
Microfluidic Production of Polymeric Core-Shell Microspheres for the Delayed Pulsatile Release of Bovine Serum Albumin as a Model Antigen

 and
and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. FITC-BSA Synthesis and Analysis
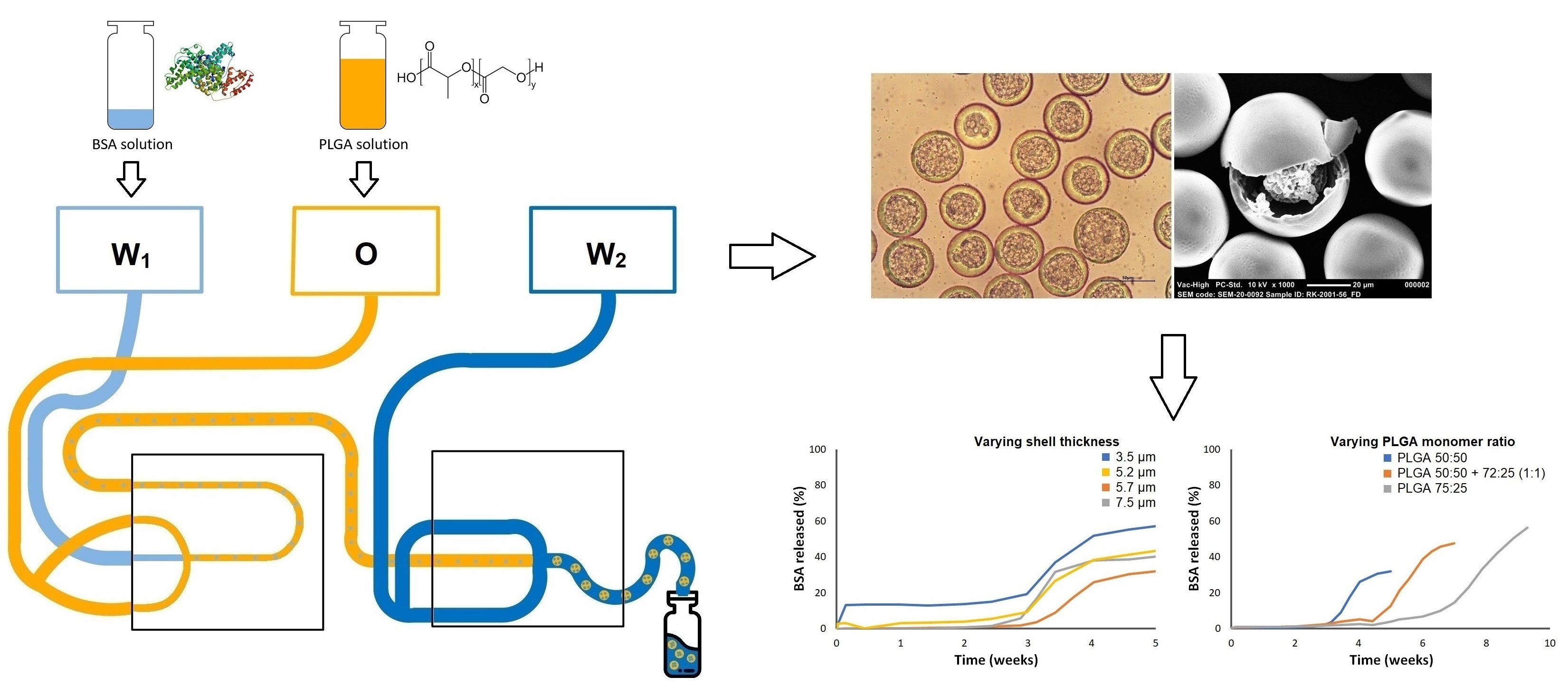
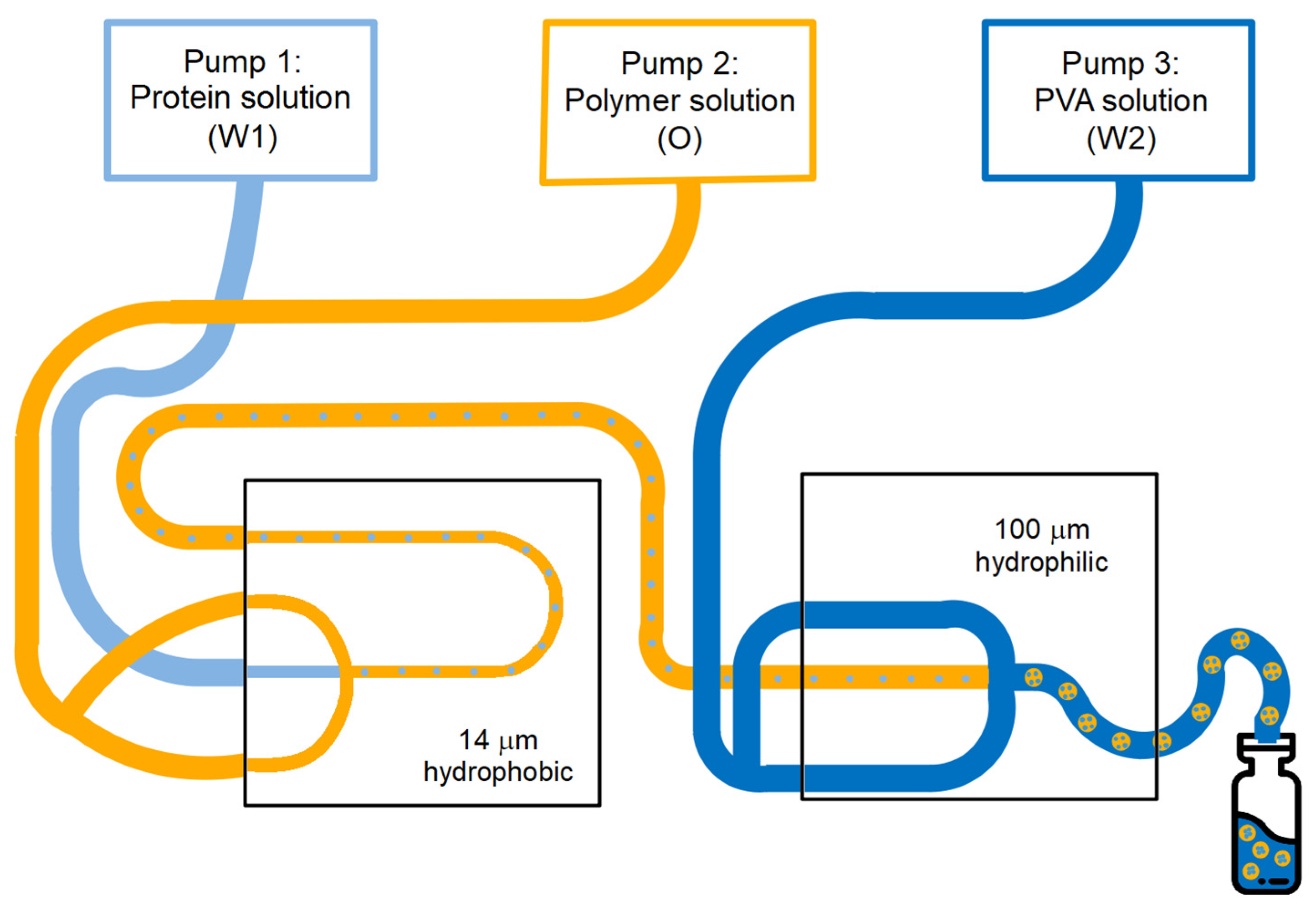
2.3. Production of Core-Shell Microspheres
2.4. Characterization of Particle Size and Morphology
2.5. FITC-BSA Localization Analysis
2.6. BSA Loading Assay
2.7. BSA In Vitro Release Assay
2.8. Statistics
3. Results and Discussion
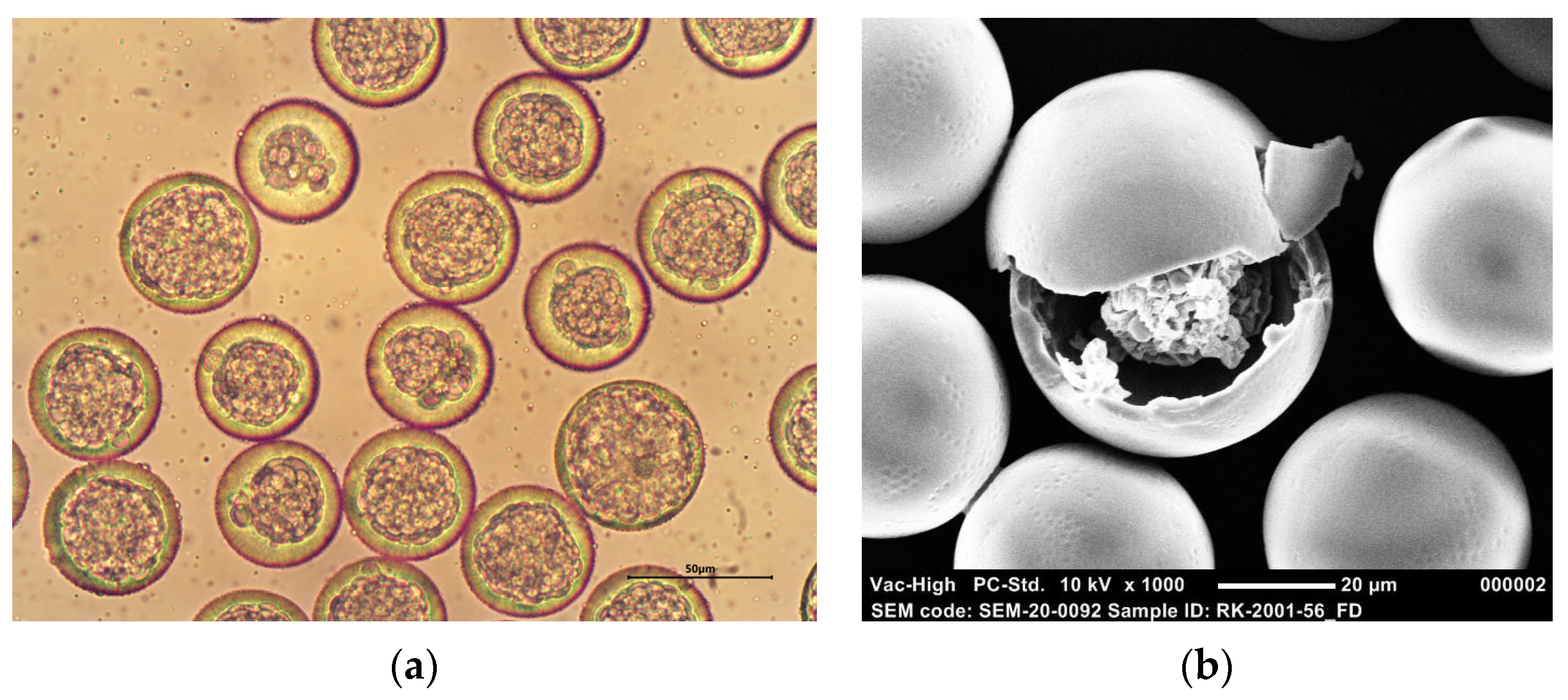
3.1. Production and Characterization of Monodisperse BSA-Loaded Core-Shell Microspheres
3.2. Effect of Production Process and Formulation Parameters on Particle Characteristics
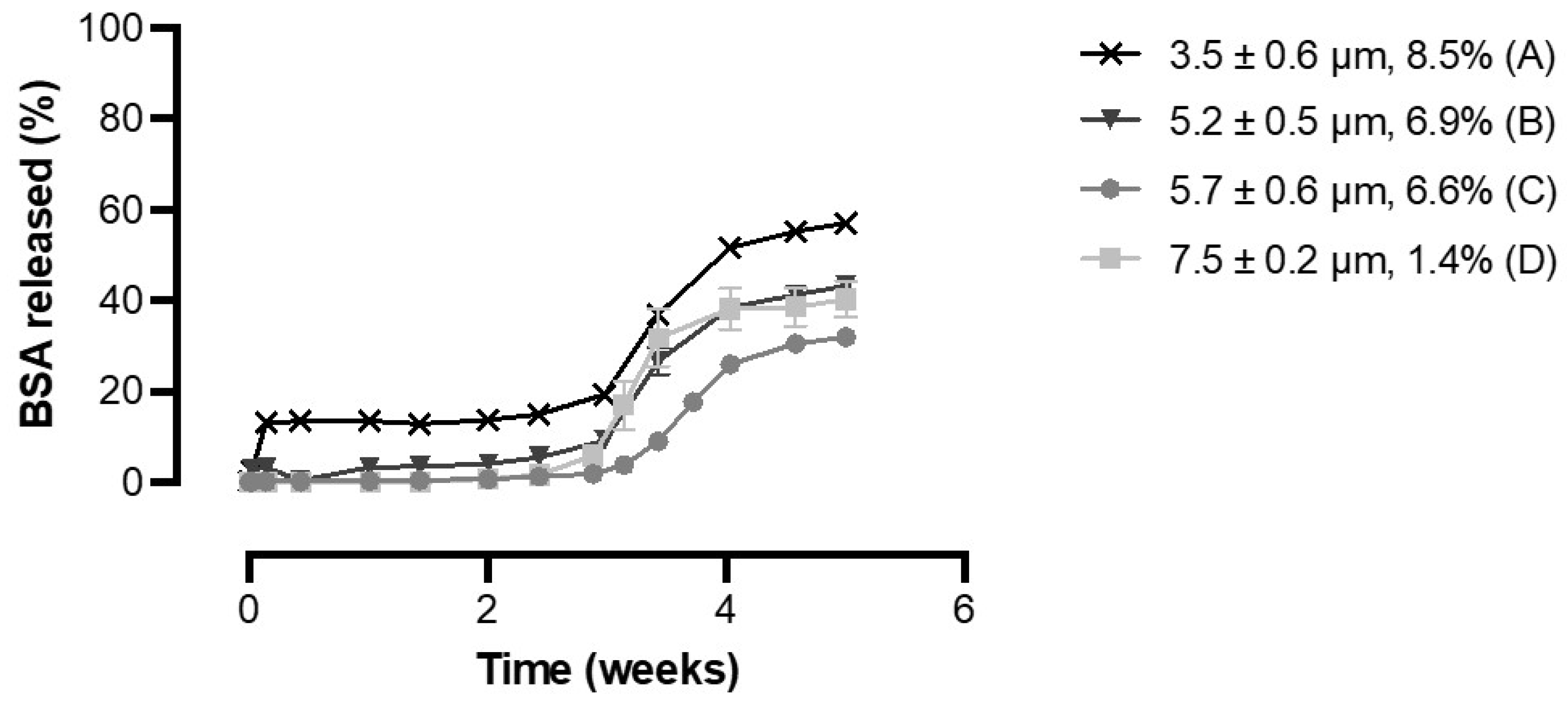
3.3. Effect of BSA Loading and Shell Thickness on the In Vitro Release of BSA from PDLG5002-Based Core-Shell Microspheres
3.4. Particle Morphology of PDLG5002-Based Core-Shell Microspheres during BSA In Vitro Release
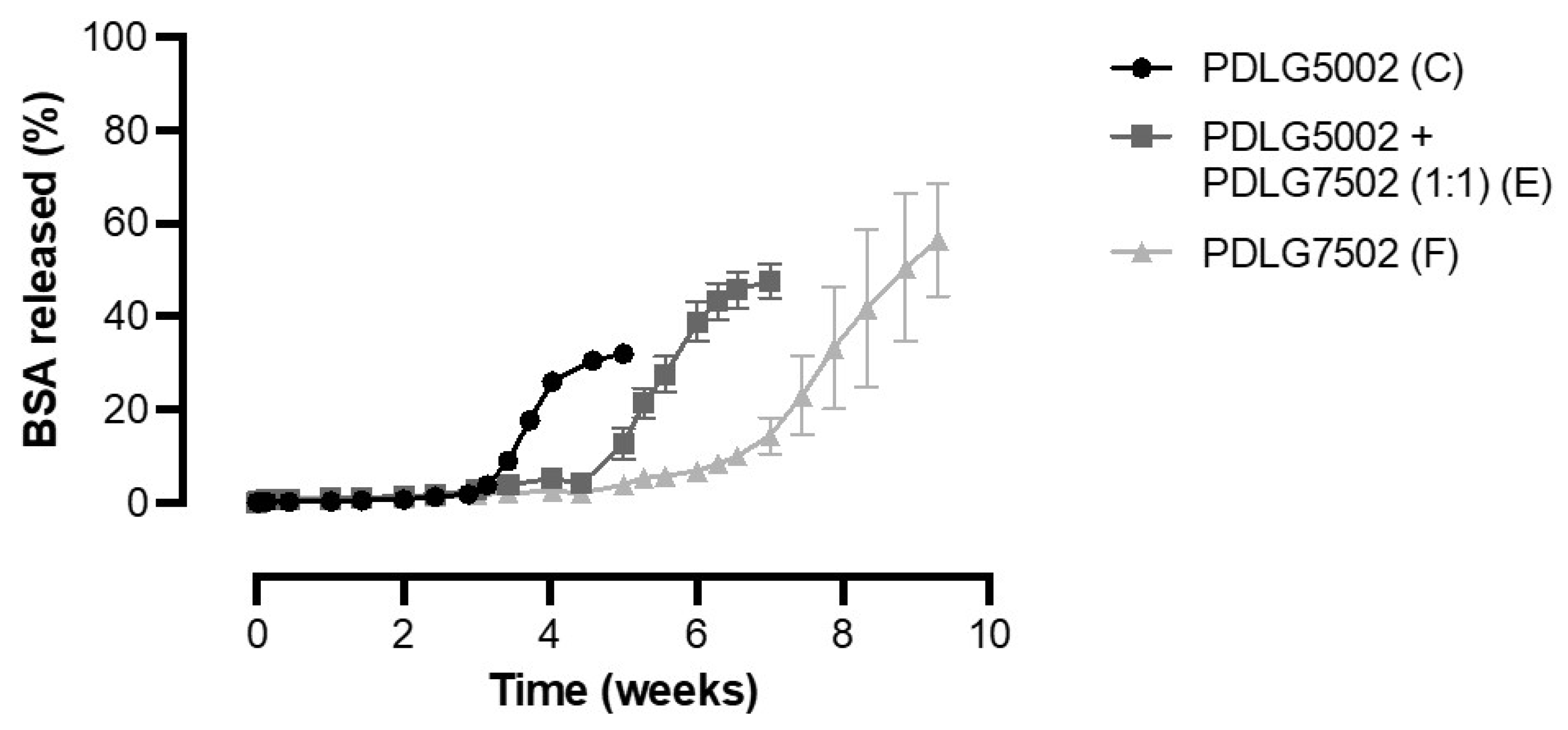
3.5. Effect of PLGA Monomer Ratio on the In Vitro Release of BSA from PLGA-Based Core-Shell Microspheres
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- WHO. Vaccines and Immunization. 2021. Available online: https://www.who.int/health-topics/vaccines-and-immuniztion (accessed on 29 September 2021).
- WHO. Immunization Coverage. 2021. Available online: https://www.who.int/news-room/fact-sheets/detail/immunization-coverage (accessed on 29 September 2021).
- Galles, N.C.; Liu, P.Y.; Updike, R.L.; Fullman, N.; Nguyen, J.; Rolfe, S.; Marks, A.; Abdoli, A.; Akalu, Y.; Gaidhane, S.; et al. Measuring routine childhood vaccination coverage in 204 countries and territories, 1980–2019: A systematic analysis for the global burden of disease study 2020, release 1. Lancet 2021, 398, 503–521. [Google Scholar] [CrossRef]
- WHO; UNICEF. Progress and Challenges with Achieving Universal Immunization Coverage. 2019. Available online: https://www.who.int/immunization/monitoring_surveillance/who-immuniz.pdf?ua=1 (accessed on 29 September 2021).
- McHugh, K.J.; Guarecuco, R.; Langer, R.; Jaklenec, A. Single-injection vaccines: Progress, challenges, and opportunities. J. Control. Release 2015, 219, 596–609. [Google Scholar] [CrossRef] [PubMed]
- Cleland, J.L. Single-administration vaccines: Controlled-release technology to mimic repeated immunizations. Trends Biotechnol. 1999, 17, 25–29. [Google Scholar] [CrossRef]
- Beugeling, M.; Grasmeijer, N.; Born, P.A.; Van Der Meulen, M.; van der Kooij, R.; Schwengle, K.; Baert, L.; Amssoms, K.; Frijlink, H.W.; Hinrichs, W.L. The mechanism behind the biphasic pulsatile drug release from physically mixed poly(DL-lactic(-co-glycolic) acid)-based compacts. Int. J. Pharm. 2018, 551, 195–202. [Google Scholar] [CrossRef]
- Amssoms, K.; Born, P.A.; Beugeling, M.; De Clerck, B.; Van Gulck, E.; Hinrichs, W.L.J.; Frijlink, H.W.; Grasmeijer, N.; Kraus, G.; Sutmuller, R.; et al. Ovalbumin-containing core-shell implants suitable to obtain a delayed IgG1 antibody response in support of a biphasic pulsatile release profile in mice. PLoS ONE 2018, 13, e0202961. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.M.; Shive, M.S. Biodegradation and biocompatibility of PLA and PLGA microspheres. Adv. Drug Deliv. Rev. 1997, 28, 5–24. [Google Scholar] [CrossRef]
- Lu, L.; Garcia, C.A.; Mikos, A.G. In vitro degradation of thin poly(DL-lactic-co-glycolic acid) films. J. Biomed. Mater. Res. 1999, 46, 236–244. [Google Scholar] [CrossRef]
- Fredenberg, S.; Wahlgren, M.; Reslow, M.; Axelsson, A. The mechanisms of drug release in poly(lactic-co-glycolic acid)-based drug delivery systems—A review. Int. J. Pharm. 2011, 415, 34–52. [Google Scholar] [CrossRef]
- Makadia, H.K.; Siegel, S.J. Poly Lactic-co-Glycolic Acid (PLGA) as biodegradable controlled drug delivery carrier. Polymers 2011, 3, 1377–1397. [Google Scholar] [CrossRef] [PubMed]
- Galogahi, F.M.; Zhu, Y.; An, H.; Nguyen, N.-T. Core-shell microparticles: Generation approaches and applications. J. Sci. Adv. Mater. Devices 2020, 5, 417–435. [Google Scholar] [CrossRef]
- Lemperle, G. Biocompatibility of injectable microspheres. Biomed. J. Sci. Tech. Res. 2018, 2, 2296–2306. [Google Scholar] [CrossRef] [Green Version]
- Ye, M.; Kim, S.; Park, K. Issues in long-term protein delivery using biodegradable microparticles. J. Control. Release 2010, 146, 241–260. [Google Scholar] [CrossRef]
- Chen, X.; Ooi, C.P.; Lim, T.H. Effect of ganciclovir on the hydrolytic degradation of poly(lactide-co-glycolide) microspheres. J. Biomater. Appl. 2006, 20, 287–302. [Google Scholar] [CrossRef] [Green Version]
- Xia, Y.; Pack, D.W. Pulsatile protein release from monodisperse liquid-core microcapsules of controllable shell thickness. Pharm. Res. 2014, 31, 3201–3210. [Google Scholar] [CrossRef] [Green Version]
- Pollauf, E.J.; Kim, K.K.; Pack, D.W. Small-molecule release from poly(D,L-Lactide)/Poly(D,L-lactide-co-glycolide) composite microparticles. J. Pharm. Sci. 2005, 94, 2013–2022. [Google Scholar] [CrossRef]
- Zheng, W. A water-in-oil-in-oil-in-water (W/O/O/W) method for producing drug-releasing, double-walled microspheres. Int. J. Pharm. 2009, 374, 90–95. [Google Scholar] [CrossRef] [PubMed]
- The, S.-Y.; Lin, R.; Hung, L.-H.; Lee, A.P. Droplet microfluidics. Lab Chip 2008, 8, 198–220. [Google Scholar]
- Shah, R.K.; Shum, H.C.; Rowat, A.C.; Lee, D.; Agresti, J.J.; Utada, A.S.; Chu, L.-Y.; Kim, J.-W.; Fernandez-Nieves, A.; Martinez, C.; et al. Designer emulsions using microfluidics. Mater. Today 2008, 11, 18–27. [Google Scholar] [CrossRef]
- Kim, S.W.; Hwangbo, K.-H.; Lee, J.H.; Cho, K.Y. Microfluidic fabrication of microparticles with multiple structures from a biodegradable polymer blend. RSC Adv. 2014, 4, 46536–46540. [Google Scholar] [CrossRef]
- Choi, S.-W.; Zhang, Y.S.; Xia, Y. Fabrication of microbeads with a controllable hollow interior and porous wall using a capillary fluidic device. Adv. Funct. Mater. 2009, 19, 2943–2949. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Dong, H.; Tang, G.; Ma, T.; Cao, X. Controllable microfluidic fabrication of Janus and microcapsule particles for drug delivery applications. RSC Adv. 2015, 5, 23181–23188. [Google Scholar] [CrossRef]
- Kong, T.; Wu, J.; Yeung, K.; To, M.K.T.; Wang, L. Microfluidic fabrication of polymeric core-shell microspheres for controlled release applications. Biomicrofluidics 2013, 7, 44128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tu, F.; Lee, D. Controlling the stability and size of double-emulsion-templated poly(lactic-co-glycolic) acid microcapsules. Langmuir 2012, 28, 9944–9952. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, A.; Gupta, R.K.; Alonso, M.J.; Siber, G.R.; Langer, R. Pulsed controlled-release system for potential use in vaccine delivery. J. Pharm. Sci. 1996, 85, 547–552. [Google Scholar] [CrossRef]
- Berkland, C.; Pollauf, E.; Varde, N.; Pack, D.W.; Kim, K. Monodisperse liquid-filled biodegradable microcapsules. Pharm. Res. 2007, 24, 1007–1013. [Google Scholar] [CrossRef] [PubMed]
- Zamani, M.; Prabhakaran, M.P.; Thian, E.S.; Ramakrishna, S. Protein encapsulated core-shell structured particles prepared by coaxial electrospraying: Investigation on material and processing variables. Int. J. Pharm. 2014, 473, 134–143. [Google Scholar] [CrossRef]
- Beugeling, M.; Amssoms, K.; Cox, F.; De Clerck, B.; Van Gulck, E.; Verwoerd, J.A.; Kraus, G.; Roymans, D.; Baert, L.; Frijlink, H.W.; et al. Development of a stable respiratory syncytial virus pre-fusion protein powder suitable for a core-shell implant with a delayed release in mice: A proof of concept study. Pharmaceutics 2019, 11, 510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berkland, C.; Pollauf, E.; Pack, D.W.; Kim, K. Uniform double-walled polymer microspheres of controllable shell thickness. J. Control. Release 2004, 96, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Han, F.Y.; Thurecht, K.J.; Whittaker, A.K.; Smith, M.T. Bioerodable PLGA-based microparticles for producing sustained-release drug formulations and strategies for improving drug loading. Front. Pharmacol. 2016, 7, 185. [Google Scholar] [CrossRef] [Green Version]
- Varde, N.K.; Pack, D.W. Microspheres for controlled release drug delivery. Expert Opin. Biol. Ther. 2004, 4, 35–51. [Google Scholar] [CrossRef]
- Ghalanbor, Z.; Koerber, M.; Bodmeier, R. Interdependency of protein-release completeness and polymer degradation in PLGA-based implants. Eur. J. Pharm. Biopharm. 2013, 85, 624–630. [Google Scholar] [CrossRef] [PubMed]
- Duque, L.; Koerber, M.; Bodmeier, R. Improving release completeness from PLGA-based implants for the acid-labile model protein ovalbumin. Int. J. Pharm. 2018, 538, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.; Mallery, S.R.; Schwendeman, S.P. Stabilization of proteins encapsulated in injectable poly (lactide- co-glycolide). Nat. Biotechnol. 2000, 18, 52–57. [Google Scholar] [CrossRef] [PubMed]
- van de Weert, M.; Hennink, W.E.; Jiskoot, W. Protein instability in poly(lactic-co-glycolic acid) microparticles. Pharm. Res. 2000, 17, 1159–1167. [Google Scholar] [CrossRef] [PubMed]
- Fu, K.; Klibanov, A.M.; Langer, R. Protein stability in controlled-release systems. Nat. Biotechnol. 2000, 18, 24–25. [Google Scholar] [CrossRef] [PubMed]
- Phua, K.K.L.; Roberts, E.R.H.; Leong, K.W. Degradable polymers. In Comprehensive Biomaterials; Ducheyne, P., Healy, K.E., Hutmacher, D.W., Grainger, D.W., Kirkpatrick, C.J., Eds.; Elsevier Ltd.: Amsterdam, The Netherlands, 2011; Volume 1, pp. 381–415. [Google Scholar]
- Kamaly, N.; Yameen, B.; Wu, J.; Farokhzad, O.C. Degradable controlled-release polymers and polymeric nanoparticles: Mechanisms of controlling drug release. Chem. Rev. 2016, 116, 2602–2663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, Y.; Pack, D.W. Uniform biodegradable microparticle systems for controlled release. Chem. Eng. Sci. 2015, 125, 129–143. [Google Scholar] [CrossRef] [Green Version]
- Centers for Disease Control and Prevention (CDC). Different COVID-19 Vaccines. 2021. Available online: https://www.cdc.gov/coronavirus/2019-ncov/vaccines/different-vaccines.html (accessed on 15 October 2021).
- Guse, C.; Koennings, S.; Blunk, T.; Siepmann, J.; Goepferich, A. Programmable implants—From pulsatile to controlled release. Int. J. Pharm. 2006, 314, 161–169. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulation | Model Compound | Polymer | Polymer Concentration (wt.%) | Flow Rates (W1–O–W2, µL/min) | Theoretical Loading (wt.%) |
|---|---|---|---|---|---|
| A | BSA | PDLG5002 | 10 | 0.58–7.8–50 | 9.3 |
| B | BSA | PDLG5002 | 10 | 0.47–7.8–50 | 7.7 |
| C | BSA | PDLG5002 | 10 | 0.40–7.8–50 | 6.7 |
| D | BSA | PDLG5002 | 10 | 0.35–7.8–50 | 5.9 |
| E | BSA | PDLG5002 + PDLG7502 (1:1) | 7.5 | 0.28–7.8–30 | 6.4 |
| F | BSA | PDLG7502 | 7.5 | 0.28–7.8–30 | 6.4 |
| G | FITC-BSA | PDLG5002 | 10 | 0.20–5.4–40 | 1.0 |
| Formulation | Actual Loading (wt.%) | EE (%) | d50particle (µm) | CVparticle (%) | d50core (µm) | CVcore (%) | Shell Thickness (µm) |
|---|---|---|---|---|---|---|---|
| A | 8.46 1 | 90.56 1 | 48.2 ± 1.8 | 3.8 | 41.3 ± 1.7 | 4.2 | 3.5 ± 0.6 |
| B | 6.91 ± 0.01 | 89.87 ± 0.17 | 43.4 ± 0.8 | 1.8 | 33.0 ± 1.2 | 3.6 | 5.2 ± 0.5 |
| C | 6.60 ± 0.06 | 98.48 ± 0.94 | 40.8 ± 1.2 | 2.9 | 29.5 ± 1.5 | 5.0 | 5.7 ± 0.6 |
| D | 1.37 ± 0.02 | 23.01 ± 0.41 | 38.1 ± 0.7 | 1.7 | 23.1 ± 0.6 | 2.6 | 7.4 ± 0.2 |
| E | 4.95 ± 0.43 | 77.74 ± 6.68 | 37.1 ± 2.8 | 7.6 | 28.5 ± 2.8 | 9.9 | 4.6 ± 1.2 |
| F | 5.73 ± 0.07 | 90.07 ± 1.03 | 46.0 ± 1.9 | 4.2 | 34.9 ± 3.9 | 11.2 | 6.3 ± 1.5 |
| G | 0.87 ± 0.04 | 86.72 ± 3.95 | 46.1 ± 2.8 | 6.1 | 35.0 ± 4.5 | 12.7 | 5.8 ± 0.9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
van der Kooij, R.S.; Steendam, R.; Zuidema, J.; Frijlink, H.W.; Hinrichs, W.L.J. Microfluidic Production of Polymeric Core-Shell Microspheres for the Delayed Pulsatile Release of Bovine Serum Albumin as a Model Antigen. Pharmaceutics 2021, 13, 1854. https://doi.org/10.3390/pharmaceutics13111854
van der Kooij RS, Steendam R, Zuidema J, Frijlink HW, Hinrichs WLJ. Microfluidic Production of Polymeric Core-Shell Microspheres for the Delayed Pulsatile Release of Bovine Serum Albumin as a Model Antigen. Pharmaceutics. 2021; 13(11):1854. https://doi.org/10.3390/pharmaceutics13111854
Chicago/Turabian Stylevan der Kooij, Renée S., Rob Steendam, Johan Zuidema, Henderik W. Frijlink, and Wouter L. J. Hinrichs. 2021. "Microfluidic Production of Polymeric Core-Shell Microspheres for the Delayed Pulsatile Release of Bovine Serum Albumin as a Model Antigen" Pharmaceutics 13, no. 11: 1854. https://doi.org/10.3390/pharmaceutics13111854