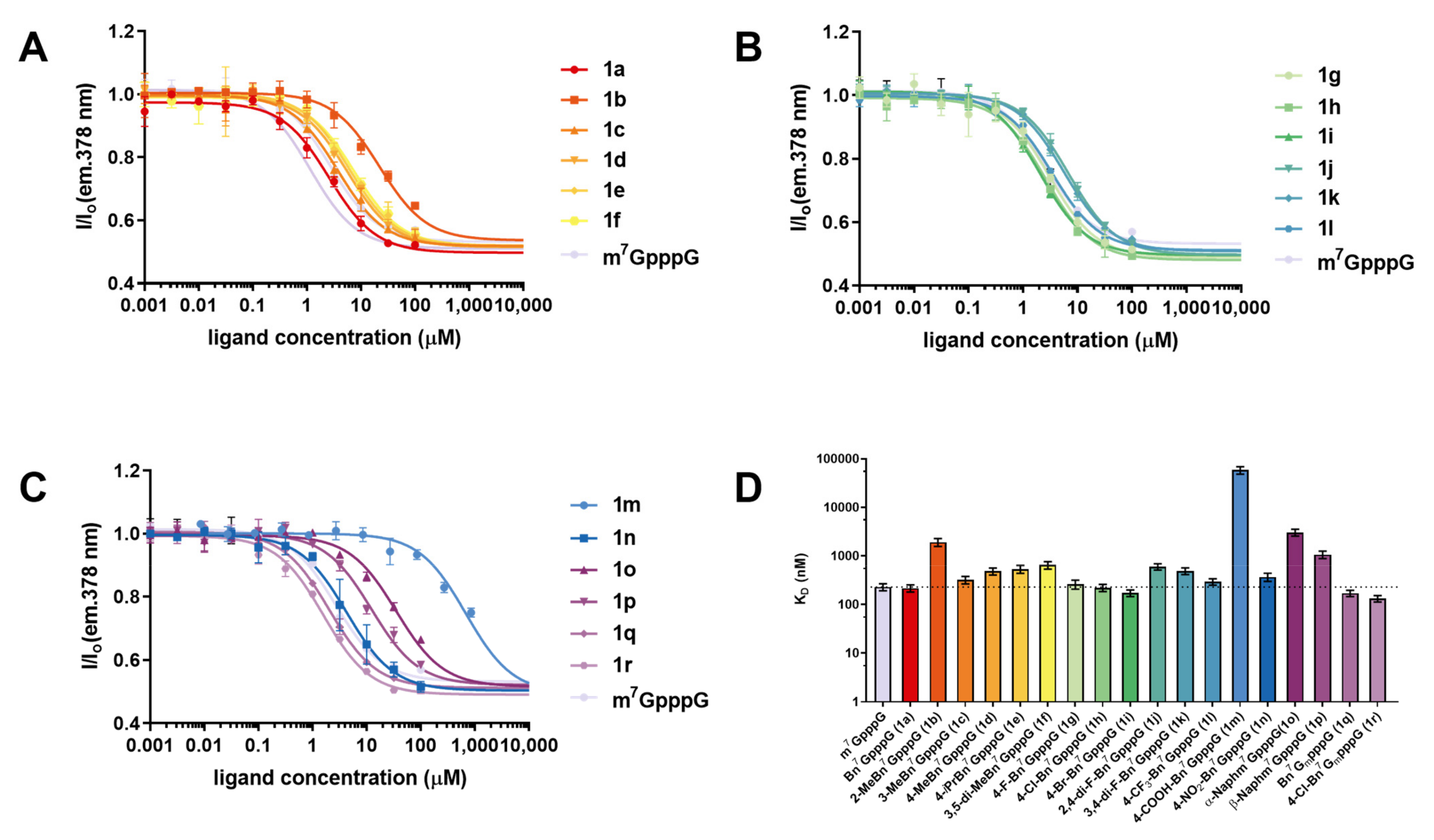
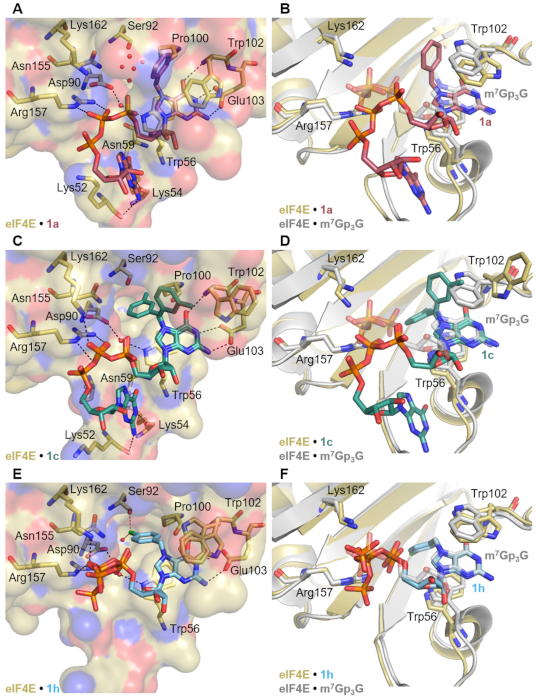
2.2.1. Interaction with eIF4E
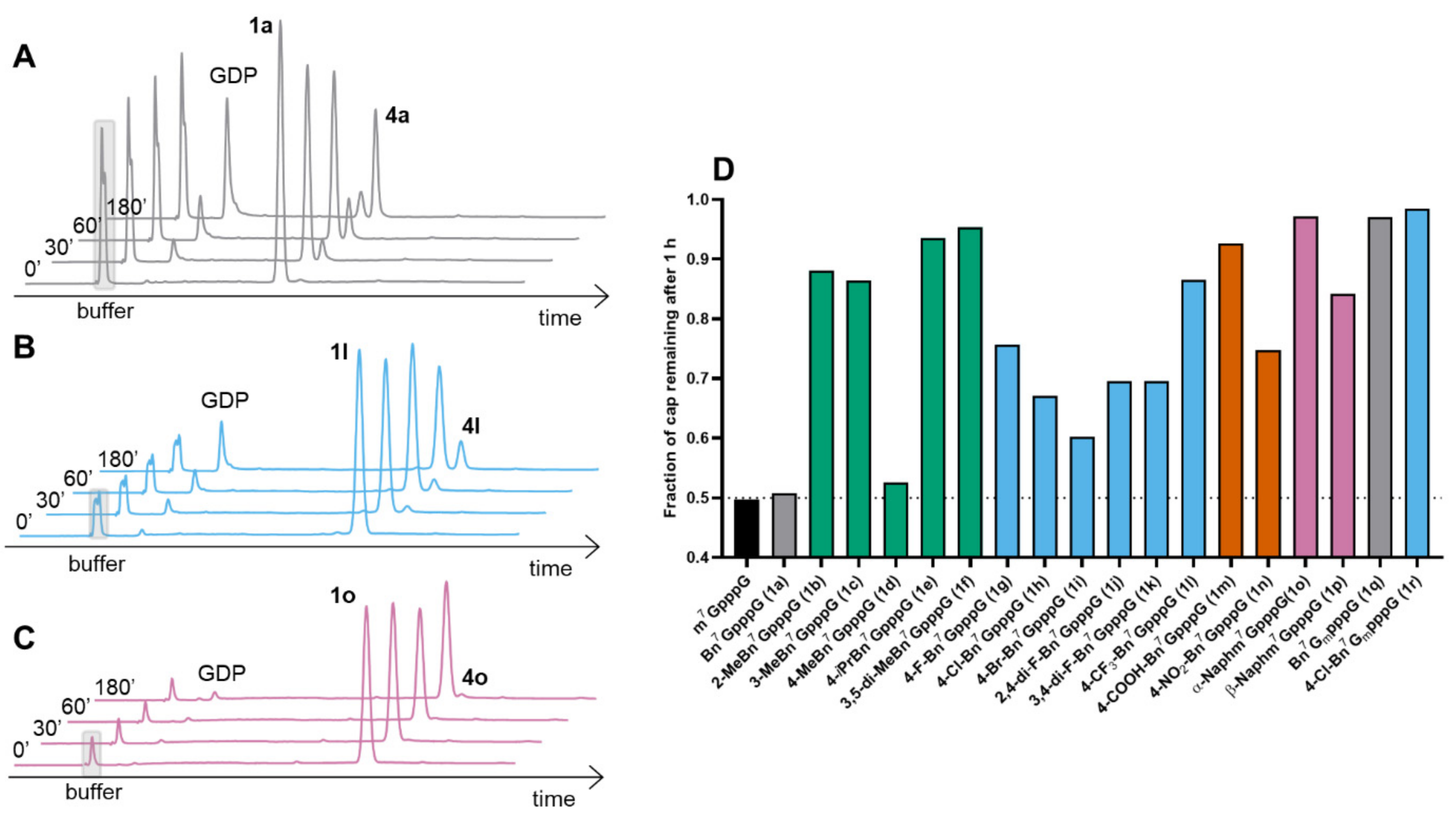
2.2.2. Susceptibility to the Human DcpS Enzyme
2.3. Inhibition of Translation by Cap Analogs in a RRL System
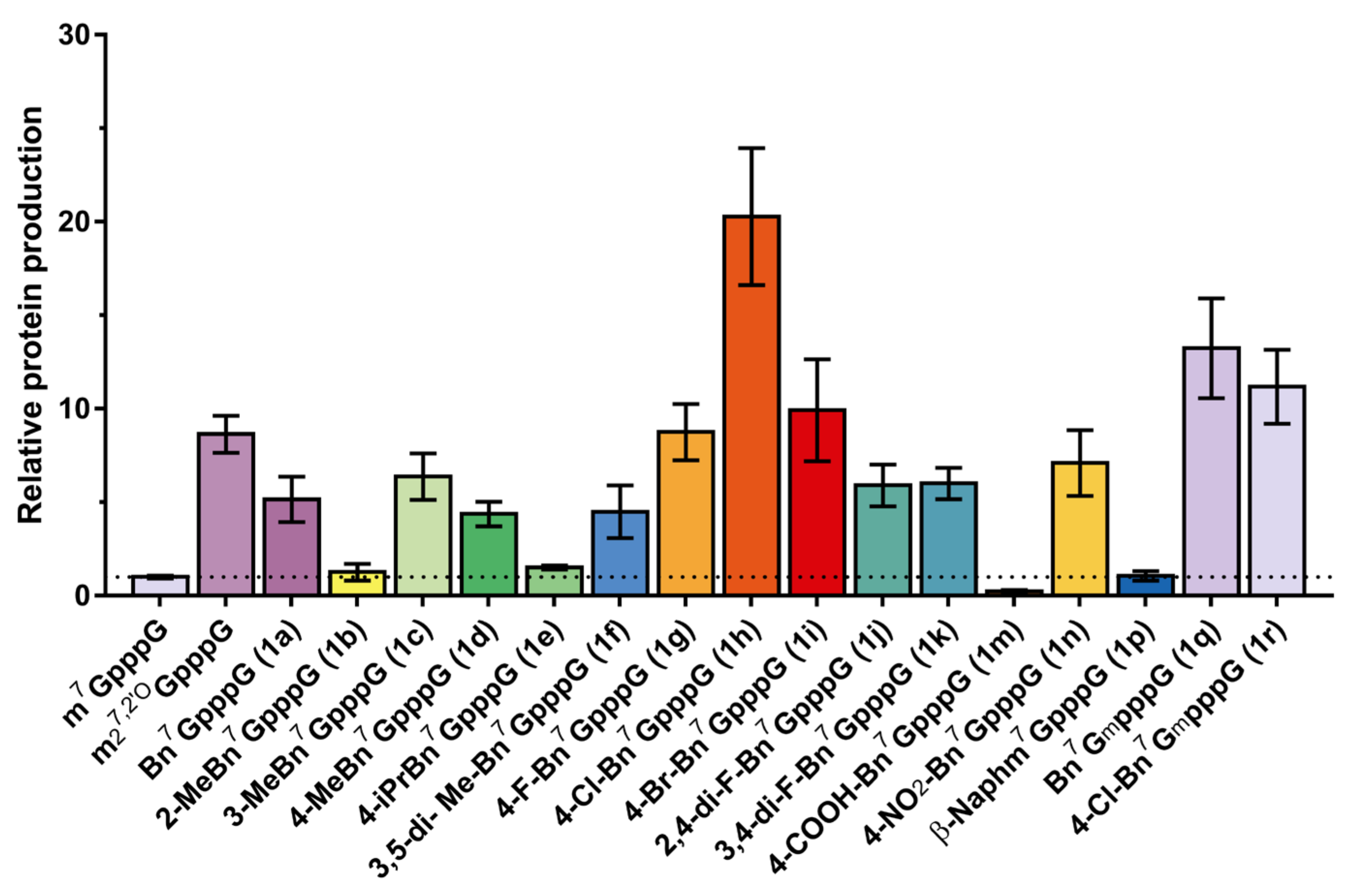
2.4. Incorporation into RNA and Translational Efficiency

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cap Analog | KD Cap-eIF4E ± SD [nM] [a] | hDcpS Assay [c] | Translation Inhibition [μM] | Rel. Protein Expression [e] |
|---|---|---|---|---|
| m7GpppG | 229 ± 37 (160.0 ± 2.6) [b] | 0.49 | 11.4 ± 2.4 (30.1 ± 9.7) [d] | 1.0 |
| m7GmpppG | n.d. | n.d. | n.d. | 8.6 ± 1.0 |
| Bn7GpppG (1a) | 217 ± 36 (107.1 ± 4.7) [b] | 0.51 | 7.4 ± 1.4 (9.6 ± 3.8) [d] | 5.2 ± 1.2 |
| 2-MeBn7GpppG (1b) | 1910 ± 360 | 0.88 | 154 ± 88 | 1.3 ± 0.4 |
| 3-MeBn7GpppG (1c) | 319 ± 55 | 0.84 | 6.6 ± 0.8 (8.2 ± 3.1) [d] | 6.4 ± 1.2 |
| 4-MeBn7GpppG (1d) | 482 ± 81 | 0.53 | 16.9 ± 3.5 | 4.4 ± 0.7 |
| 4-iPrBn7GpppG (1e) | 534 ± 100 | 0.94 | 15.2 ± 3.1 | 1.5 ± 0.1 |
| 3,5-di-MeBn7GpppG (1f) | 646 ± 114 | 0.95 | 13.2 ± 2.0 | 4.5 ± 1.4 |
| 4-F-Bn7GpppG (1g) | 261 ± 54 | 0.76 | 6.0 ± 0.9 (21.4 ± 5.1) [d] | 8.7 ± 1.5 |
| 4-Cl-Bn7GpppG (1h) | 221 ± 37 | 0.67 | 18.7 ± 3.0 (18.7 ± 3.6) [d] | 20.3 ± 3.7 |
| 4-Br-Bn7GpppG (1i) | 172 ± 26 | 0.60 | 6.4 ± 1.0 (22.2 ± 6.7) [d] | 9.9 ± 2.7 |
| 2,4-di-F-Bn7GpppG (1j) | 598 ± 87 | 0.70 | 8.4 ± 1.5 | 5.9 ± 1.1 |
| 3,4-di-F-Bn7GpppG (1k) | 489 ± 77 | 0.70 | 8.2 ± 1.1 (21.4 ± 3.6) [d] | 6.0 ± 0.8 |
| 4-CF3-Bn7GpppG (1l) | 293 ± 43 | 0.87 | 9.7 ± 2.0 (40 ± 13) [d] | n.d. |
| 4-COOH-Bn7GpppG (1m) | >50,000 | 0.93 | >500 | 0.2 ± 0.1 |
| 4-NO2-Bn7GpppG (1n) | 367 ± 71 | 0.75 | 28.2 ± 7.5 | 7.1 ± 1.8 |
| α-Naphm7GpppG (1o) | 3040 ± 490 | 0.97 | n.d. | n.d. |
| β-Naphm7GpppG (1p) | 1060 ± 190 | 0.84 | 15.9 ± 3.4 | 1.0 ± 0.3 |
| Bn7GmpppG (1q) c | 171 ± 25 | 0.97 | 3.7 ± 0.5 (8.2 ± 1.0) [d] | 13.2 ± 2.7 |
| 4-Cl-Bn7GmpppG (1r) | 132 ± 20 | 0.99 | 6.0 ± 0.9 | 11.2 ± 2.0 |
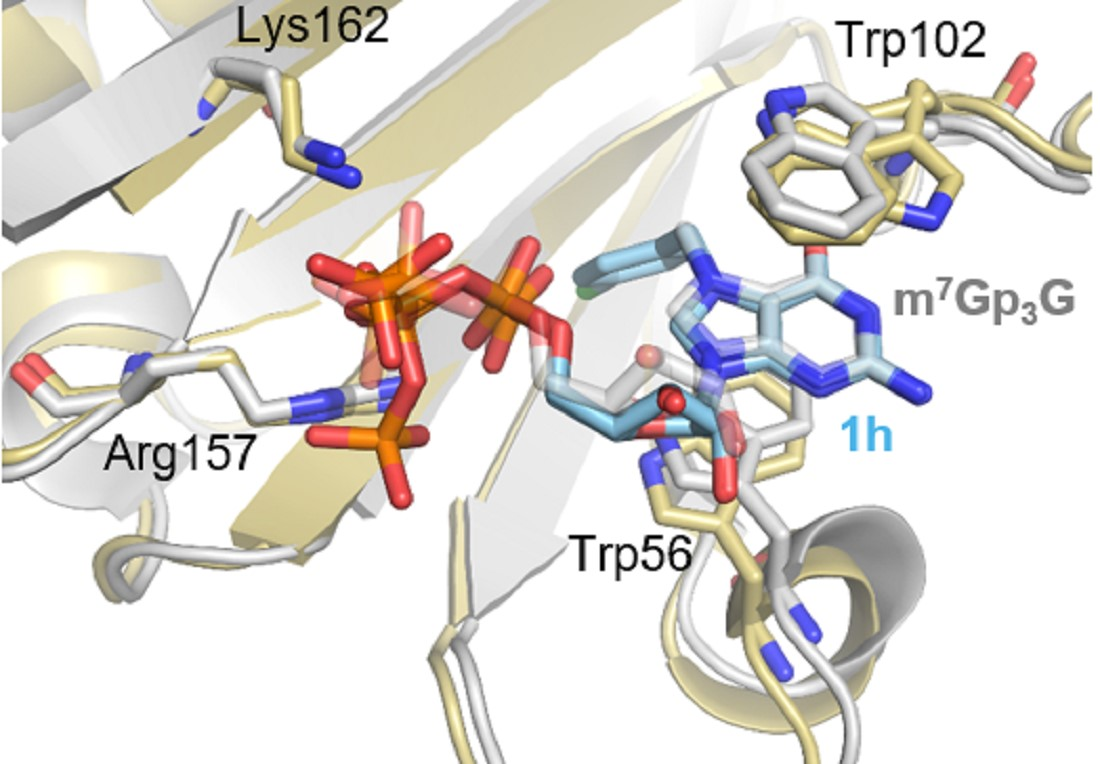
2.5. Crystallization and Structural Characterization of Murine eIF4E Complexes with Bn7GpppG, 3-MeBn7GpppG and 4-Cl-Bn7GpppG
3. Conclusions
4. Materials and Methods
4.1. Starting Materials, Chemical Reagents, Analytical Procedures
4.1.1. Nucleotide Purification via Ion-Exchange Chromatography
4.1.2. Analytical and Preparative Reversed-Phase (RP) HPLC
4.1.3. Spectroscopic Analysis of the Synthesized Compounds
4.2. Dinucleotide Stock Solutions
4.3. General Procedure A (GP-A): Synthesis of N7-Substituted Guanosine 5′-Monophosphate Analogs (X-Bn-GMP)
4.4. General Procedure B (GP-B): Synthesis of Novel Cap Analogs (X-Bn-GpppG)
4.5. Crystallization
4.6. Structure Determination and Refinement
4.7. Susceptibility to DcpS Hydrolysis
4.8. Inhibition of Translation by Cap Analogues in a RRL System
4.9. Determination of the Dissociation Constants When Studying Cap Analog–eIF4E Interactions
4.10. Determination of Translational Efficiency
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ramanathan, A.; Robb, G.B.; Chan, S.H. mRNA capping: Biological functions and applications. Nucleic Acids Res. 2016, 44, 7511–7526. [Google Scholar] [CrossRef] [PubMed]
- Furuichi, Y.; Shatkin, A.J. Viral and cellular mRNA capping: Past and prospects. Adv. Virus Res. 2000, 55, 135–184. [Google Scholar] [CrossRef] [PubMed]
- Warminski, M.; Sikorski, P.J.; Kowalska, J.; Jemielity, J. Applications of Phosphate Modification and Labeling to Study mRNA Caps. Top. Curr. Chem. 2017, 375, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grudzien-Nogalska, E.; Kowalska, J.; Su, W.; Kuhn, A.N.; Slepenkov, S.V.; Darzynkiewicz, E.; Sahin, U.; Jemielity, J.; Rhoads, R.E. Synthetic mRNAs with superior translation and stability properties. Methods Mol. Biol. 2013, 969, 55–72. [Google Scholar] [CrossRef]
- Grudzien-Nogalska, E.; Stepinski, J.; Jemielity, J.; Zuberek, J.; Stolarski, R.; Rhoads, R.E.; Darzynkiewicz, E. Synthesis of anti-reverse cap analogs (ARCAs) and their applications in mRNA translation and stability. Methods Enzymol. 2007, 431, 203–227. [Google Scholar] [CrossRef]
- Grudzien-Nogalska, E.; Jemielity, J.; Kowalska, J.; Darzynkiewicz, E.; Rhoads, R.E. Phosphorothioate cap analogs stabilize mRNA and increase translational efficiency in mammalian cells. RNA 2007, 13, 1745–1755. [Google Scholar] [CrossRef] [Green Version]
- Sahin, U.; Kariko, K.; Tureci, O. mRNA-based therapeutics—Developing a new class of drugs. Nat. Rev. Drug Discov. 2014, 13, 759–780. [Google Scholar] [CrossRef]
- Eberhardt, W.; Doller, A.; Akool, E.S.; Pfedschifter, J. Modulation of mRNA stability as a novel therapeutic approach. Pharmacol. Ther. 2007, 114, 56–73. [Google Scholar] [CrossRef]
- Pal, I.; Safari, M.; Jovanovic, M.; Bates, S.E.; Deng, C.C. Targeting Translation of mRNA as a Therapeutic Strategy in Cancer. Curr. Hematol. Malig. Rep. 2019, 14, 219–227. [Google Scholar] [CrossRef]
- Cai, A.; Jankowska-Anyszka, M.; Centers, A.; Chlebicka, L.; Stepinski, J.; Stolarski, R.; Darzynkiewicz, E.; Rhoads, R.E. Quantitative assessment of mRNA cap analogues as inhibitors of in vitro translation. Biochemistry 1999, 38, 8538–8547. [Google Scholar] [CrossRef]
- Grudzien, E.; Stepinski, J.; Jankowska-Anyszka, M.; Stolarski, R.; Darzynkiewicz, E.; Rhoads, R.E. Novel cap analogs for in vitro synthesis of mRNAs with high translational efficiency. RNA 2004, 10, 1479–1487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wojtczak, B.A.; Sikorski, P.J.; Fac-Dabrowska, K.; Nowicka, A.; Warminski, M.; Kubacka, D.; Nowak, E.; Nowotny, M.; Kowalska, J.; Jemielity, J. 5′-Phosphorothiolate Dinucleotide Cap Analogues: Reagents for Messenger RNA Modification and Potent Small-Molecular Inhibitors of Decapping Enzymes. JACS 2018, 140, 5987–5999. [Google Scholar] [CrossRef] [PubMed]
- Walczak, S.; Sikorski, P.J.; Kasprzyk, R.; Kowalska, J.; Jemielity, J. Exploring the potential of phosphotriazole 5′ mRNA cap analogues as efficient translation initiators. Org. Biomol. Chem. 2018, 16, 6741–6748. [Google Scholar] [CrossRef] [PubMed]
- Piecyk, K.; Lukaszewicz, M.; Darzynkiewicz, E.; Jankowska-Anyszka, M. Triazole-containing monophosphate mRNA cap analogs as effective translation inhibitors. RNA 2014, 20, 1539–1547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleczewska, N.; Sikorski, P.J.; Warminska, Z.; Markiewicz, L.; Kasprzyk, R.; Baran, N.; Kwapiszewska, K.; Karpinska, A.; Michalski, J.; Holyst, R.; et al. Cellular delivery of dinucleotides by conjugation with small molecules: Targeting translation initiation for anticancer applications. Chem. Sci. 2021, 12, 10242–10251. [Google Scholar] [CrossRef]
- Kalek, M.; Jemielity, J.; Darzynkiewicz, Z.M.; Bojarska, E.; Stepinski, J.; Stolarski, R.; Davis, R.E.; Darzynkiewicz, E. Enzymatically stable 5′ mRNA cap analogs: Synthesis and binding studies with human DcpS decapping enzyme. Bioorg. Med. Chem. 2006, 14, 3223–3230. [Google Scholar] [CrossRef]
- Kowalska, J.; Wypijewska del Nogal, A.; Darzynkiewicz, Z.M.; Buck, J.; Nicola, C.; Kuhn, A.N.; Lukaszewicz, M.; Zuberek, J.; Strenkowska, M.; Ziemniak, M.; et al. Synthesis, properties, and biological activity of boranophosphate analogs of the mRNA cap: Versatile tools for manipulation of therapeutically relevant cap-dependent processes. Nucleic Acids Res. 2014, 42, 10245–10264. [Google Scholar] [CrossRef] [Green Version]
- Jemielity, J.; Kowalska, J.; Rydzik, A.; Darzynkiewicz, E. Synthetic mRNA cap analogs with a modified triphosphate bridge—Synthesis, applications and prospects. New J. Chem. 2010, 34, 829–844. [Google Scholar] [CrossRef]
- Rydzik, A.M.; Warminski, M.; Sikorski, P.J.; Baranowski, M.R.; Walczak, S.; Kowalska, J.; Zuberek, J.; Lukaszewicz, M.; Nowak, E.; Claridge, T.D.W.; et al. mRNA cap analogues substituted in the tetraphosphate chain with CX2: Identification of O-to-CCl2 as the first bridging modification that confers resistance to decapping without impairing translation. Nucleic Acids Res. 2017, 45, 8661–8675. [Google Scholar] [CrossRef] [Green Version]
- Rydzik, A.M.; Kulis, M.; Lukaszewicz, M.; Kowalska, J.; Zuberek, J.; Darzynkiewicz, Z.M.; Darzynkiewicz, E.; Jemielity, J. Synthesis and properties of mRNA cap analogs containing imidodiphosphate moiety-fairly mimicking natural cap structure, yet resistant to enzymatic hydrolysis. Bioorg. Med. Chem. 2012, 20, 1699–1710. [Google Scholar] [CrossRef]
- Ferenc, G.; Padar, P.; Szolomajer, J.; Kovacs, L. N-Alkylated Guanine Derivatives. Curr. Org. Chem. 2009, 13, 1085–1135. [Google Scholar] [CrossRef] [Green Version]
- Piecyk, K.; Darzynkiewicz, Z.M.; Jankowska-Anyszka, M.; Ferenc-Mrozek, A.; Stepinski, J.; Darzynkiewicz, E.; Bojarska, E. Effect of different N7 substitution of dinucleotide cap analogs on the hydrolytic susceptibility towards scavenger decapping enzymes (DcpS). Biochem. Biophys. Res. Commun. 2015, 464, 89–93. [Google Scholar] [CrossRef]
- Jemielity, J.; Fowler, T.; Zuberek, J.; Stepinski, J.; Lewdorowicz, M.; Niedzwiecka, A.; Stolarski, R.; Darzynkiewicz, E.; Rhoads, R.E. Novel “anti-reverse” cap analogs with superior translational properties. RNA 2003, 9, 1108–1122. [Google Scholar] [CrossRef] [Green Version]
- Darzynkiewicz, E.; Stepinski, J.; Ekiel, I.; Goyer, C.; Sonenberg, N.; Temeriusz, A.; Jin, Y.X.; Sijuwade, T.; Haber, D.; Tahara, S.M. Inhibition of eukaryotic translation by nucleoside 5′-monophosphate analogs of messenger-RNA 5′-cap—Changes in N7 substituent affect analog activity. Biochemistry 1989, 28, 4771–4778. [Google Scholar] [CrossRef]
- Kasprzyk, R.; Starek, B.J.; Ciechanowicz, S.; Kubacka, D.; Kowalska, J.; Jemielity, J. Fluorescent Turn-On Probes for the Development of Binding and Hydrolytic Activity Assays for mRNA Cap-Recognizing Proteins. Chem. Eur. J. 2019, 25, 6728–6740. [Google Scholar] [CrossRef]
- Nikolovska-Coleska, Z.; Wang, R.X.; Fang, X.L.; Pan, H.G.; Tomita, Y.; Li, P.; Roller, P.P.; Krajewski, K.; Saito, N.G.; Stuckey, J.A.; et al. Development and optimization of a binding assay for the XIAP BIR3 domain using fluorescence polarization. Anal. Biochem. 2004, 332, 261–273. [Google Scholar] [CrossRef] [PubMed]
- Wojtczak, A.; Kasprzyk, R.; Warminski, M.; Ubych, K.; Kubacka, D.; Sikorski, P.J.; Jemielity, J.; Kowalska, J. Evaluation of carboxyfluorescein-labeled 7-methylguanine nucleotides as probes for studying cap-binding proteins by fluorescence anisotropy. Sci. Rep. 2021, 11, 7687–7702. [Google Scholar] [CrossRef] [PubMed]
- Kowalska, J.; Lukaszewicz, M.; Zuberek, J.; Ziemniak, M.; Darzynkiewicz, E.; Jemielity, J. Phosphorothioate analogs of m7GTP are enzymatically stable inhibitors of cap-dependent translation. Bioorganic Med. Chem. Lett. 2009, 19, 1921–1925. [Google Scholar] [CrossRef] [PubMed]
- Niedzwiecka, A.; Marcotrigiano, J.; Stepinski, J.; Jankowska-Anyszka, M.; Wyslouch-Cieszynska, A.; Dadlez, M.; Gingras, A.; Mak, P.; Darzynkiewicz, E.; Sonenberg, N.; et al. Biophysical studies of eIF4E cap-binding protein: Recognition of mRNA 5′ cap structure and synthetic fragments of eIF4G and 4E-BP1 proteins. J. Mol. Biol. 2002, 319, 615–635. [Google Scholar] [CrossRef]
- Volpon, L.; Osborne, M.J.; Topisirovic, I.; Siddiqui, N.; Borden, K.L.B. Cap-free structure of eIF4E suggests a basis for conformational regulation by its ligands. EMBO J. 2006, 25, 5138–5149. [Google Scholar] [CrossRef]
- Brown, C.J.; Verma, C.S.; Walkinshaw, M.D.; Lane, D.P. Crystallization of eIF4E complexed with eIF4GI peptide and glycerol reveals distinct structural differences around the cap-binding site. Cell Cycle 2009, 8, 1905–1911. [Google Scholar] [CrossRef] [Green Version]
- Lama, D.; Pradhan, M.R.; Brown, C.J.; Eapen, R.S.; Joseph, T.L.; Kwoh, C.K.; Lane, D.P.; Verma, C.S. Water-Bridge Mediates Recognition of mRNA Cap in eIF4E. Structure 2017, 25, 188–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, C.J.; McNae, I.; Fischer, P.M.; Walkinshaw, M.D. Crystallographic and mass spectrometric characterisation of elF4E with N-7-alkylated cap derivatives. J. Mol. Biol. 2007, 372, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Chiu, T.L.; Amin, E.A.; Polunovsky, V.; Bitterman, P.B.; Wagner, C.R. Design, synthesis and evaluation of analogs of initiation factor 4E (eIF4E) cap-binding antagonist Bn-7-GMP. Eur. J. Med. Chem. 2010, 45, 1304–1313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warminski, M.; Kowalska, J.; Nowak, E.; Kubacka, D.; Tibble, R.; Kasprzyk, R.; Sikorski, P.J.; Gross, J.D.; Nowotny, M.; Jemielity, J. Structural Insights into the Interaction of Clinically Relevant Phosphorothioate mRNA Cap Analogs with Translation Initiation Factor 4E Reveal Stabilization via Electrostatic Thio-Effect. ACS Chem. Biol. 2021, 16, 334–343. [Google Scholar] [CrossRef]
- Kabsch, W. XDS. Acta Crystallogr. D 2010, 66, 125–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mccoy, A.; Grosse-Kunstleve, R.; Adams, P.; Winn, M.; Storoni, L.; Read, R. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. [Google Scholar] [CrossRef] [Green Version]
- Schuttelkopf, A.W.; van Aalten, D.M.F. PRODRG: A tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr. D 2004, 60, 1355–1363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. D 2010, 66, 486–501. [Google Scholar] [CrossRef] [Green Version]
- Adams, P.; Afonine, P.; Bunkoczi, G.; Chen, V.; Davis, I.; Echols, N.; Headd, J.; Hung, L.; Kapral, G.; Grosse-Kunstleve, R.; et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D 2010, 66, 213–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, C.J.; Headd, J.J.; Moriarty, N.W.; Prisant, M.G.; Videau, L.L.; Deis, L.N.; Verma, V.; Keedy, D.A.; Hintze, B.J.; Chen, V.B.; et al. MolProbity: More and better reference data for improved all-atom structure validation. Protein Sci. 2018, 27, 293–315. [Google Scholar] [CrossRef] [PubMed]








 | ||||
|---|---|---|---|---|
| Entry | Starting Material | Product | R2 | Yield % a (%) b |
| m7GpppG analogs (R1 = H) | ||||
| 1 | 4a | 1a |  | 51 (83) |
| 2 | 4b | 1b |  | 39 (82) |
| 3 | 4c | 1c |  | 36 (58) |
| 4 | 4d | 1d |  | 58 (76) |
| 5 | 4e | 1e |  | 42 (74) |
| 6 | 4f | 1f |  | 62 (84) |
| 7 | 4g | 1g |  | 44 (82) |
| 8 | 4h | 1h |  | 22 (75) |
| 9 | 4i | 1i |  | 56 (88) |
| 10 | 4j | 1j |  | 36 (80) |
| 11 | 4k | 1k |  | 41 (66) |
| 12 | 4l | 1l |  | 27 (70) |
| 13 | 4m | 1m |  | 22 (81) |
| 14 | 4n | 1n |  | 43 (90) |
| 15 | 4o | 1o |  | 57 (89) |
| 16 | 4p | 1p |  | 45 (81) |
| Anti-reverse cap analogs (ARCAs) (R1 = CH3) | ||||
| 17 | 4q | 1q |  | 33 (49) |
| 18 | 4r | 1r |  | 42 (86) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wojcik, R.; Baranowski, M.R.; Markiewicz, L.; Kubacka, D.; Bednarczyk, M.; Baran, N.; Wojtczak, A.; Sikorski, P.J.; Zuberek, J.; Kowalska, J.; et al. Novel N7-Arylmethyl Substituted Dinucleotide mRNA 5′ cap Analogs: Synthesis and Evaluation as Modulators of Translation. Pharmaceutics 2021, 13, 1941. https://doi.org/10.3390/pharmaceutics13111941
Wojcik R, Baranowski MR, Markiewicz L, Kubacka D, Bednarczyk M, Baran N, Wojtczak A, Sikorski PJ, Zuberek J, Kowalska J, et al. Novel N7-Arylmethyl Substituted Dinucleotide mRNA 5′ cap Analogs: Synthesis and Evaluation as Modulators of Translation. Pharmaceutics. 2021; 13(11):1941. https://doi.org/10.3390/pharmaceutics13111941
Chicago/Turabian StyleWojcik, Radoslaw, Marek R. Baranowski, Lukasz Markiewicz, Dorota Kubacka, Marcelina Bednarczyk, Natalia Baran, Anna Wojtczak, Pawel J. Sikorski, Joanna Zuberek, Joanna Kowalska, and et al. 2021. "Novel N7-Arylmethyl Substituted Dinucleotide mRNA 5′ cap Analogs: Synthesis and Evaluation as Modulators of Translation" Pharmaceutics 13, no. 11: 1941. https://doi.org/10.3390/pharmaceutics13111941