
RNA-Based Therapeutics: Current Developments in Targeted Molecular Therapy of Triple-Negative Breast Cancer


Abstract
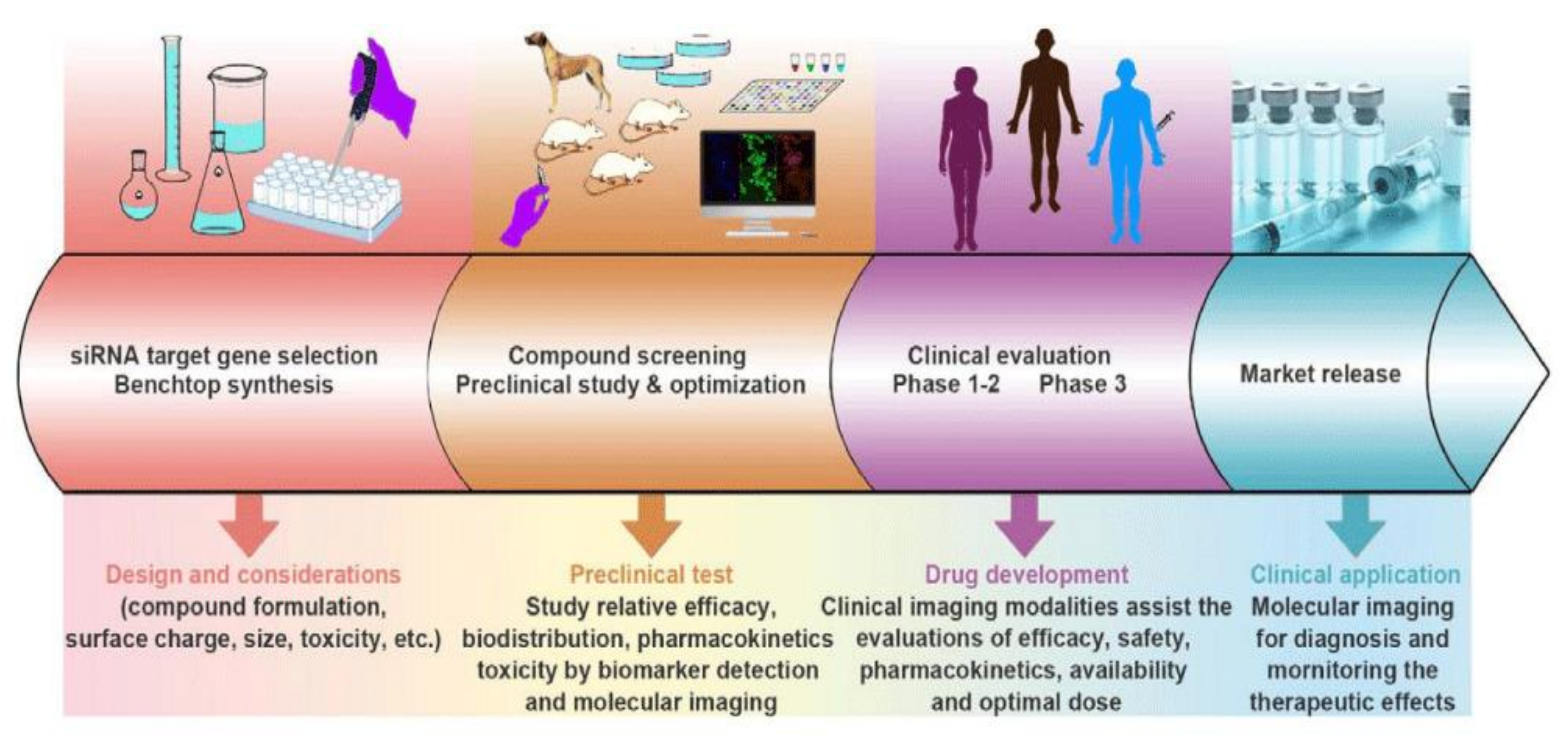
:1. Introduction
1.1. Triple-Negative Breast Cancer
1.1.1. Clinical Significance of TNBC
1.1.2. Molecular Pathogenesis and Description of Key Molecular Targets
1.1.3. Current Therapies
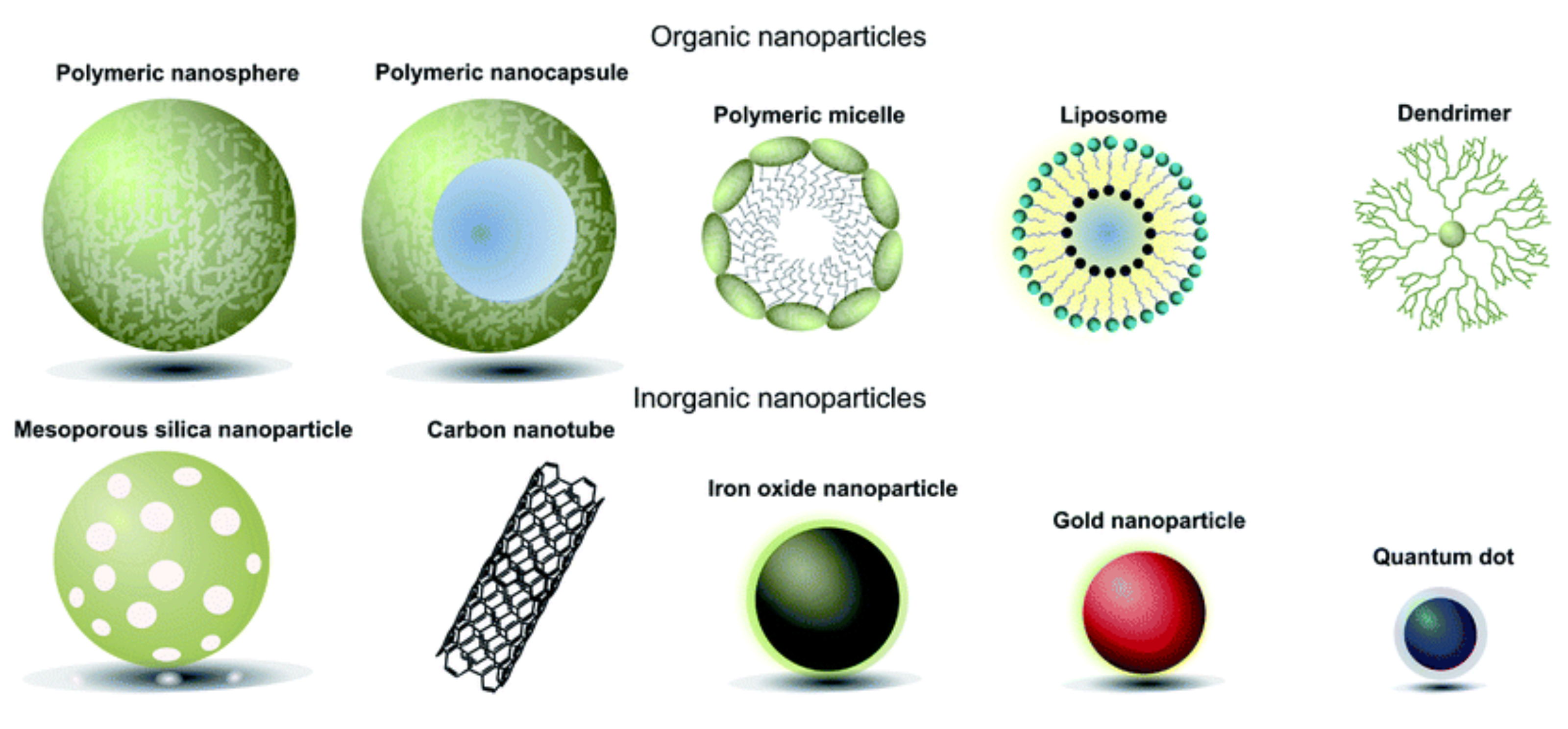
1.2. Gene Therapies Using Non-Coding RNAs
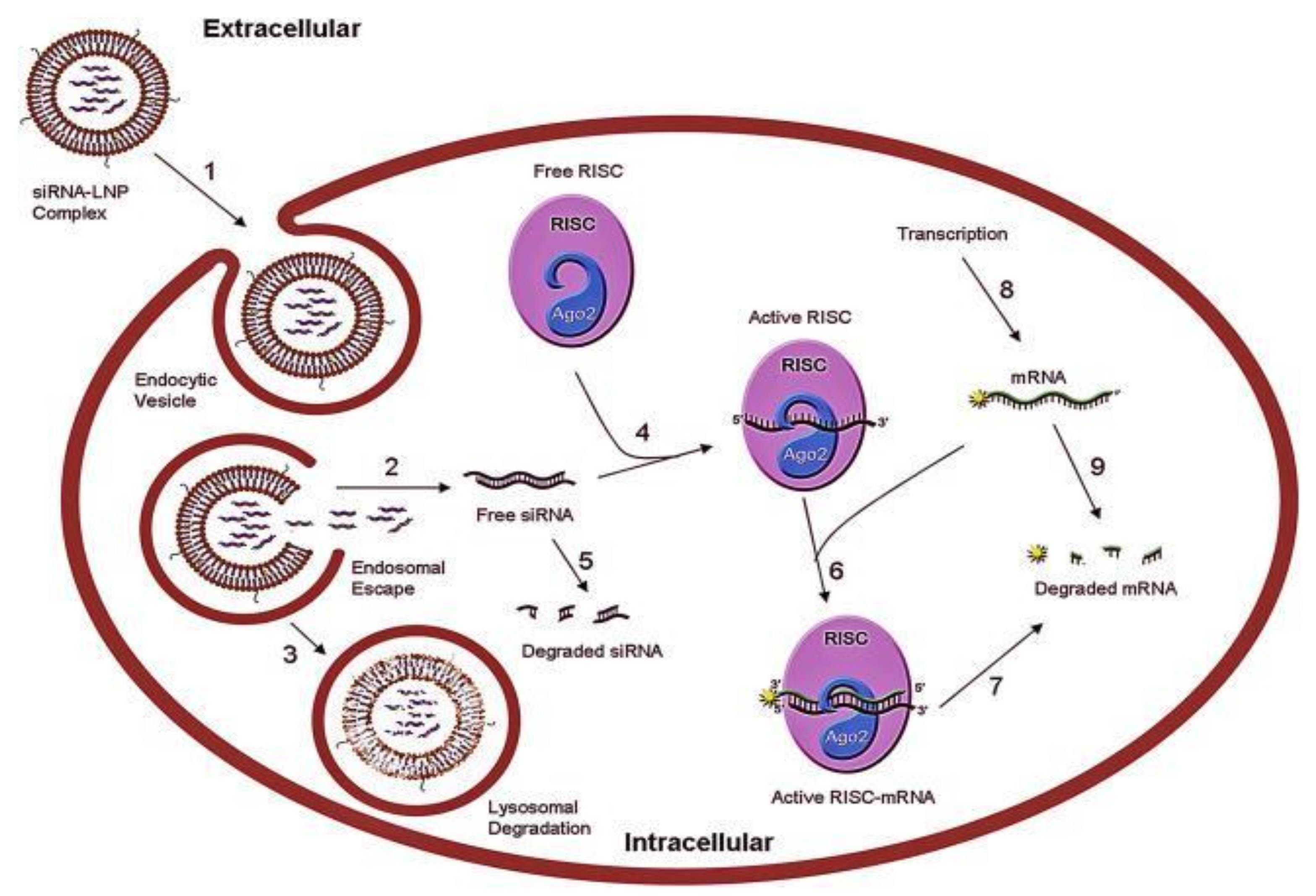
RNA-Interference Therapies
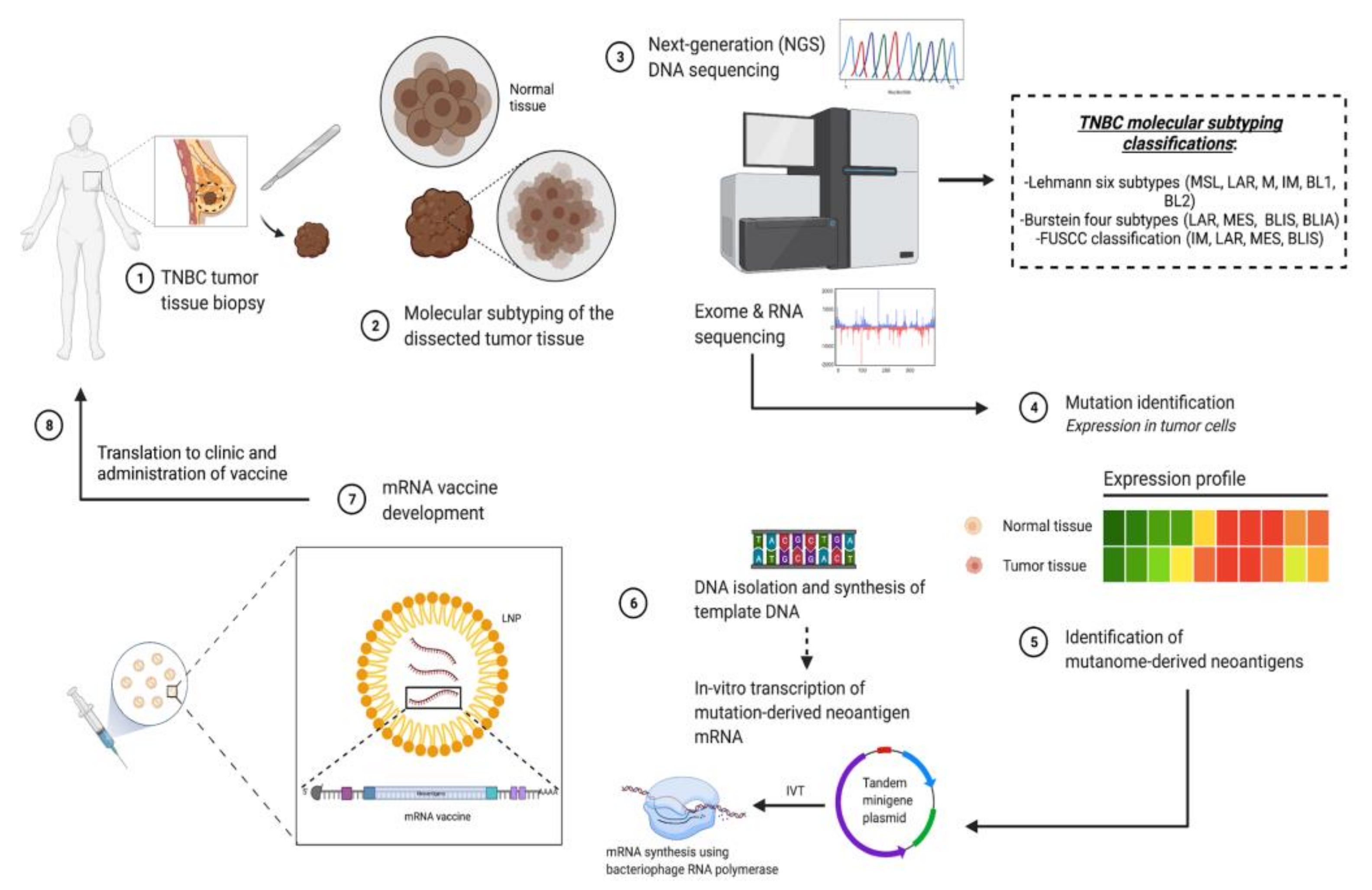
1.3. RNA-Immunotherapy
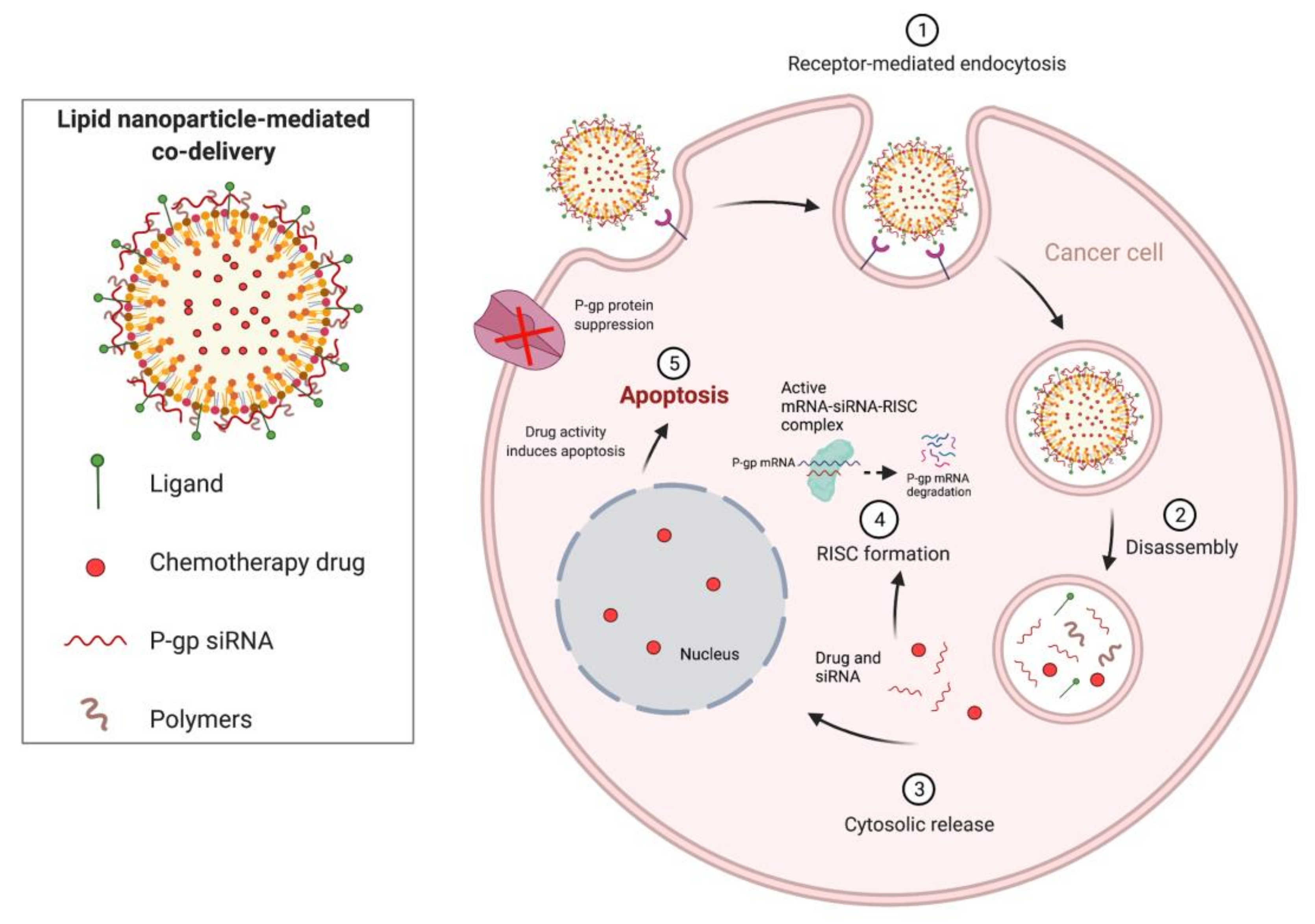
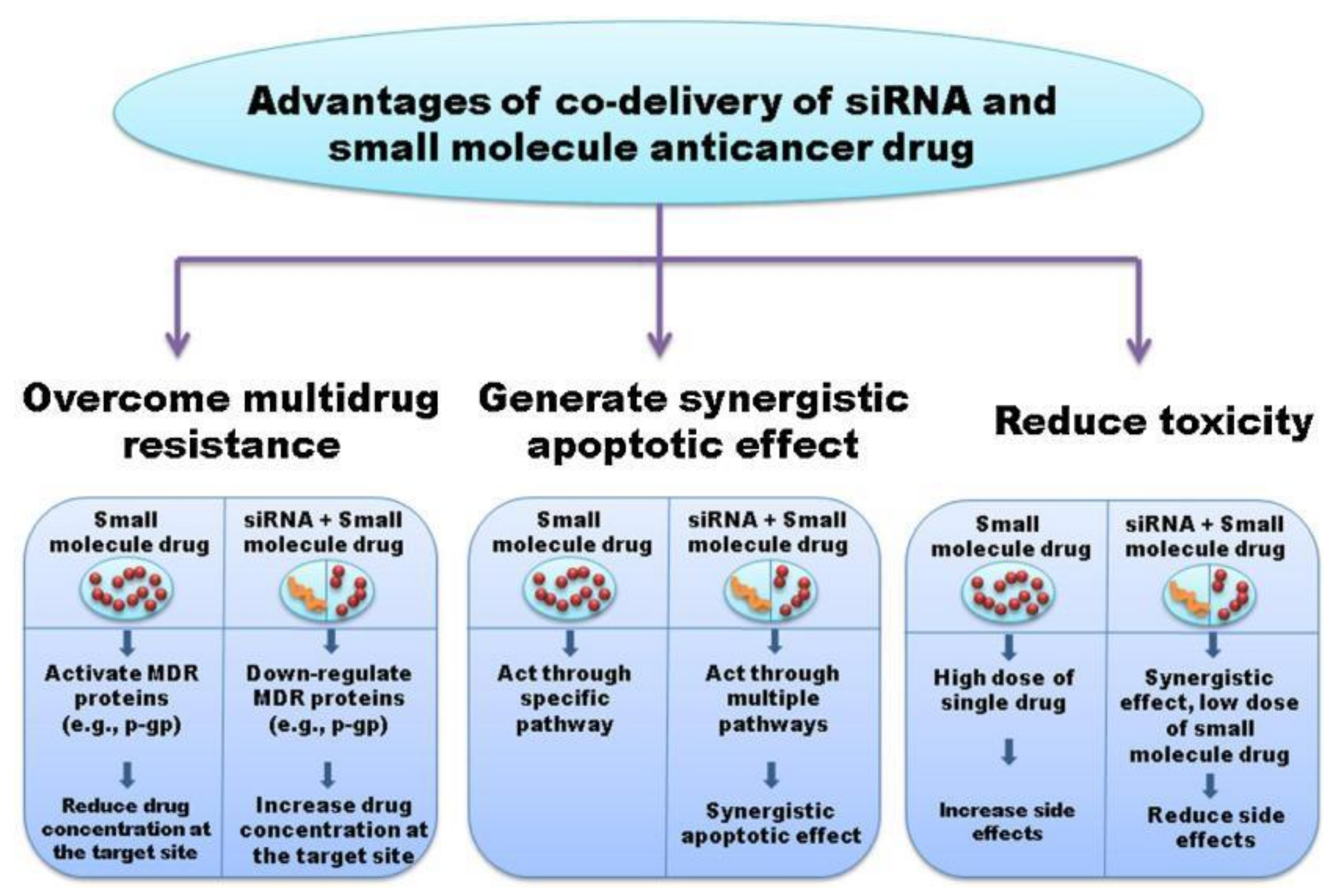
1.4. Combination Therapies Using Chemotherapeutics and RNA-Based Therapies
2. Therapeutic Applications of RNA-Based Methods for Treatment of TNBC
2.1. Pre-Clinical Progress of RNA-Based Therapies
2.2. Current Clinical Trials Using RNA-Based Therapies
3. Future Perspectives and Conclusions
3.1. Ongoing Challenges with siRNA, miRNA, and mRNA Therapies
3.2. Future Use in TNBC Molecular Therapy
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prat, A.; Parker, J.S.; Karginova, O.; Fan, C.; Livasy, C.; Herschkowitz, J.I.; He, X.; Perou, C.M. Phenotypic and Molecular Characterization of the Claudin-Low Intrinsic Subtype of Breast Cancer. Breast Cancer Res. 2010, 12, R68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torre, L.A.; Islami, F.; Siegel, R.L.; Ward, E.M.; Jemal, A. Global Cancer in Women: Burden and Trends. Cancer Epidemiol. Biomark. Prev. 2017, 26, 444–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prat, A.; Perou, C.M. Deconstructing the Molecular Portraits of Breast Cancer. Mol. Oncol. 2011, 5, 5–23. [Google Scholar] [CrossRef] [PubMed]
- Randhawa, J.; Park, T.; Malhotra, A.; Charif, M. Two Cases of Possible Remission in Metastatic Triple-Negative Breast Cancer. J. Community Support Oncol. 2017, 15, e176–e178. [Google Scholar] [CrossRef]
- Dent, R.; Trudeau, M.; Pritchard, K.I.; Hanna, W.M.; Kahn, H.K.; Sawka, C.A.; Lickley, L.A.; Rawlinson, E.; Sun, P.; Narod, S.A. Triple-Negative Breast Cancer: Clinical Features and Patterns of Recurrence. Clin. Cancer Res. 2007, 13, 4429–4434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, S.-Y.; Park, S.; Kwon, Y. Recent Therapeutic Trends and Promising Targets in Triple Negative Breast Cancer. Pharmacol. Ther. 2019, 199, 30–57. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of Human Triple-Negative Breast Cancer Subtypes and Preclinical Models for Selection of Targeted Therapies. J. Clin. Investig. 2011, 121, 2750–2767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehmann, B.D.; Pietenpol, J.A. Identification and Use of Biomarkers in Treatment Strategies for Triple-Negative Breast Cancer Subtypes. J. Pathol. 2014, 232, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Gerratana, L.; Basile, D.; Buono, G.; De Placido, S.; Giuliano, M.; Minichillo, S.; Coinu, A.; Martorana, F.; De Santo, I.; Del Mastro, L.; et al. Androgen Receptor in Triple Negative Breast Cancer: A Potential Target for the Targetless Subtype. Cancer Treat. Rev. 2018, 68, 102–110. [Google Scholar] [CrossRef]
- Sporikova, Z.; Koudelakova, V.; Trojanec, R.; Hajduch, M. Genetic Markers in Triple-Negative Breast Cancer. Clin. Breast Cancer 2018, 18, e841–e850. [Google Scholar] [CrossRef] [PubMed]
- Yin, L.; Duan, J.-J.; Bian, X.-W.; Yu, S.-C. Triple-Negative Breast Cancer Molecular Subtyping and Treatment Progress. Breast Cancer Res. 2020, 22, 61. [Google Scholar] [CrossRef]
- Lehmann, B.D.; Pietenpol, J.A.; Tan, A.R. Triple-Negative Breast Cancer: Molecular Subtypes and New Targets for Therapy. Am. Soc. Clin. Oncol Educ. Book 2015, 35, e31–e39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.-Y.; Jiang, Z.; Ben-David, Y.; Woodgett, J.R.; Zacksenhaus, E. Molecular Stratification within Triple-Negative Breast Cancer Subtypes. Sci. Rep. 2019, 9, 19107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pohl, S.-G.; Brook, N.; Agostino, M.; Arfuso, F.; Kumar, A.P.; Dharmarajan, A. Wnt Signaling in Triple-Negative Breast Cancer. Oncogenesis 2017, 6, e310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bender, R.J.; Mac Gabhann, F. Expression of VEGF and Semaphorin Genes Define Subgroups of Triple Negative Breast Cancer. PLoS ONE 2013, 8, e61788. [Google Scholar] [CrossRef] [Green Version]
- Abramson, V.G.; Lehmann, B.D.; Ballinger, T.J.; Pietenpol, J.A. Subtyping of Triple-Negative Breast Cancer: Implications for Therapy. Cancer 2015, 121, 8–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, Y.; Ou, X.; Li, X.; Zeng, Y.; Gao, G.; Lyu, N.; Liu, P. LGR6 Promotes Tumor Proliferation and Metastasis through Wnt/β-Catenin Signaling in Triple-Negative Breast Cancer. Mol. Ther.-Oncolytics 2020, 18, 351–359. [Google Scholar] [CrossRef] [PubMed]
- Dalton, A.C.; Howe, P.H. Epithelial to Mesenchymal Transition. In Reference Module in Biomedical Sciences; Elsevier Inc.: Amsterdam, The Netherlands, 2021; ISBN 978-0-12-801238-3. [Google Scholar] [CrossRef]
- Fruman, D.A.; Chiu, H.; Hopkins, B.D.; Bagrodia, S.; Cantley, L.C.; Abraham, R.T. The PI3K Pathway in Human Disease. Cell 2017, 170, 605–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denkert, C.; Liedtke, C.; Tutt, A.; von Minckwitz, G. Molecular Alterations in Triple-Negative Breast Cancer-the Road to New Treatment Strategies. Lancet 2017, 389, 2430–2442. [Google Scholar] [CrossRef] [Green Version]
- Xue, Z.; Vis, D.J.; Bruna, A.; Sustic, T.; van Wageningen, S.; Batra, A.S.; Rueda, O.M.; Bosdriesz, E.; Caldas, C.; Wessels, L.F.A.; et al. MAP3K1 and MAP2K4 Mutations Are Associated with Sensitivity to MEK Inhibitors in Multiple Cancer Models. Cell Res. 2018, 28, 719–729. [Google Scholar] [CrossRef] [PubMed]
- Feys, L. Radiation-Induced Collateral Damage: Impact on Metastasis. 2016. Available online: https://www.researchgate.net/publication/298882469_Radiation-induced_collateral_damage_Impact_on_metastasis (accessed on 19 February 2021).
- Vaddepally, R.K.; Kharel, P.; Pandey, R.; Garje, R.; Chandra, A.B. Review of Indications of FDA-Approved Immune Checkpoint Inhibitors per NCCN Guidelines with the Level of Evidence. Cancers 2020, 12, 738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, M.J.; Foy, K.C.; Kaumaya, P.T.P. Cancer Immunotherapy: Present Status, Future Perspective, and a New Paradigm of Peptide Immunotherapeutics. Discov. Med. 2013, 15, 166–176. [Google Scholar]
- Martino, E.; Casamassima, G.; Castiglione, S.; Cellupica, E.; Pantalone, S.; Papagni, F.; Rui, M.; Siciliano, A.M.; Collina, S. Vinca Alkaloids and Analogues as Anti-Cancer Agents: Looking Back, Peering Ahead. Bioorg. Med. Chem. Lett. 2018, 28, 2816–2826. [Google Scholar] [CrossRef]
- Scoles, D.R.; Minikel, E.V.; Pulst, S.M. Antisense Oligonucleotides: A Primer. Neurol. Genet. 2019, 5, e323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, X.; Li, D.; Leng, S.; Zhu, X. RNA-Based Pharmacotherapy for Tumors: From Bench to Clinic and Back. Biomed. Pharm. 2020, 125, 109997. [Google Scholar] [CrossRef]
- Lam, J.K.W.; Chow, M.Y.T.; Zhang, Y.; Leung, S.W.S. siRNA Versus miRNA as Therapeutics for Gene Silencing. Mol. Ther.-Nucleic Acids 2015, 4, e252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, D.; Gonzalez-Duarte, A.; O’Riordan, W.D.; Yang, C.-C.; Ueda, M.; Kristen, A.V.; Tournev, I.; Schmidt, H.H.; Coelho, T.; Berk, J.L.; et al. Patisiran, an RNAi Therapeutic, for Hereditary Transthyretin Amyloidosis. N. Engl. J. Med. 2018, 379, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Scott, L.J. Givosiran: First Approval. Drugs 2020, 80, 335–339. [Google Scholar] [CrossRef] [PubMed]
- Mihaila, R.; Ruhela, D.; Keough, E.; Cherkaev, E.; Chang, S.; Galinski, B.; Bartz, R.; Brown, D.; Howell, B.; Cunningham, J.J. Mathematical Modeling: A Tool for Optimization of Lipid Nanoparticle-Mediated Delivery of siRNA. Mol. Ther.-Nucleic Acids 2017, 7, 246–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gebert, L.F.R.; Rebhan, M.A.E.; Crivelli, S.E.M.; Denzler, R.; Stoffel, M.; Hall, J. Miravirsen (SPC3649) Can Inhibit the Biogenesis of MiR-122. Nucleic Acids Res. 2014, 42, 609–621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmadzada, T.; Reid, G.; McKenzie, D.R. Fundamentals of siRNA and miRNA Therapeutics and a Review of Targeted Nanoparticle Delivery Systems in Breast Cancer. Biophys. Rev. 2018, 10, 69–86. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Goel, V.; Attarwala, H.; Sweetser, M.T.; Clausen, V.A.; Robbie, G.J. Patisiran Pharmacokinetics, Pharmacodynamics, and Exposure-Response Analyses in the Phase 3 APOLLO Trial in Patients with Hereditary Transthyretin-Mediated (HATTR) Amyloidosis. J. Clin. Pharm. 2020, 60, 37–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eygeris, Y.; Patel, S.; Jozic, A.; Sahay, G. Deconvoluting Lipid Nanoparticle Structure for Messenger RNA Delivery. Nano Lett. 2020, 20, 4543–4549. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Qin, J.; Jiang, Y.; Duncan, R.G.; Brigham, B.; Fishman, S.; Nair, J.K.; Akinc, A.; Barros, S.A.; Kasperkovitz, P.V. Shielding of Lipid Nanoparticles for siRNA Delivery: Impact on Physicochemical Properties, Cytokine Induction, and Efficacy. Mol. Ther. Nucleic Acids 2014, 3, e210. [Google Scholar] [CrossRef] [PubMed]
- Mohanty, A.; Uthaman, S.; Park, I.-K. Utilization of Polymer-Lipid Hybrid Nanoparticles for Targeted Anti-Cancer Therapy. Molecules 2020, 25, 4377. [Google Scholar] [CrossRef] [PubMed]
- Richards, D.A.; Maruani, A.; Chudasama, V. Antibody Fragments as Nanoparticle Targeting Ligands: A Step in the Right Direction. Chem. Sci. 2016, 8, 63–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azam, M.A.; Zulkapli, N.N.; Nawi, Z.M.; Azren, N.M. Systematic Review of Catalyst Nanoparticles Synthesized by Solution Process: Towards Efficient Carbon Nanotube Growth. J. Sol.-Gel. Sci. Technol. 2015, 73, 484–500. [Google Scholar] [CrossRef]
- Mohd Amin, M.C.I.; Butt, A.M.; Amjad, M.; Kesharwani, P. Chapter 5: Polymeric Micelles for Drug Targeting and Delivery. In Nanotechnology-Based Approaches for Targeting and Delivery of Drugs and Genes, 1st ed.; Mishra, V., Kesharwani, P., Amin, M.C., Iyer, A., Eds.; Academic Press: Cambridge, MA, USA, 2017; pp. 167–202. ISBN 978-0-12-809717-5. [Google Scholar] [CrossRef]
- Bastakoti, B.P.; Ishihara, S.; Leo, S.-Y.; Ariga, K.; Wu, K.C.-W.; Yamauchi, Y. Polymeric Micelle Assembly for Preparation of Large-Sized Mesoporous Metal Oxides with Various Compositions. Langmuir 2014, 30, 651–659. [Google Scholar] [CrossRef] [PubMed]
- Kondiah, P.P.D.; Choonara, Y.; Kondiah, P.; Marimuthu, T.; Kumar, P.; du Toit, L.; Modi, G.; Pillay, V. Chapter 17: Nanocomposites for Therapeutic Application in Multiple Sclerosis. In Applications of Nanocomposite Materials in Drug Delivery, 1st ed.; Inamuddin Asiri, A.M., Mohammad, A., Eds.; Woodhead Publishing: Sawston, UK, 2018; pp. 391–408. ISBN 978-0-12-813741-3. [Google Scholar] [CrossRef]
- Simões, S.M.N.; Figueiras, A.R.; Veiga, F.; Concheiro, A.; Alvarez-Lorenzo, C. Polymeric Micelles for Oral Drug Administration Enabling Locoregional and Systemic Treatments. Expert Opin. Drug Deliv. 2015, 12, 297–318. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Xiong, G.; Guo, S.; Xu, C.; Xu, R.; Guo, P.; Shu, D. Delivery of Anti-miRNA for Triple-Negative Breast Cancer Therapy Using RNA Nanoparticles Targeting Stem Cell Marker CD133. Mol. Ther. 2019, 27, 1252–1261. [Google Scholar] [CrossRef] [Green Version]
- Dees, S.; Ganesan, R.; Singh, S.; Grewal, I.S. Emerging CAR-T Cell Therapy for the Treatment of Triple-Negative Breast Cancer. Mol. Cancer Ther. 2020, 19, 2409–2421. [Google Scholar] [CrossRef]
- McNamara, M.A.; Nair, S.K.; Holl, E.K. RNA-Based Vaccines in Cancer Immunotherapy. J. Immunol. Res. 2015, 2015, 794528. [Google Scholar] [CrossRef] [Green Version]
- Jaiswal, M.; Dudhe, R.; Sharma, P.K. Nanoemulsion: An Advanced Mode of Drug Delivery System. 3 Biotech 2015, 5, 123–127. [Google Scholar] [CrossRef] [Green Version]
- Van Tendeloo, V.F.; Ponsaerts, P.; Lardon, F.; Nijs, G.; Lenjou, M.; Van Broeckhoven, C.; Van Bockstaele, D.R.; Berneman, Z.N. Highly Efficient Gene Delivery by mRNA Electroporation in Human Hematopoietic Cells: Superiority to Lipofection and Passive Pulsing of mRNA and to Electroporation of Plasmid CDNA for Tumor Antigen Loading of Dendritic Cells. Blood 2001, 98, 49–56. [Google Scholar] [CrossRef]
- Pastor, F.; Berraondo, P.; Etxeberria, I.; Frederick, J.; Sahin, U.; Gilboa, E.; Melero, I. An RNA Toolbox for Cancer Immunotherapy. Nat. Rev. Drug Discov. 2018, 17, 751–767. [Google Scholar] [CrossRef]
- Stagg, J.; Allard, B. Immunotherapeutic Approaches in Triple-Negative Breast Cancer: Latest Research and Clinical Prospects. Adv. Med. Oncol. 2013, 5, 169–181. [Google Scholar] [CrossRef] [Green Version]
- Kannan, A.; Philley, J.V.; Hertweck, K.L.; Ndetan, H.; Singh, K.P.; Sivakumar, S.; Wells, R.B.; Vadlamudi, R.K.; Dasgupta, S. Cancer Testis Antigen Promotes Triple Negative Breast Cancer Metastasis and Is Traceable in the Circulating Extracellular Vesicles. Sci. Rep. 2019, 9, 11632. [Google Scholar] [CrossRef]
- Jirapongwattana, N.; Thongchot, S.; Thuwajit, C. The Overexpressed Antigens in Triple Negative Breast Cancer and the Application in Immunotherapy. Genom. Genet. 2020, 13, 19–32. [Google Scholar]
- Almanzar, G.; Olkhanud, P.B.; Bodogai, M.; Dell’agnola, C.; Baatar, D.; Hewitt, S.M.; Ghimenton, C.; Tummala, M.K.; Weeraratna, A.T.; Hoek, K.S.; et al. Sperm-Derived SPANX-B Is a Clinically Relevant Tumor Antigen That Is Expressed in Human Tumors and Readily Recognized by Human CD4+ and CD8+ T Cells. Clin. Cancer Res. 2009, 15, 1954–1963. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Wang, Y.; Miao, L.; Liu, Q.; Musetti, S.; Li, J.; Huang, L. Combination Immunotherapy of MUC1 mRNA Nano-Vaccine and CTLA-4 Blockade Effectively Inhibits Growth of Triple Negative Breast Cancer. Mol. Ther. 2018, 26, 45–55. [Google Scholar] [CrossRef] [Green Version]
- Barros, S.A.; Gollob, J.A. Safety Profile of RNAi Nanomedicines. Adv. Drug Deliv. Rev. 2012, 64, 1730–1737. [Google Scholar] [CrossRef]
- Pardi, N.; Hogan, M.J.; Porter, F.W.; Weissman, D. mRNA Vaccines—A New Era in Vaccinology. Nat. Rev. Drug Discov. 2018, 17, 261–279. [Google Scholar] [CrossRef] [Green Version]
- Lee, E.; Oh, C.; Kim, I.-S.; Kwon, I.C.; Kim, S. Co-Delivery of Chemosensitizing siRNA and an Anticancer Agent via Multiple Monocomplexation-Induced Hydrophobic Association. J. Control. Release 2015, 210, 105–114. [Google Scholar] [CrossRef]
- Zhou, Z.; Kennell, C.; Lee, J.-Y.; Leung, Y.-K.; Tarapore, P. Calcium Phosphate-Polymer Hybrid Nanoparticles for Enhanced Triple Negative Breast Cancer Treatment via Co-Delivery of Paclitaxel and MiR-221/222 Inhibitors. Nanomedicine 2017, 13, 403–410. [Google Scholar] [CrossRef] [Green Version]
- Bhargava-Shah, A.; Foygel, K.; Devulapally, R.; Paulmurugan, R. Orlistat and Antisense-miRNA-Loaded PLGA-PEG Nanoparticles for Enhanced Triple Negative Breast Cancer Therapy. Nanomedicine 2016, 11, 235–247. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.; Li, Y.; Liu, H.; Jain, A.; Patel, P.V.; Cheng, K. Co-Delivery of IKBKE siRNA and Cabazitaxel by Hybrid Nanocomplex Inhibits Invasiveness and Growth of Triple-Negative Breast Cancer. Sci Adv. 2020, 6, eabb0616. [Google Scholar] [CrossRef]
- Gong, C.; Tian, J.; Wang, Z.; Gao, Y.; Wu, X.; Ding, X.; Qiang, L.; Li, G.; Han, Z.; Yuan, Y.; et al. Functional Exosome-Mediated Co-Delivery of Doxorubicin and Hydrophobically Modified MicroRNA 159 for Triple-Negative Breast Cancer Therapy. J. Nanobiotechnol. 2019, 17, 93. [Google Scholar] [CrossRef] [Green Version]
- Deng, X.; Cao, M.; Zhang, J.; Hu, K.; Yin, Z.; Zhou, Z.; Xiao, X.; Yang, Y.; Sheng, W.; Wu, Y.; et al. Hyaluronic Acid-Chitosan Nanoparticles for Co-Delivery of MiR-34a and Doxorubicin in Therapy against Triple Negative Breast Cancer. Biomaterials 2014, 35, 4333–4344. [Google Scholar] [CrossRef]
- Wang, S.; Zhang, J.; Wang, Y.; Chen, M. Hyaluronic Acid-Coated PEI-PLGA Nanoparticles Mediated Co-Delivery of Doxorubicin and MiR-542-3p for Triple Negative Breast Cancer Therapy. Nanomedicine 2016, 12, 411–420. [Google Scholar] [CrossRef]
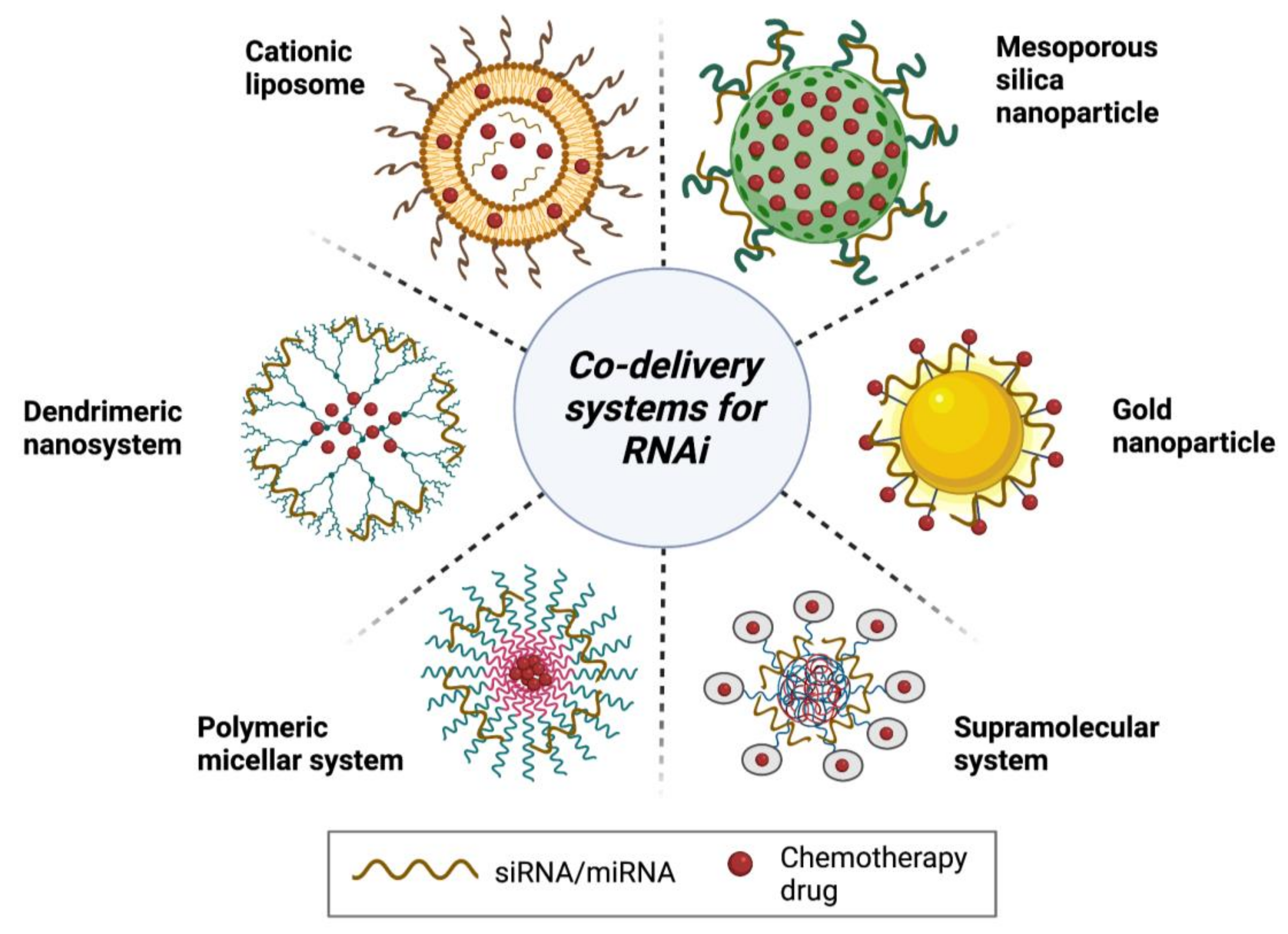
- Babu, A.; Munshi, A.; Ramesh, R. Combinatorial Therapeutic Approaches with RNAi and Anticancer Drugs Using Nanodrug Delivery Systems. Drug Dev. Ind. Pharm. 2017, 43, 1391–1401. [Google Scholar] [CrossRef]
- Saraswathy, M.; Gong, S. Recent Developments in the Co-Delivery of siRNA and Small Molecule Anticancer Drugs for Cancer Treatment. Mater. Today 2014, 17, 298–306. [Google Scholar] [CrossRef]
- Olusanya, T.O.B.; Haj Ahmad, R.R.; Ibegbu, D.M.; Smith, J.R.; Elkordy, A.A. Liposomal Drug Delivery Systems and Anticancer Drugs. Molecules 2018, 23, 907. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Wang, J.; Li, B.; Meng, L.; Tian, Z. Recent Advances in Mechanism-Based Chemotherapy Drug-siRNA Pairs in Co-Delivery Systems for Cancer: A Review. Colloids Surf. B Biointerfaces 2017, 157, 297–308. [Google Scholar] [CrossRef]
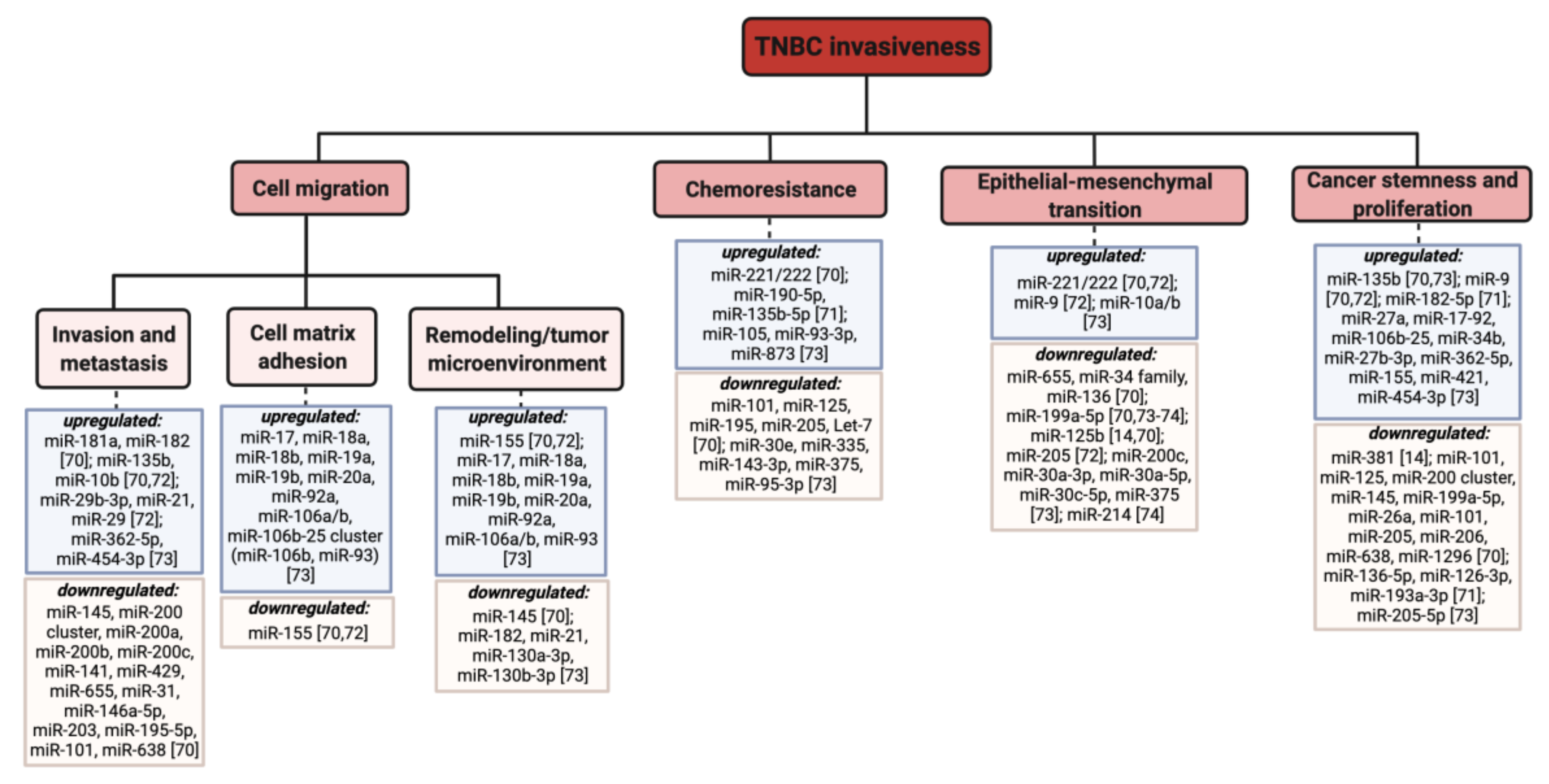
- Paszek, S.; Gabło, N.; Barnaś, E.; Szybka, M.; Morawiec, J.; Kołacińska, A.; Zawlik, I. Dysregulation of MicroRNAs in Triple-Negative Breast Cancer. Ginekol. Pol. 2017, 88, 530–536. [Google Scholar] [CrossRef] [Green Version]
- Piasecka, D.; Braun, M.; Kordek, R.; Sadej, R.; Romanska, H. MicroRNAs in Regulation of Triple-Negative Breast Cancer Progression. J. Cancer Res. Clin. Oncol. 2018, 144, 1401–1411. [Google Scholar] [CrossRef] [Green Version]
- Angius, A.; Cossu-Rocca, P.; Arru, C.; Muroni, M.R.; Rallo, V.; Carru, C.; Uva, P.; Pira, G.; Orrù, S.; De Miglio, M.R. Modulatory Role of MicroRNAs in Triple Negative Breast Cancer with Basal-Like Phenotype. Cancers 2020, 12, 3298. [Google Scholar] [CrossRef]
- Qattan, A. Novel MiRNA Targets and Therapies in the Triple-Negative Breast Cancer Microenvironment: An Emerging Hope for a Challenging Disease. Int. J. Mol. Sci. 2020, 21, 8905. [Google Scholar] [CrossRef]
- Fridrichova, I.; Zmetakova, I. MicroRNAs Contribute to Breast Cancer Invasiveness. Cells 2019, 8, 1361. [Google Scholar] [CrossRef] [Green Version]
- Volovat, S.R.; Volovat, C.; Hordila, I.; Hordila, D.-A.; Mirestean, C.C.; Miron, O.T.; Lungulescu, C.; Scripcariu, D.V.; Stolniceanu, C.R.; Konsoulova-Kirova, A.A.; et al. MiRNA and LncRNA as Potential Biomarkers in Triple-Negative Breast Cancer: A Review. Front. Oncol. 2020, 10, 2423. [Google Scholar] [CrossRef]
- Moore, C.B.; Guthrie, E.H.; Huang, M.T.-H.; Taxman, D.J. Short Hairpin RNA (shRNA): Design, Delivery, and Assessment of Gene Knockdown. Methods Mol. Biol. 2010, 629, 141–158. [Google Scholar] [CrossRef] [Green Version]
- Miao, Y.; Zhang, L.-F.; Guo, R.; Liang, S.; Zhang, M.; Shi, S.; Shang-Guan, C.-F.; Liu, M.-F.; Li, B. (18)F-FDG PET/CT for Monitoring the Response of Breast Cancer to MiR-143-Based Therapeutics by Targeting Tumor Glycolysis. Mol. Ther. -Nucleic Acids 2016, 5, e357. [Google Scholar] [CrossRef] [Green Version]
- Umeh-Garcia, M.; Simion, C.; Ho, P.-Y.; Batra, N.; Berg, A.L.; Carraway, K.L.; Yu, A.; Sweeney, C. A Novel Bioengineered MiR-127 Prodrug Suppresses the Growth and Metastatic Potential of Triple-Negative Breast Cancer Cells. Cancer Res. 2020, 80, 418–429. [Google Scholar] [CrossRef] [Green Version]
- Taipaleenmäki, H.; Farina, N.H.; van Wijnen, A.J.; Stein, J.L.; Hesse, E.; Stein, G.S.; Lian, J.B. Antagonizing MiR-218-5p Attenuates Wnt Signaling and Reduces Metastatic Bone Disease of Triple Negative Breast Cancer Cells. Oncotarget 2016, 7, 79032–79046. [Google Scholar] [CrossRef] [Green Version]
- Deci, M.B.; Liu, M.; Gonya, J.; Lee, C.J.; Li, T.; Ferguson, S.W.; Bonacquisti, E.E.; Wang, J.; Nguyen, J. Carrier-Free CXCR4-Targeted Nanoplexes Designed for Polarizing Macrophages to Suppress Tumor Growth. Cell Mol. Bioeng. 2019, 12, 375–388. [Google Scholar] [CrossRef]
- Bayraktar, R.; Ivan, C.; Bayraktar, E.; Kanlikilicer, P.; Kabil, N.N.; Kahraman, N.; Mokhlis, H.A.; Karakas, D.; Rodriguez-Aguayo, C.; Arslan, A.; et al. Dual Suppressive Effect of MiR-34a on the FOXM1/EEF2-Kinase Axis Regulates Triple-Negative Breast Cancer Growth and Invasion. Clin. Cancer Res. 2018, 24, 4225–4241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almanza, G.; Rodvold, J.J.; Tsui, B.; Jepsen, K.; Carter, H.; Zanetti, M. Extracellular Vesicles Produced in B Cells Deliver Tumor Suppressor MiR-335 to Breast Cancer Cells Disrupting Oncogenic Programming in Vitro and in Vivo. Sci Rep. 2018, 8, 17581. [Google Scholar] [CrossRef]
- Bayraktar, R.; Pichler, M.; Kanlikilicer, P.; Ivan, C.; Bayraktar, E.; Kahraman, N.; Aslan, B.; Oguztuzun, S.; Ulasli, M.; Arslan, A.; et al. MicroRNA 603 Acts as a Tumor Suppressor and Inhibits Triple-Negative Breast Cancer Tumorigenesis by Targeting Elongation Factor 2 Kinase. Oncotarget 2017, 8, 11641–11658. [Google Scholar] [CrossRef] [Green Version]
- D’Ippolito, E.; Plantamura, I.; Bongiovanni, L.; Casalini, P.; Baroni, S.; Piovan, C.; Orlandi, R.; Gualeni, A.V.; Gloghini, A.; Rossini, A.; et al. MiR-9 and MiR-200 Regulate PDGFRβ-Mediated Endothelial Differentiation of Tumor Cells in Triple-Negative Breast Cancer. Cancer Res. 2016, 76, 5562–5572. [Google Scholar] [CrossRef] [Green Version]
- O’Brien, K.; Lowry, M.C.; Corcoran, C.; Martinez, V.G.; Daly, M.; Rani, S.; Gallagher, W.M.; Radomski, M.W.; MacLeod, R.A.F.; O’Driscoll, L. MiR-134 in Extracellular Vesicles Reduces Triple-Negative Breast Cancer Aggression and Increases Drug Sensitivity. Oncotarget 2015, 6, 32774–32789. [Google Scholar] [CrossRef] [Green Version]
- Ramchandani, D.; Lee, S.K.; Yomtoubian, S.; Han, M.S.; Tung, C.-H.; Mittal, V. Nanoparticle Delivery of MiR-708 Mimetic Impairs Breast Cancer Metastasis. Mol. Cancer Ther. 2019, 18, 579–591. [Google Scholar] [CrossRef] [Green Version]
- Vu, L.T.; Peng, B.; Zhang, D.X.; Ma, V.; Mathey-Andrews, C.A.; Lam, C.K.; Kiomourtzis, T.; Jin, J.; McReynolds, L.; Huang, L.; et al. Tumor-Secreted Extracellular Vesicles Promote the Activation of Cancer-Associated Fibroblasts via the Transfer of MicroRNA-125b. J. Extracell Vesicles 2019, 8, 1599680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, R.; Cho, W.C.S.; Ma, V.; Cheuk, W.; So, Y.-K.; Wong, S.C.C.; Zhang, M.; Li, C.; Sun, Y.; Zhang, H.; et al. ADAM9 Mediates Triple-Negative Breast Cancer Progression via AKT/NF-ΚB Pathway. Front. Med. 2020, 7, 214. [Google Scholar] [CrossRef] [PubMed]
- Rafael, D.; Gener, P.; Andrade, F.; Seras-Franzoso, J.; Montero, S.; Fernández, Y.; Hidalgo, M.; Arango, D.; Sayós, J.; Florindo, H.F.; et al. AKT2 siRNA Delivery with Amphiphilic-Based Polymeric Micelles Show Efficacy against Cancer Stem Cells. Drug Deliv. 2018, 25, 961–972. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.W.; Jeong, H.Y.; Kang, S.J.; Jeong, I.H.; Choi, M.J.; You, Y.M.; Im, C.S.; Song, I.H.; Lee, T.S.; Lee, J.S.; et al. Anti-EGF Receptor Aptamer-Guided Co-Delivery of Anti-Cancer siRNAs and Quantum Dots for Theranostics of Triple-Negative Breast Cancer. Theranostics 2019, 9, 837–852. [Google Scholar] [CrossRef] [PubMed]
- Alshaer, W.; Hillaireau, H.; Vergnaud, J.; Mura, S.; Deloménie, C.; Sauvage, F.; Ismail, S.; Fattal, E. Aptamer-Guided siRNA-Loaded Nanomedicines for Systemic Gene Silencing in CD-44 Expressing Murine Triple-Negative Breast Cancer Model. J. Control. Release 2018, 271, 98–106. [Google Scholar] [CrossRef]
- Zhang, C.; Yuan, W.; Wu, Y.; Wan, X.; Gong, Y. Co-Delivery of EGFR and BRD4 siRNA by Cell-Penetrating Peptides-Modified Redox-Responsive Complex in Triple Negative Breast Cancer Cells. Life Sci. 2021, 266, 118886. [Google Scholar] [CrossRef] [PubMed]
- Guo, P.; Yang, J.; Jia, D.; Moses, M.A.; Auguste, D.T. ICAM-1-Targeted, Lcn2 siRNA-Encapsulating Liposomes Are Potent Anti-angiogenic Agents for Triple Negative Breast Cancer. Theranostics 2016, 6, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parmar, M.B.; Arteaga Ballesteros, B.E.; Fu, T.; KC, R.B.; Montazeri Aliabadi, H.; Hugh, J.C.; Löbenberg, R.; Uludağ, H. Multiple siRNA Delivery against Cell Cycle and Anti-Apoptosis Proteins Using Lipid-Substituted Polyethylenimine in Triple-Negative Breast Cancer and Nonmalignant Cells. J. Biomed. Mater. Res. A 2016, 104, 3031–3044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Liu, Y.; Li, Y.; Wang, H.; Stewart, S.; Van der Jeught, K.; Agarwal, P.; Zhang, Y.; Liu, S.; Zhao, G.; et al. Precise Targeting of POLR2A as a Therapeutic Strategy for Human Triple Negative Breast Cancer. Nat. Nanotechnol. 2019, 14, 388–397. [Google Scholar] [CrossRef]
- Werfel, T.A.; Wang, S.; Jackson, M.A.; Kavanaugh, T.E.; Joly, M.M.; Lee, L.H.; Hicks, D.J.; Sanchez, V.; Ericsson, P.G.; Kilchrist, K.V.; et al. Selective MTORC2 Inhibitor Therapeutically Blocks Breast Cancer Cell Growth and Survival. Cancer Res. 2018, 78, 1845–1858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitobe, Y.; Ikeda, K.; Sato, W.; Kodama, Y.; Naito, M.; Gotoh, N.; Miyata, K.; Kataoka, K.; Sasaki, H.; Horie-Inoue, K.; et al. Proliferation-Associated Long Noncoding RNA, TMPO-AS1, Is a Potential Therapeutic Target for Triple-Negative Breast Cancer. Cancer Sci. 2020, 111, 2440–2450. [Google Scholar] [CrossRef]
- Vaidya, A.M.; Sun, Z.; Ayat, N.; Schilb, A.; Liu, X.; Jiang, H.; Sun, D.; Scheidt, J.; Qian, V.; He, S.; et al. Systemic Delivery of Tumor-Targeting siRNA Nanoparticles against an Oncogenic LncRNA Facilitates Effective Triple-Negative Breast Cancer Therapy. Bioconjug. Chem. 2019, 30, 907–919. [Google Scholar] [CrossRef] [PubMed]
- Fu, W.; Sun, H.; Zhao, Y.; Chen, M.; Yang, L.; Yang, X.; Jin, W. Targeted Delivery of CD44s-siRNA by ScFv Overcomes de Novo Resistance to Cetuximab in Triple Negative Breast Cancer. Mol. Immunol. 2018, 99, 124–133. [Google Scholar] [CrossRef] [PubMed]
- Gujrati, M.; Vaidya, A.M.; Mack, M.; Snyder, D.; Malamas, A.; Lu, Z.-R. Targeted Dual PH-Sensitive Lipid ECO/siRNA Self-Assembly Nanoparticles Facilitate In Vivo Cytosolic SieIF4E Delivery and Overcome Paclitaxel Resistance in Breast Cancer Therapy. Adv. Healthc. Mater. 2016, 5, 2882–2895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, P.; Xiao, Z.; Wang, S.; Zhang, M.; Wei, Y.; Hang, Q.; Kim, J.; Yao, F.; Rodriguez-Aguayo, C.; Ton, B.N.; et al. ZRANB1 Is an EZH2 Deubiquitinase and a Potential Therapeutic Target in Breast Cancer. Cell Rep. 2018, 23, 823–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nam, K.; Son, S.-H.; Oh, S.; Jeon, D.; Kim, H.; Noh, D.-Y.; Kim, S.; Shin, I. Binding of Galectin-1 to Integrin Β1 Potentiates Drug Resistance by Promoting Survivin Expression in Breast Cancer Cells. Oncotarget 2017, 8, 35804–35823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Liu, X.; Wang, H.; Zhou, Q.; Liang, Y.; Sui, A.; Yao, R.; Zhao, B.; Sun, M. Lentiviral Vector-Mediated Doxycycline-Inducible USP39 shRNA or cDNA Expression in Triple-Negative Breast Cancer Cells. Oncol. Rep. 2015, 33, 2477–2483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Mi, P.; Lin, G.; Wáng, Y.X.J.; Liu, G.; Chen, X. Imaging Guided Delivery of RNAi for Anticancer Treatment. Adv. Drug Deliv. Rev. 2016, 104, 44–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinclair, C.; Revenko, A.S.; Johnson, R.B.; Peter, A.; Taylor, M.A.; Hettrick, L.A.; Klein, S.; Solanki, A.; Chapman, M.; Yates, J.; et al. Abstract 2713: Discovery and Characterization of AZD8701, a High Affinity Antisense Oligonucleotide Targeting FOXP3 to Relieve Immunosuppression in Cancer. Cancer Res. 2019, 79, 2713. [Google Scholar] [CrossRef]
- Syed, Y.Y. Durvalumab: First Global Approval. Drugs 2017, 77, 1369–1376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, M.R.; Bauer, T.M.; Jimeno, A.; Wang, D.; LoRusso, P.; Do, K.T.; Stemmer, S.M.; Maurice-Dror, C.; Geva, R.; Zacharek, S.; et al. A Phase I Study of mRNA-2752, a Lipid Nanoparticle Encapsulating mRNAs Encoding Human OX40L, IL-23, and IL-36γ, for Intratumoral (ITu) Injection Alone and in Combination with Durvalumab. J. Clin. Oncol. 2020, 38, 3092. [Google Scholar] [CrossRef]
- Sajid, M.I.; Moazzam, M.; Kato, S.; Yeseom Cho, K.; Tiwari, R.K. Overcoming Barriers for siRNA Therapeutics: From Bench to Bedside. Pharmaceuticals 2020, 13, 294. [Google Scholar] [CrossRef]
- Wang, J.; Lu, Z.; Wientjes, M.G.; Au, J.L.-S. Delivery of siRNA Therapeutics: Barriers and Carriers. AAPS J. 2010, 12, 492–503. [Google Scholar] [CrossRef] [PubMed]
- Stevens, D.; Ingels, J.; Van Lint, S.; Vandekerckhove, B.; Vermaelen, K. Dendritic Cell-Based Immunotherapy in Lung Cancer. Front. Immunol. 2020, 11, 620374. [Google Scholar] [CrossRef]
- Suschak, J.J.; Williams, J.A.; Schmaljohn, C.S. Advancements in DNA Vaccine Vectors, Non-Mechanical Delivery Methods, and Molecular Adjuvants to Increase Immunogenicity. Hum. Vaccin Immunother. 2017, 13, 2837–2848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, P.M.; Butterfield, L.H. Dendritic Cell-Based Cancer Vaccines. J. Immunol. 2018, 200, 443–449. [Google Scholar] [CrossRef] [PubMed]
- Catuogno, S.; Esposito, C.L.; Quintavalle, C.; Condorelli, G.; de Franciscis, V.; Cerchia, L. Nucleic Acids in Human Glioma Treatment: Innovative Approaches and Recent Results. J. Signal. Transduct. 2012, 2012, 735135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campeau, E.; Gobeil, S. RNA Interference in Mammals: Behind the Screen. Brief. Funct. Genom. 2011, 10, 215–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gousseinov, E. RNA-Based Therapeutics and Vaccines. Available online: https://www.genengnews.com/insights/rna-based-therapeutics-and-vaccines/ (accessed on 20 April 2021).
- Maruggi, G.; Zhang, C.; Li, J.; Ulmer, J.B.; Yu, D. mRNA as a Transformative Technology for Vaccine Development to Control Infectious Diseases. Mol. Ther. 2019, 27, 757–772. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Maruggi, G.; Shan, H.; Li, J. Advances in mRNA Vaccines for Infectious Diseases. Front. Immunol. 2019, 10, 594. [Google Scholar] [CrossRef] [Green Version]
- Lopes, A.; Vandermeulen, G.; Préat, V. Cancer DNA Vaccines: Current Preclinical and Clinical Developments and Future Perspectives. J. Exp. Clin. Cancer Res. 2019, 38, 146. [Google Scholar] [CrossRef] [PubMed]
- Howard, F.; Muthana, M. Designer Nanocarriers for Navigating the Systemic Delivery of Oncolytic Viruses. Nanomedicine 2020, 15, 93–110. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Liu, S.; Han, D.; Tang, B.; Ma, J. Delivery and Biosafety of Oncolytic Virotherapy. Front. Oncol. 2020, 10, 475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, X.; Tan, C. Combination of MicroRNA Therapeutics with Small-Molecule Anticancer Drugs: Mechanism of Action and Co-Delivery Nanocarriers. Adv. Drug Deliv. Rev. 2015, 81, 184–197. [Google Scholar] [CrossRef] [PubMed]
- Polack, F.P.; Thomas, S.J.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Perez, J.L.; Pérez Marc, G.; Moreira, E.D.; Zerbini, C.; et al. Safety and Efficacy of the BNT162b2 MRNA Covid-19 Vaccine. N. Engl. J. Med. 2020, 383, 2603–2615. [Google Scholar] [CrossRef] [PubMed]
- Han, H.J.; Nwagwu, C.; Anyim, O.; Ekweremadu, C.; Kim, S. COVID-19 and Cancer: From Basic Mechanisms to Vaccine Development Using Nanotechnology. Int. Immunopharmacol. 2021, 90, 107247. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TNBC Molecular Subtype | Intrinsic BCa Subtype | Signaling Pathway(s) | Molecular Genetic Target(s) |
|---|---|---|---|
| BL1 (basal-like 1) | Basal-like | DNA-damage response | RAD51, CHEK1 [10]; ATR [11]; BRCA2 [11,12]; CTNND1, TOP2B, CAMK1G [12]; TP53 [12,14] |
| Cell division | NRAS (N-Ras) [11]; RB1 [12,14] | ||
| Cell proliferation | PARP1, PLK1, TTK, AURKA/B [10]; MKI67 (Ki-67) [11]; BRCA1 [11,12]; MAPK13, SMAD4, STAT4, CDKN2A, PIK3CA (PI3K), MDC1, PPAR1 [12]; MYC [11,12,14]; PTEN, TP53, RB1 [12,14]; CDCA4, RHOA, AKT1 [14] | ||
| Cell cycle gene expression | BRCA1 [11,12]; KDM6A (UTX), STAT4 [12]; CDCA4 [14] | ||
| BL2 (basal-like 2) | Basal-like | DNA-damage response | MTOR [10]; TP53 [11,12] |
| Cell division | EGFR [11]; RB1 [12]; PTEN [12,17] | ||
| Cell proliferation | TP63 [10]; EGFR [11]; MET (c-Met), EPHA2 [10,11]; TP53 [11,12]; RB1, IGF1R, MYC, BRCA1 [12]; AKT1, TGFB1, E2F2, RHOA, CXCL8 [14]; PIK3CA (PI3K), PIK3R1 [17] | ||
| Glycolysis, gluconeogenesis | CDKN2A, KDM6A (UTX) [12]; TP53 [11,12] | ||
| Growth factor signaling | EGF/EGFR [10]; BRCA1 [12]; TGFB1 [14] | ||
| M (mesenchymal-like) | Unclassified/luminal B | Cell motility | PIK3CA (PI3K) [10,12]; WNT [11]; PTEN, RB1, TP53 [12,14]; PDGFA/B, KIT [15] |
| DNA-damage response | MTOR [10] | ||
| Cell differentiation | FGFR1 [10]; PIK3CA (PI3K) [10,12]; ALK, TGFB1 (TGF-β1) [11]; KIT, CTNNB1 (β-catenin), SFRP4, TCF4 [15] | ||
| Epithelial–mesenchymal transition | TP53 [12,14]; MMP2, TWIST, SNAI2, TCF4 [15] | ||
| Growth factor signaling | PDGFRA (PDGFRα) [10]; TGFB1 (TGF-β1) [11]; TP53 [12,14]; PDGFA/B, IGF1 [15] | ||
| Cell proliferation | IGF1R, SRC, PDGFRA/B [10]; WNT, ALK, TGFB1 (TGF-β1) [11]; MYC [12]; PIK3CA (PI3K) [10,12]; RB1 [12,14]; SNAI2, PDGFA/B, IGF1, KIT, CTNNB1 (β-catenin), DKK2/3, SFRP4, TCF7L2, FZD4 [15]; LGR6 [18] | ||
| MSL (mesenchymal stem-like) | Unclassified | Cell motility and adhesion | TGFBR3 [10]; RB1, TP53, VCAM1 [12]; PIK3CA (PI3K) [10,12]; CCL2 [14]; WNT, KIT, PDGFA/B [15] |
| Cell differentiation | MAP2K1, FGFR1 [10]; MAPK1/3 [11]; PIK3CA (PI3K) [10,12]; KIT, CTNNB1 (β-catenin), SFRP4, TCF4 [15] | ||
| Cell proliferation/stemness | SRC, MAP2K1, NFKB1, IGF1R [10]; EGFR [11]; TP53, NF1, RB1, ABCA8, PROCR, ENG, PER1, ABCB1, TERT2IP, BCL2, BMP2, THY, HOXA5, HOXA10, MEIS1, MEIS2, MEOX1, MEOX2, MSX1, THY1, BRCA1 [12]; PDGFRA/B [10,11,12]; PIK3CA (PI3K) [10,12]; SNAI2, WNT, PDGFA/B, IGF1, KIT, CTNNB1 (β-catenin), DKK2/3, SFRP4, TCF7L2, FZD4 [15]; ALDHA1 [12,15] | ||
| DNA-damage response | MTOR [10] | ||
| Epithelial–mesenchymal transition | TP53 [12]; MMP2, TWIST, SNAI2, TCF4 [15] | ||
| Angiogenesis | PDGFRA/B [10,11,12]; PIK3CA (PI3K) [10,12]; TP53, HRAS, BRAF, CDKN2A, KRAS, NF2, ENG, ITGAV, NT5E [12]; CCL2 [14]; PDGFA/B [15]; VEGFC [16] | ||
| Growth factor signaling | MAP2K2 [10]; TP53, BMP2, NGFR, BRCA1 [12]; KDR (VEGFR2) [11,12]; EGFR [11,14]; PDGFA/B, IGF1 [15] | ||
| LAR (luminal androgen receptor) | Luminal A | Androgen, estrogen metabolism, porphyrin, and lipid metabolism | AR [10,11]; FOXA1, XBP1, KRT18 [11]; APOD, CDH1 [12]; PIK3CA (PI3K) [10,12] |
| Steroid synthesis/regulation | PTEN, DHCR24, FASN, FKBP5 [12] | ||
| Cell proliferation | HSP90AB1, FGFR4 [10]; ALCAM, PIP, SPDEF, CLDN8, RB1, MYC [12]; PIK3CA (PI3K) [10,12]; AKT1 [14]; CCND1 [15] | ||
| Molecular apocrine subtype | PIK3CA (PI3K) [10,12]; RB1, TP53, PTEN [12] | ||
| IM (immunomodulatory) | Luminal B | Immune signaling | BTK [10]; JAK1/2, STAT1/4, IRF1/7/8 [10,11]; TNF [11]; TP53, CTNNA1, DDX18, HUWE1, NFKBIA, APC, BRAF, MAP2K4, RB1 [12]; PDCD1 (PD-1), CTLA4, CD274 (PD-L1), IFNG, IFNA1, PTEN [14] |
| Cell proliferation | NFKB1 [10]; TNF [11]; TP53, HUWE1, APC, MAP2K4, RB1, MYC [12]; AKT1, RHOA [14] | ||
| Cell differentiation | CTNNA1, HUWE1, BRAF, MAP2K4 [12] | ||
| Cell division | DDX18, APC, BRAF, RB1 [12] | ||
| DNA-damage response, growth factor signaling | LYN [10]; TP53 [12] | ||
| UNS (unstable) | Claudin-low | DNA-damage response | RAD51, CHEK1, PARP1 [10] |
| Cell proliferation | PARP1, PLK1, TTK, AURKA/B [10] |
| TNBC Molecular Subtype | Therapeutic Strategies | Targeted Therapies |
| BL1 (basal-like 1) | -Inhibit cell proliferation and DNA damage-response | -Antimitotic agents -Cytostatic agents -PARP inhibitors -DNA synthesis inhibitors -PI3K inhibitors -MYC inhibitors -Aurora kinase inhibitors |
| BL2 (basal-like 2) | -Inhibit growth factor signaling (EGF/EGFR) -Inhibit cell division and proliferation | -Antimitotic agents -Cytostatic agents -PARP inhibitors -Growth factor receptor inhibitors -mTOR inhibitors -PI3K/Akt inhibitors -IGF1R inhibitors -RAS/MAPK inhibitors |
| M (mesenchymal-like) | -Inhibit cell migration -Inhibit Wnt/β-catenin signaling pathway | -Tyrosine kinase inhibitors (TKIs) -mTOR inhibitors -Growth factor receptor inhibitors -PI3K inhibitors -Src inhibitors -Eribulin mesylate -Wnt/β-catenin inhibitors |
| MSL (mesenchymal stem-like) | -Inhibit cell migration -Inhibit PDGFRα/β -Inhibit EGF/EGFR/VEGFR2 | -Src inhibitors -Growth factor receptor inhibitors -mTOR inhibitors -PI3K inhibitors -MAPK inhibitors -Wnt/β-catenin inhibitors -Angiogenesis inhibitors (antiangiogenics) |
| LAR (luminal androgen receptor) | -Inhibit AR signaling -Inhibit FOXA1 and targets involved in cell differentiation (ERBB4) | -PI3K inhibitors -AR antagonists (steroidal antiandrogens) -Nonsteroidal antiandrogens -mTOR inhibitors -Hsp90 inhibitors |
| IM (immunomodulatory) | -Inhibit immune checkpoint proteins including PD-1/PD-L1 and CTLA-4 | -MEK inhibitors -Cytostatic agents -PARP inhibitors -Immune checkpoint inhibitors |
| UNS (unstable) | -Inhibit PLK1/TTK amplification | -PLK1 inhibitors -TTK inhibitors |
| Select Examples of Current Chemotherapy Drugs | ||
| ||
| miRNA Name | miRNA Class | Key Function(s) | Reference(s) |
| miR-143 | Oncosuppressor | Suppression of cell proliferation, migration, and glycolytic pathway; apoptosis induction; tumor growth inhibition Target gene(s): MUC1 [71]; HK2 [76] | [76] |
| miR-127 | Oncosuppressor | Suppression of cell proliferation, migration, and invasion Target gene(s): CERK, NANOS1, FOXO6, SOX11, SOX12, FASN, SUSD2 | [77] |
| miR-218-5p | Oncogenic | Promotion of cell proliferation, bone metastasis, and tumor growth in the bone marrow; osteoclast activation and bone resorption; mediating acquisition of osteomimetic properties of bone metastatic BCa cellsTarget gene(s): SOST, SFRP2 | [78] |
| miR-127-5p | Oncosuppressor | Mediation of M1 macrophage polarization Target gene(s): CXCR4 | [79] |
| miR-34a | Oncosuppressor | Regulation of eEF2K gene; inhibition of tumor cell proliferation and growth, cell migration, and invasion Target gene(s): NOTCH1, BCL2, CD44, SIRT1, RAC1, FOSL1 (Fra-1) [63]; TP53, NOTCH2 [74]; EEF2K, FOXM1 [80] | [63,80] |
| miR-335 | Oncosuppressor | Inhibition of tumor re-initiation, cell growth and proliferation, and tumor metastasis Target gene(s): SOX4 | [81] |
| miR-159 | Oncosuppressor | Inhibition of cell proliferation and decrease in overall BCa incidence and progression Target gene(s): TCF7 | [62] |
| miR-542-3p | Oncosuppressor | Tumor suppression; regulator of p53 tumor suppressor and anti-apoptotic protein survivin; apoptosis promotion Target gene(s): TP53, BIRC5 | [64] |
| miR-603 | Oncosuppressor | Tumor suppression; cell proliferation, migration/invasion, and tumorigenesis through regulation of eEF2K gene Target gene(s): EEF2K | [82] |
| miR-200 family | Oncosuppressor | Inhibition of tube formation ability through PDGFRβ gene repression; regulation of tumor-mediated vasculogenesis and EMT; inhibition of tumor proliferation and metastasis; suppression of cell migration, invasion, and stemness; overcoming resistance to standard therapies Target gene(s): PRKCA [74]; ZEB1/2, SNAI1/2 [71,74]; JAG1, MAML2/3, PIK3CA [70]; PDGFRB [83] | [83] |
| miR-9 | Oncogenic | EMT progression; formation of vascular-like structures Target genes(s): CHN1 [70,71]; PDGFRB [71,83] | [83] |
| miR-134 | Oncosuppressor | Inhibition of cell migration and invasion; direct regulator of STAT5B gene and indirectly of Hsp90 Target gene(s): STAT5B | [84] |
| miR-708 | Oncosuppressor | Inhibition of tumor cell migration and metastasis Target gene(s): NNAT | [85] |
| miR-125b | Oncogenic | Promotion of cell proliferation and tumorigenesis; fibroblast activation through suppression of TP53 gene Target gene(s): TP53, TP53INP1 | [86] |
| miR-21 | Oncogenic | Antiapoptotic activity, tumor proliferation, and drug resistance; enhancing migration and invasion Target gene(s): HIF1A (HIF1α), TIMP3, TM1 [70]; PDCD4, PTEN [70,71]; TPM1, TGFBR2 [71] | [60] |
| siRNA/shRNA Molecular Genetic Targets | |||
| Study Name and Clinical Trial Identifier | Sponsor/Collaborator | Intervention/Treatment Method | Study Recruitment Status |
|---|---|---|---|
| NCT03739931: Dose Escalation Study of mRNA-2752 for Intratumoral Injection to Participants With Advanced Malignancies | ModernaTX, Inc./AstraZeneca | Biological: mRNA-2752 Biological: Durvalumab (MEDI4736) | Recruiting; Phase 1 |
| NCT02316457: RNA-Immunotherapy of IVAC_W_bre1_uID and IVAC_M_uID (TNBC-MERIT) | BioNTech SE/Seventh Framework Programme | Biological: IVAC_W_bre1_uID Biological: IVAC_W_bre1_uID/IVAC_M_uID | Active, not recruiting; Phase 1 |
| NCT01837602: cMet CAR RNA T Cells Targeting Breast Cancer | University of Pennsylvania | Biological: c-Met RNA CAR-T cells | Completed; Phase 1 |
| NCT04504669: First Time in Human Study of AZD8701 With or Without Durvalumab in Participants With Advanced Solid Tumors | AstraZeneca | Drug: AZD8701 Biological: Durvalumab (MEDI4736) | Recruiting; Phase 1 |
| Therapeutic/Prophylactic Approach | Delivery Method(s) | Advantages | Disadvantages | |
|---|---|---|---|---|
| RNAi | miRNA | -LNP delivery -Biopolymers -Covalent conjugation -Liposome -Polymeric, carbon, silica, and gold NPs -Dendrimer and micelle systems -Naked delivery -CPPs -Nanoplexes | -Dual mimetic/agonistic and replacement/antagonistic functions -Chemically synthesized and readily chemically modifiable -Less immunogenic than proteins -Small size | -Unstable -Nuclease-mediated degradation -Off-target effects -Not yet developed in clinical trials -More technologically challenging -Complementarity to target mRNA is not exact; lack of specificity |
| siRNA | -More stable than miRNA -Easily and rapidly generated -Chemically synthesized and readily chemically modifiable -Less immunogenic than proteins -Small size -Already developed in clinical trials -100% complementarity to target mRNA—can knock down specific genes -High transfection efficiency | -Potential minor off-target exceptions and transient effects -Induction of nonspecific immune responses | ||
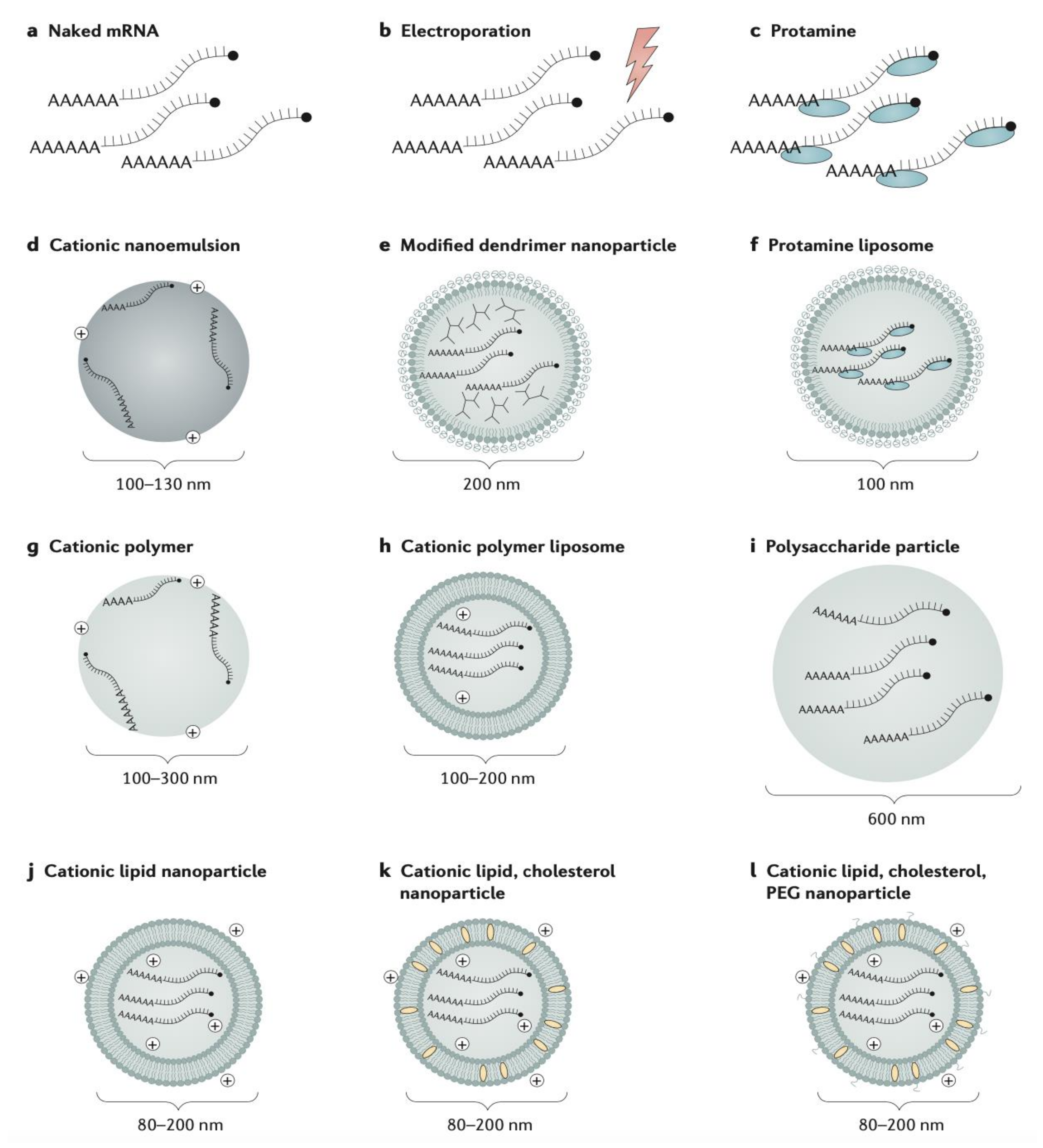
| Vaccine immunotherapy | mRNA vaccine | -Encapsulated LNP delivery-Naked delivery -Cationic liposomes -Cationic nanoemulsions -Dendrimer nanoparticles -Protamine liposome -Polysaccharide particle -Electroporation -Cationic polymer | -Thermally stable -Synthetic production (egg and cell-free) -Rapid and scalable production -Non-infectious, non-integrating, and naturally degraded -Expression in situ to produce antigens with structure unaltered by in vivo manufacturing process | -Concerns with instability (particularly unstable in plasma) -Limited immunogenicity data in humans -Potential toxic effect of free extracellular mRNA -Inflammation due to enhanced type I IFN activation |
| DC (cell-based) vaccine | -Direct infusion of DCs through subcutaneous, intradermal, intranodal, intralymphatic, or intravenous routes | -High immunogenicity -Control of antigen presentation | -Expensive and difficult to produce -Vascular injury/electrolyte imbalances may occur after leukapheresis | |
| Gene-based vaccine (DNA/RNA) | -Lipid and polymeric micro- and nanoparticles -Cationic liposomes -Microspheres -Liposome-derived nanovesicles | -Easy delivery of multiple antigens -Induction of adaptive, and humoral immunity (B and T cell immune responses) -Stimulation of innate immune response -Non-infectious -Egg and cell-free -Not restricted to HLA-patient type -Rapid and scalable production -DNA vaccines: chemically stable -RNA vaccines: non-integrating, natural degradation | -Specific transportation/storage conditions for RNA vaccines -Both vaccines poorly immunogenic in humans -Potential integration of DNA vaccines into human genome -Chemical instability of RNA vaccines | |
| Viral vectored vaccines (oncolytic virotherapy) | -Liposomal, polymeric, or nanoparticle (magnetic and metallic NPs) delivery through intravenous, intratumoral, or intraperitoneal routes -Cell carriers | -Induction of adaptive, and humoral immunity; stimulation of innate immune response -High immunogenicity -Easy to produce on a large scale | -Induction of anti-vector immunity -Cell-based manufacturing -Potential high toxicity -Risk of undesired infections | |
| Combination therapy | RNAi/small-molecule chemotherapeutics | -Nanoparticles (gold and mesoporous silica) -Cationic liposome -Micelle system -Dendrimer/supramolecular systems | -Overcome multidrug resistance -Produce additive or synergistic anti-cancer effects (promote apoptosis and autophagy) -Reduce drug-related toxicities -Increased spectrum of activity -Revert EMT -Suppress tumor angiogenesis -Downregulate MDR proteins, including ABC transporters and P-gp | -Potential risk for development of novel adverse reactions/increased toxicity -Increased risk for unfavorable interactions -Increased cost -Antagonistic effects |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Haque, S.; Cook, K.; Sahay, G.; Sun, C. RNA-Based Therapeutics: Current Developments in Targeted Molecular Therapy of Triple-Negative Breast Cancer. Pharmaceutics 2021, 13, 1694. https://doi.org/10.3390/pharmaceutics13101694
Haque S, Cook K, Sahay G, Sun C. RNA-Based Therapeutics: Current Developments in Targeted Molecular Therapy of Triple-Negative Breast Cancer. Pharmaceutics. 2021; 13(10):1694. https://doi.org/10.3390/pharmaceutics13101694
Chicago/Turabian StyleHaque, Sakib, Kiri Cook, Gaurav Sahay, and Conroy Sun. 2021. "RNA-Based Therapeutics: Current Developments in Targeted Molecular Therapy of Triple-Negative Breast Cancer" Pharmaceutics 13, no. 10: 1694. https://doi.org/10.3390/pharmaceutics13101694