
Lipid Nanoparticles for Organ-Specific mRNA Therapeutic Delivery

Abstract
:Simple Summary
Abstract
1. Introduction
2. mRNA Delivery Methods
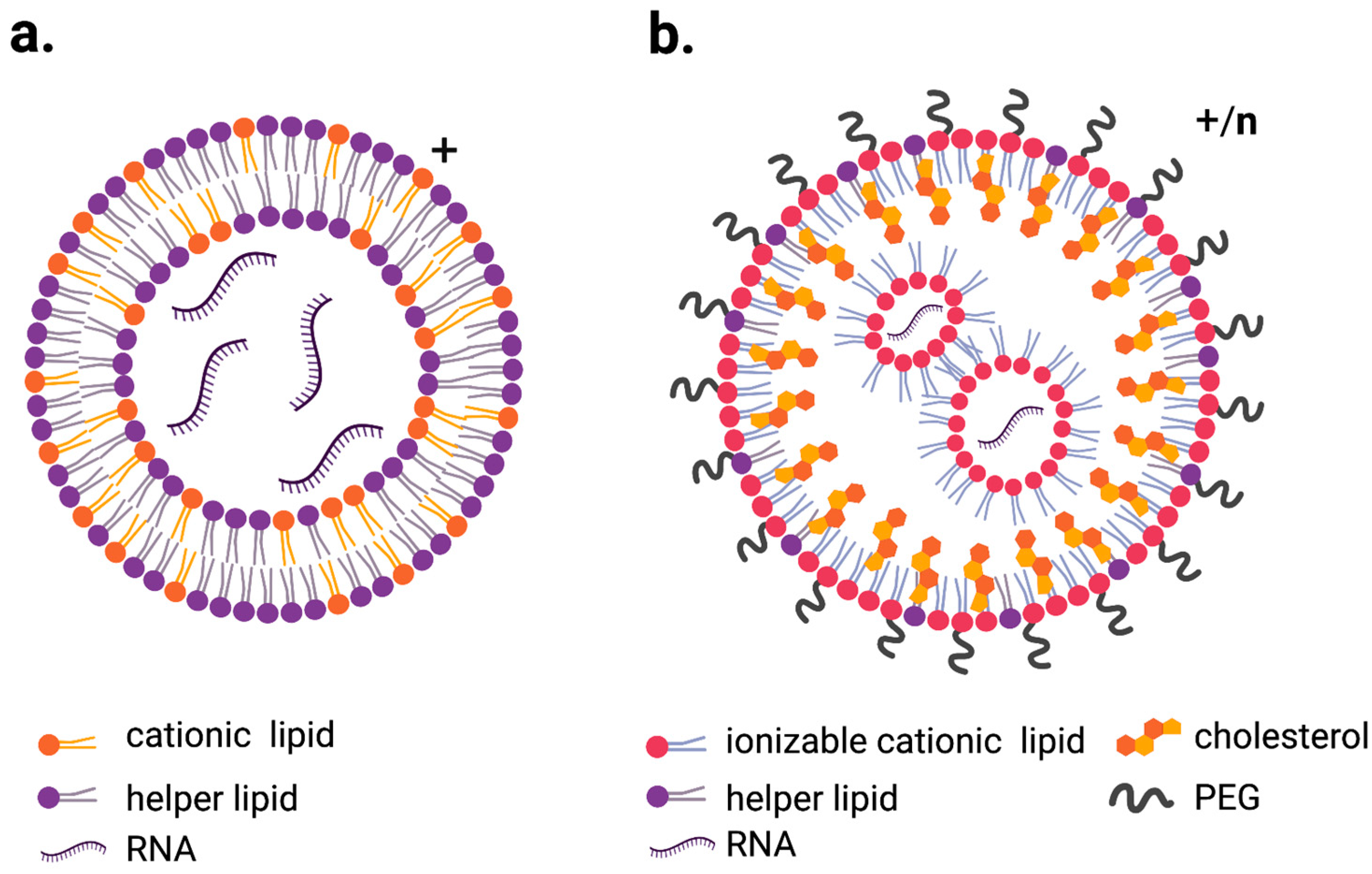
3. Lipid Nanoparticles (LNPs)
3.1. Cationic LNPs
3.2. Ionizable Cationic Lipids LNPs
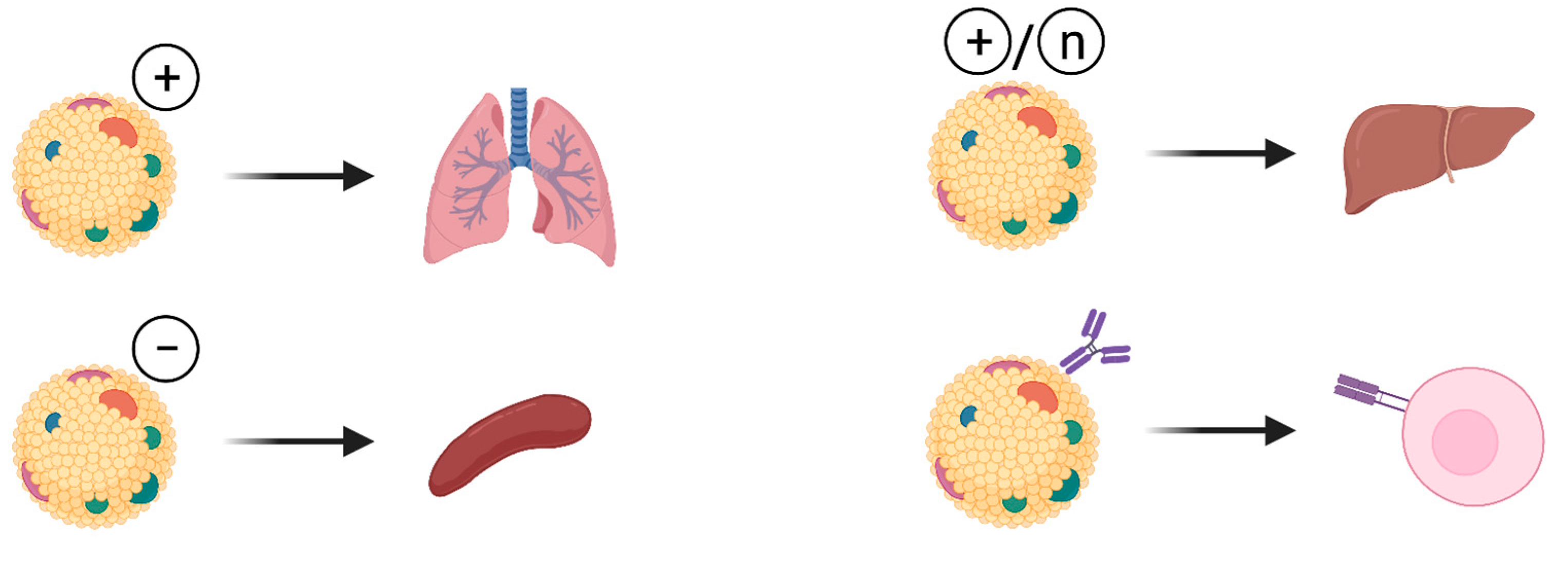
3.3. Organ-Specific LNPs
4. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Sahin, U.; Karikó, K.; Türeci, Ö. MRNA-based therapeutics-developing a new class of drugs. Nat. Rev. Drug Discov. 2014, 13, 759–780. [Google Scholar] [CrossRef]
- Brenner, B.D.S.; Meselson, D.M. An unstable intermediate carrying information from genes to ribosomes for protein synthesis. Def. At. Support Agency Rep. DASA-532 B 1957, 186, 2528. [Google Scholar] [CrossRef]
- Wolff, J.A.; Malone, R.W.; Williams, P.; Chong, W.; Acsadi, G.; Jani, A.; Felgner, P.L. Direct Gene Transfer into Mouse Muscle in Vivo. Science 1990, 247, 1465–1468. [Google Scholar] [CrossRef] [PubMed]
- Jirikowski, G.F.; Sanna, P.P.; Maciejewski-Lenoir, D.; Bloom, F.E. Reversal of diabetes insipidus in Brattleboro tats: Intrahypothalamic injection of vasopressin mRNA. Science 1992, 255, 996–998. [Google Scholar] [CrossRef] [PubMed]
- Heil, F.; Hemmi, H.; Hochrein, H.; Ampenberger, F.; Kirschning, C.; Akira, S.; Lipford, G.; Wagner, H.; Bauer, S. Species-Specific Recognition of Single-Stranded RNA via Toll-like Receptor 7 and 8. Science 2004, 303, 1526–1529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pichlmair, A.; Schulz, O.; Tan, C.-P.; Rehwinkel, J.; Kato, H.; Takeuchi, O.; Akira, S.; Way, M.; Schiavo, G.; e Sousa, C.R. Activation of MDA5 Requires Higher-Order RNA Structures Generated during Virus Infection. J. Virol. 2009, 83, 10761–10769. [Google Scholar] [CrossRef] [Green Version]
- Schlee, M.; Roth, A.; Hornung, V.; Hagmann, C.A.; Wimmenauer, V.; Barchet, W.; Coch, C.; Janke, M.; Mihailovic, A.; Wardle, G.; et al. Recognition of 5′ Triphosphate by RIG-I Helicase Requires Short Blunt Double-Stranded RNA as Contained in Panhandle of Negative-Strand Virus. Immunity 2009, 31, 25–34. [Google Scholar] [CrossRef] [Green Version]
- Rigby, R.; Rehwinkel, J. RNA degradation in antiviral immunity and autoimmunity. Trends Immunol. 2015, 36, 179–188. [Google Scholar] [CrossRef] [Green Version]
- Dyer, K.D.; Rosenberg, H.F. The RNase a superfamily: Generation of diversity and innate host defense. Mol. Divers. 2006, 10, 585–597. [Google Scholar] [CrossRef] [Green Version]
- Karikó, K.; Muramatsu, H.; Welsh, F.A.; Ludwig, J.; Kato, H.; Akira, S.; Weissman, D. Incorporation of Pseudouridine Into mRNA Yields Superior Nonimmunogenic Vector with Increased Translational Capacity and Biological Stability. Mol. Ther. 2008, 16, 1833–1840. [Google Scholar] [CrossRef]
- Karikó, K.; Buckstein, M.; Ni, H.; Weissman, D. Suppression of RNA Recognition by Toll-like Receptors: The Impact of Nucleoside Modification and the Evolutionary Origin of RNA. Immunity 2005, 23, 165–175. [Google Scholar] [CrossRef] [Green Version]
- Sultana, N.; Magadum, A.; Hadas, Y.; Kondrat, J.; Singh, N.; Youssef, E.; Calderon, D.; Chepurko, E.; Dubois, N.; Hajjar, R.J.; et al. Optimizing Cardiac Delivery of Modified mRNA. Mol. Ther. 2017, 25, 1306–1315. [Google Scholar] [CrossRef] [Green Version]
- Eyler, D.E.; Franco, M.K.; Batool, Z.; Wu, M.Z.; Dubuke, M.L.; Dobosz-Bartoszek, M.; Jones, J.; Polikanov, Y.S.; Roy, B.; Koutmou, K.S. Pseudouridinylation of mRNA coding sequences alters translation. Proc. Natl. Acad. Sci. USA 2019, 116, 23068–23074. [Google Scholar] [CrossRef]
- Baden, L.R.; El Sahly, H.M.; Essink, B.; Kotloff, K.; Frey, S.; Novak, R.; Diemert, D.; Spector, S.A.; Rouphael, N.; Creech, C.B.; et al. Efficacy and Safety of the mRNA-1273 SARS-CoV-2 Vaccine. N. Engl. J. Med. 2021, 384, 403–416. [Google Scholar] [CrossRef]
- Polack, F.P.; Thomas, S.J.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Perez, J.L.; Marc, G.P.; Moreira, E.D.; Zerbini, C.; et al. Safety and Efficacy of the BNT162b2 mRNA Covid-19 Vaccine. N. Engl. J. Med. 2020, 383, 2603–2615. [Google Scholar] [CrossRef]
- Adams, D.; Gonzalez-Duarte, A.; O’Riordan, W.D.; Yang, C.-C.; Ueda, M.; Kristen, A.V.; Tournev, I.; Schmidt, H.H.; Coelho, T.; Berk, J.L.; et al. Patisiran, an RNAi Therapeutic, for Hereditary Transthyretin Amyloidosis. N. Engl. J. Med. 2018, 379, 11–21. [Google Scholar] [CrossRef]
- Moss, K.H.; Popova, P.; Hadrup, S.R.; Astakhova, K.; Taskova, M. Lipid Nanoparticles for Delivery of Therapeutic RNA Oligonucleotides. Mol. Pharm. 2019, 16, 2265–2277. [Google Scholar] [CrossRef]
- Piotrowski-Daspit, A.S.; Kauffman, A.C.; Bracaglia, L.G.; Saltzman, W.M. Polymeric vehicles for nucleic acid delivery. Adv. Drug Deliv. Rev. 2020, 156, 119–132. [Google Scholar] [CrossRef] [PubMed]
- Ulkoski, D.; Bak, A.; Wilson, J.T.; Krishnamurthy, V.R. Recent advances in polymeric materials for the delivery of RNA therapeutics. Expert Opin. Drug Deliv. 2019, 16, 1149–1167. [Google Scholar] [CrossRef] [PubMed]
- Di Gioia, S.; Conese, M. Polyethylenimine-mediated gene delivery to the lung and therapeutic applications. Drug Des. Devel. Ther. 2008, 2, 163. [Google Scholar]
- Moghimi, S.M.; Symonds, P.; Murray, J.C.; Hunter, A.; Debska, G.; Szewczyk, A. A two-stage poly(ethylenimine)-mediated cytotoxicity: Implications for gene transfer/therapy. Mol. Ther. 2005, 11, 990–995. [Google Scholar] [CrossRef]
- Yan, Y.; Xiong, H.; Zhang, X.; Cheng, Q.; Siegwart, D.J. Systemic mRNA Delivery to the Lungs by Functional Polyester-based Carriers. Biomacromolecules 2017, 18, 4307–4315. [Google Scholar] [CrossRef]
- Golan, T.; Khvalevsky, E.Z.; Hubert, A.; Gabai, R.M.; Hen, N.; Segal, A.; Domb, A.J.; Harari, G.; Ben-David, E.; Raskin, S.; et al. RNAi therapy targeting KRAS in combination with chemotherapy for locally advanced pancreatic cancer patients. Oncotarget 2015, 6, 24560–24570. [Google Scholar] [CrossRef] [Green Version]
- Ramot, Y.; Rotkopf, S.; Gabai, R.M.; Khvalevsky, E.Z.; Muravnik, S.; Marzoli, G.A.; Domb, A.J.; Shemi, A.; Nyska, A. Preclinical Safety Evaluation in Rats of a Polymeric Matrix Containing an siRNA Drug Used as a Local and Prolonged Delivery System for Pancreatic Cancer Therapy. Toxicol. Pathol. 2016, 44, 856–865. [Google Scholar] [CrossRef] [Green Version]
- Cao, Y.; Tan, Y.F.; Wong, Y.S.; Liew, M.W.J.; Venkatraman, S. Recent Advances in Chitosan-Based Carriers for Gene Delivery. Mar. Drugs 2019, 17, 381. [Google Scholar] [CrossRef] [Green Version]
- Van Meer, G.; Voelker, D.R.; Feigenson, G.W. Membrane lipids: Where they are and how they behave. Nat. Rev. Mol. Cell Biol. 2008, 9, 112–124. [Google Scholar] [CrossRef]
- Cullis, P.R.; Hope, M.J. Lipid Nanoparticle Systems for Enabling Gene Therapies. Mol. Ther. 2017, 25, 1467–1475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bessodes, M.; Dhotel, H.; Mignet, N. Lipids for Nucleic Acid Delivery: Cationic or Neutral Lipoplexes, Synthesis, and Particle Formation. Methods Mol. Biol. 2019, 1943, 123–139. [Google Scholar] [PubMed]
- Xue, H.; Guo, P.; Wen, W.-C.; Wong, H. Lipid-Based Nanocarriers for RNA Delivery. Curr. Pharm. Des. 2015, 21, 3140–3147. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.J.; Tam, Y.K. Controlling Protein Expression by Delivery of RNA Therapeutics Using Lipid Nanoparticles. In Nucleic Acid Nanotheranostics: Biomedical Applications; Elsevier: Amsterdam, The Netherlands, 2019; pp. 277–310. [Google Scholar]
- Suk, J.S.; Xu, Q.; Kim, N.; Hanes, J.; Ensign, L.M. PEGylation as a strategy for improving nanoparticle-based drug and gene delivery. Adv. Drug Deliv. Rev. 2015, 99, 28–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaczmarek, J.C.; Kowalski, P.; Anderson, D.G. Advances in the delivery of RNA therapeutics: From concept to clinical reality. Genome Med. 2017, 9, 60. [Google Scholar] [CrossRef] [Green Version]
- Vlatkovic, I. Non-Immunotherapy Application of LNP-mRNA: Maximizing Efficacy and Safety. Biomedicines 2021, 9, 530. [Google Scholar] [CrossRef]
- Belliveau, N.; Huft, J.; Lin, P.J.; Chen, S.; Leung, A.K.; Leaver, T.J.; Wild, A.W.; Lee, J.B.; Taylor, R.J.; Tam, Y.K.; et al. Microfluidic Synthesis of Highly Potent Limit-size Lipid Nanoparticles for In Vivo Delivery of siRNA. Mol. Ther. Nucleic Acids 2012, 1, e37. [Google Scholar] [CrossRef]
- Ryals, R.C.; Patel, S.; Acosta, C.; McKinney, M.; Pennesi, M.E.; Sahay, G. The effects of PEGylation on LNP based mRNA delivery to the eye. PLoS ONE 2020, 15, e0241006. [Google Scholar] [CrossRef]
- Kumar, V.; Qin, J.; Jiang, Y.; Duncan, R.G.; Brigham, B.; Fishman, S.; Nair, J.K.; Akinc, A.; Barros, S.A.; Kasperkovitz, P.V. Shielding of Lipid Nanoparticles for siRNA Delivery: Impact on Physicochemical Properties, Cytokine Induction, and Efficacy. Mol. Ther. Nucleic Acids 2014, 3, e210. [Google Scholar] [CrossRef]
- Samaridou, E.; Heyes, J.; Lutwyche, P. Lipid nanoparticles for nucleic acid delivery: Current perspectives. Adv. Drug Deliv. Rev. 2020, 154–155, 37–63. [Google Scholar] [CrossRef] [PubMed]
- Judge, A.; McClintock, K.; Phelps, J.R.; Maclachlan, I. Hypersensitivity and loss of disease site targeting caused by antibody responses to PEGylated liposomes. Mol. Ther. 2006, 13, 328–337. [Google Scholar] [CrossRef]
- Suzuki, T.; Suzuki, Y.; Hihara, T.; Kubara, K.; Kondo, K.; Hyodo, K.; Yamazaki, K.; Ishida, T.; Ishihara, H. PEG shedding-rate-dependent blood clearance of PEGylated lipid nanoparticles in mice: Faster PEG shedding attenuates anti-PEG IgM production. Int. J. Pharm. 2020, 588, 119792. [Google Scholar] [CrossRef]
- Eygeris, Y.; Patel, S.; Jozic, A.; Sahay, G. Deconvoluting Lipid Nanoparticle Structure for Messenger RNA Delivery. Nano Lett. 2020, 20, 4543–4549. [Google Scholar] [CrossRef] [PubMed]
- Malone, R.W.; Felgner, P.L.; Verma, I.M. Cationic liposome-mediated RNA transfection [cationic lipid vesicies/N-[1-(2,3-dioleyloxy)propyl]-NNN-timethylammonium chloride (DOTMA)/translationj. Proc. Nati. Acad. Sci. USA 1989, 86, 6077–6081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dokka, S.; Toledo, D.; Shi, X.; Castranova, V.; Rojanasakul, Y. Oxygen Radical-Mediated Pulmonary Toxicity Induced by Some Cationic Liposomes. Pharm. Res. 2000, 17, 521–525. [Google Scholar] [CrossRef]
- Semple, S.C.; Akinc, A.; Chen, J.; Sandhu, A.P.; Mui, B.L.; Cho, C.K.; Sah, D.W.Y.; Stebbing, D.; Crosley, E.J.; Yaworski, E.; et al. Rational design of cationic lipids for siRNA delivery. Nat. Biotechnol. 2010, 28, 172–176. [Google Scholar] [CrossRef] [PubMed]
- Hoekstra, D.; Scherphof, G. Effect of fetal calf serum and serum protein fractions on the uptake of liposomal phosphatidylcholine by rat hepatocytes in primary monolayer culture. Biochim. Biophys. Acta BBA Biomembr. 1979, 551, 109–121. [Google Scholar] [CrossRef]
- Chonn, A.; Semple, S.; Cullis, P. Association of blood proteins with large unilamellar liposomes in vivo. Relation to circulation lifetimes. J. Biol. Chem. 1992, 267, 18759–18765. [Google Scholar] [CrossRef]
- Filion, M.C.; Phillips, N.C. Toxicity and immunomodulatory activity of liposomal vectors formulated with cationic lipids toward immune effector cells. Biochim. Biophys. Acta BBA Biomembr. 1997, 1329, 345–356. [Google Scholar] [CrossRef] [Green Version]
- Kedmi, R.; Ben-Arie, N.; Peer, D. The systemic toxicity of positively charged lipid nanoparticles and the role of Toll-like receptor 4 in immune activation. Biomaterials 2010, 31, 6867–6875. [Google Scholar] [CrossRef]
- Lonez, C.; Bessodes, M.; Scherman, D.; Vandenbranden, M.; Escriou, V.; Ruysschaert, J.-M. Cationic lipid nanocarriers activate Toll-like receptor 2 and NLRP3 inflammasome pathways. Nanomed. Nanotechnol. Biol. Med. 2014, 10, 775–782. [Google Scholar] [CrossRef]
- Shirai, S.; Kawai, A.; Shibuya, M.; Munakata, L.; Omata, D.; Suzuki, R.; Yoshioka, Y. Lipid Nanoparticle Acts as a Potential Adjuvant for Influenza Split Vaccine without Inducing Inflammatory Responses. Vaccines 2020, 8, 433. [Google Scholar] [CrossRef]
- Sato, Y.; Hatakeyama, H.; Sakurai, Y.; Hyodo, M.; Akita, H.; Harashima, H. A pH-sensitive cationic lipid facilitates the delivery of liposomal siRNA and gene silencing activity in vitro and in vivo. J. Control. Release 2012, 163, 267–276. [Google Scholar] [CrossRef] [Green Version]
- Akinc, A.; Maier, M.A.; Manoharan, M.; Fitzgerald, K.; Jayaraman, M.; Barros, S.; Ansell, S.; Du, X.; Hope, M.J.; Madden, T.D.; et al. The Onpattro story and the clinical translation of nanomedicines containing nucleic acid-based drugs. Nat. Nanotechnol. 2019, 14, 1084–1087. [Google Scholar] [CrossRef]
- Kulkarni, J.; Cullis, P.R.; van der Meel, R. Lipid Nanoparticles Enabling Gene Therapies: From Concepts to Clinical Utility. Nucleic Acid Ther. 2018, 28, 146–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlich, M.; Palomba, R.; Costabile, G.; Mizrahy, S.; Pannuzzo, M.; Peer, D.; Decuzzi, P. Cytosolic delivery of nucleic acids: The case of ionizable lipid nanoparticles. Bioeng. Transl. Med. 2021, 6, e10213. [Google Scholar] [CrossRef]
- Sayers, E.; Peel, S.E.; Schantz, A.; England, R.M.; Beano, M.; Bates, S.M.; Desai, A.S.; Puri, S.; Ashford, M.B.; Jones, A.T. Endocytic Profiling of Cancer Cell Models Reveals Critical Factors Influencing LNP-Mediated mRNA Delivery and Protein Expression. Mol. Ther. 2019, 27, 1950–1962. [Google Scholar] [CrossRef]
- Maugeri, M.; Nawaz, M.; Papadimitriou, A.; Angerfors, A.; Camponeschi, A.; Na, M.; Hölttä, M.; Skantze, P.; Johansson, S.; Sundqvist, M.; et al. Linkage between endosomal escape of LNP-mRNA and loading into EVs for transport to other cells. Nat. Commun. 2019, 10, 4333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeffs, L.B.; Palmer, L.R.; Ambegia, E.G.; Giesbrecht, C.; Ewanick, S.; MacLachlan, I. A Scalable, Extrusion-Free Method for Efficient Liposomal Encapsulation of Plasmid DNA. Pharm. Res. 2005, 22, 362–372. [Google Scholar] [CrossRef] [Green Version]
- Semple, S.C.; Klimuk, S.K.; Harasym, T.O.; Dos Santos, N.; Ansell, S.M.; Wong, K.F.; Maurer, N.; Stark, H.; Cullis, P.R.; Hope, M.J.; et al. Efficient encapsulation of antisense oligonucleotides in lipid vesicles using ionizable aminolipids: Formation of novel small multilamellar vesicle structures. Biochim. Biophys. Acta BBA Biomembr. 2001, 1510, 152–166. [Google Scholar] [CrossRef] [Green Version]
- Terada, T.; Kulkarni, J.A.; Huynh, A.; Chen, S.; Van Der Meel, R.; Tam, Y.Y.C.; Cullis, P.R. Characterization of Lipid Nanoparticles Containing Ionizable Cationic Lipids Using Design-of-Experiments Approach. Langmuir 2021, 37, 1120–1128. [Google Scholar] [CrossRef]
- Pardi, N.; Hogan, M.; Pelc, R.; Muramatsu, H.; Andersen, H.; DeMaso, C.R.; Dowd, K.A.; Sutherland, L.L.; Scearce, R.M.; Parks, R.; et al. Zika virus protection by a single low-dose nucleoside-modified mRNA vaccination. Nature 2017, 543, 248–251. [Google Scholar] [CrossRef]
- Pardi, N.; LaBranche, C.C.; Ferrari, G.; Cain, D.W.; Tombácz, I.; Parks, R.J.; Muramatsu, H.; Mui, B.L.; Tam, Y.K.; Karikó, K.; et al. Characterization of HIV-1 Nucleoside-Modified mRNA Vaccines in Rabbits and Rhesus Macaques. Mol. Ther. Nucleic Acids 2019, 15, 36–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pardi, N.; Hogan, M.J.; Naradikian, M.S.; Parkhouse, K.; Cain, D.W.; Jones, L.; Moody, M.A.; Verkerke, H.P.; Myles, A.; Willis, E.; et al. Nucleoside-modified mRNA vaccines induce potent T follicular helper and germinal center B cell responses. J. Exp. Med. 2018, 215, 1571–1588. [Google Scholar] [CrossRef]
- Richner, J.; Himansu, S.; Dowd, K.A.; Butler, S.L.; Salazar, V.; Fox, J.; Julander, J.G.; Tang, W.; Shresta, S.; Pierson, T.C.; et al. Modified mRNA Vaccines Protect against Zika Virus Infection. Cell 2017, 168, 1114–1125.e10. [Google Scholar] [CrossRef] [Green Version]
- Bahl, K.; Senn, J.J.; Yuzhakov, O.; Bulychev, A.; Brito, L.A.; Hassett, K.J.; Laska, M.E.; Smith, M.; Almarsson, O.; Thompson, J.; et al. Preclinical and Clinical Demonstration of Immunogenicity by mRNA Vaccines against H10N8 and H7N9 Influenza Viruses. Mol. Ther. 2017, 25, 1316–1327. [Google Scholar] [CrossRef] [Green Version]
- Liang, F.; Lindgren, G.; Lin, A.; Thompson, E.A.; Ols, S.; Röhss, J.; John, S.; Hassett, K.; Yuzhakov, O.; Bahl, K.; et al. Efficient Targeting and Activation of Antigen-Presenting Cells In Vivo after Modified mRNA Vaccine Administration in Rhesus Macaques. Mol. Ther. 2017, 25, 2635–2647. [Google Scholar] [CrossRef] [Green Version]
- Patel, S.; Ashwanikumar, N.; Robinson, E.; DuRoss, A.; Sun, C.; Murphy-Benenato, K.E.; Mihai, C.; Almarsson, Ö.; Sahay, G. Boosting Intracellular Delivery of Lipid Nanoparticle-Encapsulated mRNA. Nano Lett. 2017, 17, 5711–5718. [Google Scholar] [CrossRef] [PubMed]
- John, S.; Yuzhakov, O.; Woods, A.; Deterling, J.; Hassett, K.; Shaw, C.A.; Ciaramella, G. Multi-antigenic human cytomegalovirus mRNA vaccines that elicit potent humoral and cell-mediated immunity. Vaccine 2018, 36, 1689–1699. [Google Scholar] [CrossRef]
- Pardi, N.; Tuyishime, S.; Muramatsu, H.; Kariko, K.; Mui, B.L.; Tam, Y.K.; Madden, T.D.; Hope, M.J.; Weissman, D. Expression kinetics of nucleoside-modified mRNA delivered in lipid nanoparticles to mice by various routes. J. Control. Release 2015, 217, 345–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hassett, K.J.; Benenato, K.E.; Jacquinet, E.; Lee, A.; Woods, A.; Yuzhakov, O.; Himansu, S.; Deterling, J.; Geilich, B.M.; Ketova, T.; et al. Optimization of Lipid Nanoparticles for Intramuscular Administration of mRNA Vaccines. Mol. Ther. Nucleic Acids 2019, 15, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miao, L.; Li, L.; Huang, Y.; Delcassian, D.; Chahal, J.; Han, J.; Shi, Y.; Sadtler, K.; Gao, W.; Lin, J.; et al. Delivery of mRNA vaccines with heterocyclic lipids increases anti-tumor efficacy by STING-mediated immune cell activation. Nat. Biotechnol. 2019, 37, 1174–1185. [Google Scholar] [CrossRef]
- Getz, G.S.; Reardon, C.A. Apoprotein E as a lipid transport and signaling protein in the blood, liver, and artery wall. J. Lipid Res. 2009, 50, S156–S161. [Google Scholar] [CrossRef] [Green Version]
- Hauser, P.S.; Narayanaswami, V.; Ryan, R.O. Apolipoprotein E: From lipid transport to neurobiology. Prog. Lipid Res. 2011, 50, 62–74. [Google Scholar] [CrossRef] [Green Version]
- Sebastiani, F.; Yanez Arteta, M.; Lerche, M.; Porcar, L.; Lang, C.; Bragg, R.A.; Elmore, C.S.; Krishnamurthy, V.R.; Russell, R.A.; Darwish, T.; et al. Apolipoprotein E Binding Drives Structural and Compositional Rearrangement of mRNA-Containing Lipid Nanoparticles. ACS Nano 2021, 15, 6709–6722. [Google Scholar] [CrossRef]
- Akinc, A.; Querbes, W.; De, S.; Qin, J.; Frank-Kamenetsky, M.; Jayaprakash, K.N.; Jayaraman, M.; Rajeev, K.G.; Cantley, W.L.; Dorkin, J.R.; et al. Targeted Delivery of RNAi Therapeutics with Endogenous and Exogenous Ligand-Based Mechanisms. Mol. Ther. 2010, 18, 1357–1364. [Google Scholar] [CrossRef]
- Cheng, Q.; Wei, T.; Farbiak, L.; Johnson, L.T.; Dilliard, S.A.; Siegwart, D.J. Selective organ targeting (SORT) nanoparticles for tissue-specific mRNA delivery and CRISPR–Cas gene editing. Nat. Nanotechnol. 2020, 15, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Kranz, L.; Diken, M.; Haas, H.; Kreiter, S.; Loquai, C.; Reuter, K.C.; Meng, M.; Fritz, D.; Vascotto, F.; Hefesha, H.; et al. Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immunotherapy. Nature 2016, 534, 396–401. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.B.; Zhang, S.; Kos, P.; Xiong, H.; Zhou, K.; Perelman, S.S.; Zhu, H.; Siegwart, D.J. Non-Viral CRISPR/Cas Gene Editing In Vitro and In Vivo Enabled by Synthetic Nanoparticle Co-Delivery of Cas9 mRNA and sgRNA. Angew. Chem. Int. Ed. 2016, 56, 1059–1063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fehring, V.; Schaeper, U.; Ahrens, K.; Santel, A.; Keil, O.; Eisermann, M.; Giese, K.; Kaufmann, J. Delivery of Therapeutic siRNA to the Lung Endothelium via Novel Lipoplex Formulation DACC. Mol. Ther. 2014, 22, 811–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Cheng, Q.; Wei, T.; Yu, X.; Johnson, L.T.; Farbiak, L.; Siegwart, D.J. Membrane-destabilizing ionizable phospholipids for organ-selective mRNA delivery and CRISPR—Cas gene editing. Nat. Mater. 2021, 20, 701–710. [Google Scholar] [CrossRef]
- Kim, M.; Jeong, M.; Hur, S.; Cho, Y.; Park, J.; Jung, H.; Seo, Y.; Woo, H.A.; Nam, K.T.; Lee, K.; et al. Engineered ionizable lipid nanoparticles for targeted delivery of RNA therapeutics into different types of cells in the liver. Sci. Adv. 2021, 7, eabf4398. [Google Scholar] [CrossRef]
- Bray, F.; Me, J.F.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Yin, H.; Kanasty, R.L.; Eltoukhy, A.A.; Vegas, A.J.; Dorkin, J.R.; Anderson, D.G. Non-viral vectors for gene-based therapy. Nat. Rev. Genet. 2014, 15, 541–555. [Google Scholar] [CrossRef]
- Rosenblum, D.; Gutkin, A.; Kedmi, R.; Ramishetti, S.; Veiga, N.; Jacobi, A.M.; Schubert, M.S.; Friedmann-Morvinski, D.; Cohen, Z.R.; Behlke, M.A.; et al. CRISPR-Cas9 genome editing using targeted lipid nanoparticles for cancer therapy. Sci. Adv. 2020, 6, eabc9450. [Google Scholar] [CrossRef] [PubMed]
- Kedmi, R.; Veiga, N.; Ramishetti, S.; Goldsmith, M.; Rosenblum, D.; Dammes, N.; Hazan-Halevy, I.; Nahary, L.; Leviatan-Ben-Arye, S.; Harlev, M.; et al. A modular platform for targeted RNAi therapeutics. Nat. Nanotechnol. 2018, 13, 214–219. [Google Scholar] [CrossRef]
- Gottschalk, N.; Kimmig, R.; Lang, S.; Singh, M.; Brandau, S. Anti-epidermal growth factor receptor (EGFR) antibodies overcome resistance of ovarian cancer cells to targeted therapy and natural cytotoxicity. Int. J. Mol. Sci. 2012, 13, 12000–12016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, H.; Liu, S.; Wei, T.; Cheng, Q.; Siegwart, D.J. Theranostic dendrimer-based lipid nanoparticles containing PEGylated BODIPY dyes for tumor imaging and systemic mRNA delivery in vivo. J. Control. Release 2020, 325, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Xiong, H.; Kos, P.; Yan, Y.; Zhou, K.; Miller, J.B.; Elkassih, S.; Siegwart, D.J. Activatable Water-Soluble Probes Enhance Tumor Imaging by Responding to Dysregulated pH and Exhibiting High Tumor-to-Liver Fluorescence Emission Contrast. Bioconjugate Chem. 2016, 27, 1737–1744. [Google Scholar] [CrossRef]
- Xiong, H.; Zuo, H.; Yan, Y.; Occhialini, G.; Zhou, K.; Wan, Y.; Siegwart, D.J. High-Contrast Fluorescence Detection of Metastatic Breast Cancer Including Bone and Liver Micrometastases via Size-Controlled pH-Activatable Water-Soluble Probes. Adv. Mater. 2017, 29, 1700131. [Google Scholar] [CrossRef]
- Krienke, C.; Kolb, L.; Diken, E.; Streuber, M.; Kirchhoff, S.; Bukur, T.; Akilli-Öztürk, O.; Kranz, L.M.; Berger, H.; Petschenka, J.; et al. A noninflammatory mRNA vaccine for treatment of experimental autoimmune encephalomyelitis. Science 2021, 371, 145–153. [Google Scholar] [CrossRef]
- Miller, S.D.; Turley, D.M.; Podojil, J.R. Antigen-specific tolerance strategies for the prevention and treatment of autoimmune disease. Nat. Rev. Immunol. 2007, 7, 665–677. [Google Scholar] [CrossRef]
- Karikó, K.; Muramatsu, H.; Ludwig, J.; Weissman, D. Generating the optimal mRNA for therapy: HPLC purification eliminates immune activation and improves translation of nucleoside-modified, protein-encoding mRNA. Nucleic Acids Res. 2011, 39, e142. [Google Scholar] [CrossRef] [Green Version]
- Veiga, N.; Goldsmith, M.; Granot, Y.; Rosenblum, D.; Dammes, N.; Kedmi, R.; Ramishetti, S.; Peer, D. Cell specific delivery of modified mRNA expressing therapeutic proteins to leukocytes. Nat. Commun. 2018, 9, 4493. [Google Scholar] [CrossRef]
- Magadum, A.; Kurian, A.A.; Chepurko, E.; Sassi, Y.; Hajjar, R.J.; Zangi, L. Specific Modified mRNA Translation System. Circulation 2020, 142, 2485–2488. [Google Scholar] [CrossRef] [PubMed]
- Lagos-Quintana, M.; Rauhut, R.; Yalcin, A.; Meyer, J.; Lendeckel, W.; Tuschl, T. Identification of Tissue-Specific MicroRNAs from Mouse. Curr. Biol. 2002, 12, 735–739. [Google Scholar] [CrossRef] [Green Version]
- Callis, T.E.; Pandya, K.; Seok, H.Y.; Tang, R.; Tatsuguchi, M.; Huang, Z.; Chen, J.-F.; Deng, Z.; Gunn, B.; Shumate, J.; et al. MicroRNA-208a is a regulator of cardiac hypertrophy and conduction in mice. J. Clin. Investig. 2009, 119, 2772–2786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tam, Y.K.; Madden, T.D.; Hope, M.J. Pieter Cullis’ quest for a lipid-based, fusogenic delivery system for nucleic acid therapeutics: Success with siRNA so what about mRNA? J. Drug Target. 2016, 24, 774–779. [Google Scholar] [CrossRef] [PubMed]
- Kauffman, K.J.; Dorkin, J.R.; Yang, J.H.; Heartlein, M.W.; DeRosa, F.; Mir, F.F.; Fenton, O.S.; Anderson, D.G. Optimization of Lipid Nanoparticle Formulations for mRNA Delivery in Vivo with Fractional Factorial and Definitive Screening Designs. Nano Lett. 2015, 15, 7300–7306. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Luo, X.; Deng, B.; Wang, J.; McComb, D.W.; Shi, Y.; Gaensler, K.M.L.; Tan, X.; Dunn, A.; Kerlin, B.; et al. An Orthogonal Array Optimization of Lipid-like Nanoparticles for mRNA Delivery in Vivo. Nano Lett. 2015, 15, 8099–8107. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, M.J.; Billingsley, M.M.; Haley, R.M.; Wechsler, M.E.; Peppas, N.A.; Langer, R. Engineering precision nanoparticles for drug delivery. Nat. Rev. Drug Discov. 2020, 20, 101–124. [Google Scholar] [CrossRef] [PubMed]
- Ashby, B. pH Studies in human malignant tumours. Lancet 1966, 288, 312–315. [Google Scholar] [CrossRef]
- Tannock, I.F.; Rotin, D. Acid pH in tumors and its potential for therapeutic exploitation. Cancer Res. 1989, 49, 4373–4384. [Google Scholar]
- Feng, L.; Dong, Z.; Tao, D.; Zhang, Y.; Liu, Z. The acidic tumor microenvironment: A target for smart cancer nano-theranostics. Natl. Sci. Rev. 2017, 5, 269–286. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| AAV | Modified mRNA | miRNA/siRNA | Small Molecules | Protein | |
|---|---|---|---|---|---|
 |  |  |  |  | |
| Route of Administration | Local or I.V | Local or I.V | Local or I.V | I.V or oral | Local |
| Limitation of Gene Size | Yes (4.5 Kb) | No | N/A | N/A | No |
| Pharmacokinetics | Long term | Short term | Short term | Long or short term | Short term |
| Multiple Administration | No | Yes | Yes | Yes | Yes |
| Compromised DNA Integrity | Yes | No | No | No | No |
| Controlled Expression | No | Yes | Yes | Yes | Yes |
| Gene Expression Regulation | Up or down | Up or down | Mostly down | Mostly down | Mostly up |
| Organ Specificity | Yes | No/possibly | No | No | No |
| Cell Specificity | Yes | No/possibly | No | No | No |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Żak, M.M.; Zangi, L. Lipid Nanoparticles for Organ-Specific mRNA Therapeutic Delivery. Pharmaceutics 2021, 13, 1675. https://doi.org/10.3390/pharmaceutics13101675
Żak MM, Zangi L. Lipid Nanoparticles for Organ-Specific mRNA Therapeutic Delivery. Pharmaceutics. 2021; 13(10):1675. https://doi.org/10.3390/pharmaceutics13101675
Chicago/Turabian StyleŻak, Magdalena M., and Lior Zangi. 2021. "Lipid Nanoparticles for Organ-Specific mRNA Therapeutic Delivery" Pharmaceutics 13, no. 10: 1675. https://doi.org/10.3390/pharmaceutics13101675