
Utilising Co-Axial Electrospinning as a Taste-Masking Technology for Paediatric Drug Delivery

 ,
,  ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of Precursor Solutions
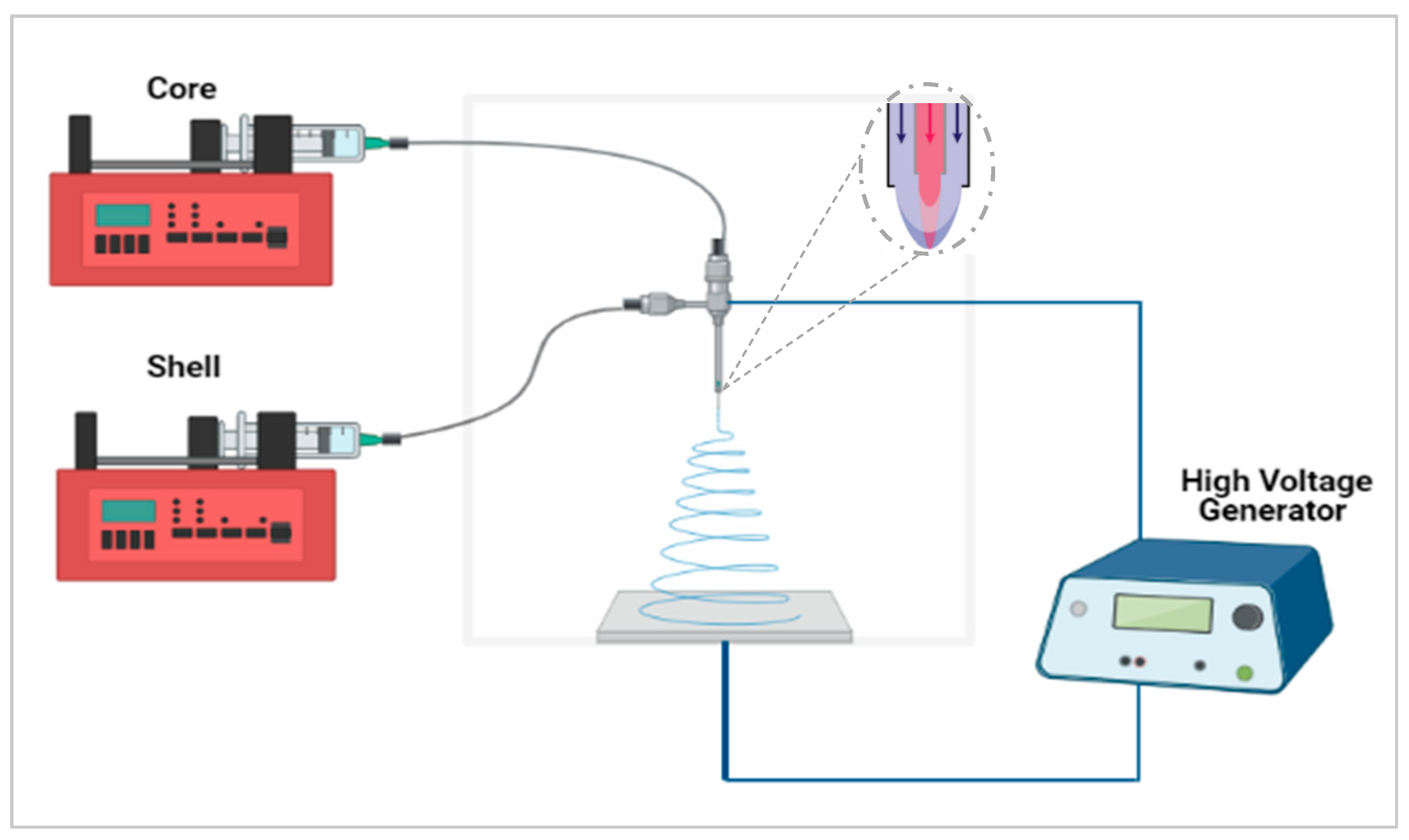
2.3. Electrospinning
Co-Axial Electrospinning
2.4. Fluorescence Microscopy
2.5. Scanning Electron Microscopy
2.6. Transmission Electron Microscopy
2.7. Differential Scanning Calorimetry
2.8. UV Spectroscopy and Drug-Loading
2.9. X-ray Diffraction
2.10. Fourier-Transform Infrared Spectroscopy
2.11. E-Tongue Taste-Assessment
- Measurement of reference potential (Vr) in reference solution;
- Measurement of electric potential (Vs) in sample (initial taste);
- Lightly washing of sensors in reference solution;
- Measurement of electric potential (Vr1) in reference solution again (aftertaste or CPA);
- Refreshing of sensors in alcohol solution to give them a complete wash before the measurement of the next sample.
2.11.1. Bitterness Threshold
2.11.2. Sample Preparation of the Fibres
2.11.3. Data Analysis
2.12. In-Vitro Dissolution
2.13. Film Thickness and Folding Endurance
3. Results and Discussion
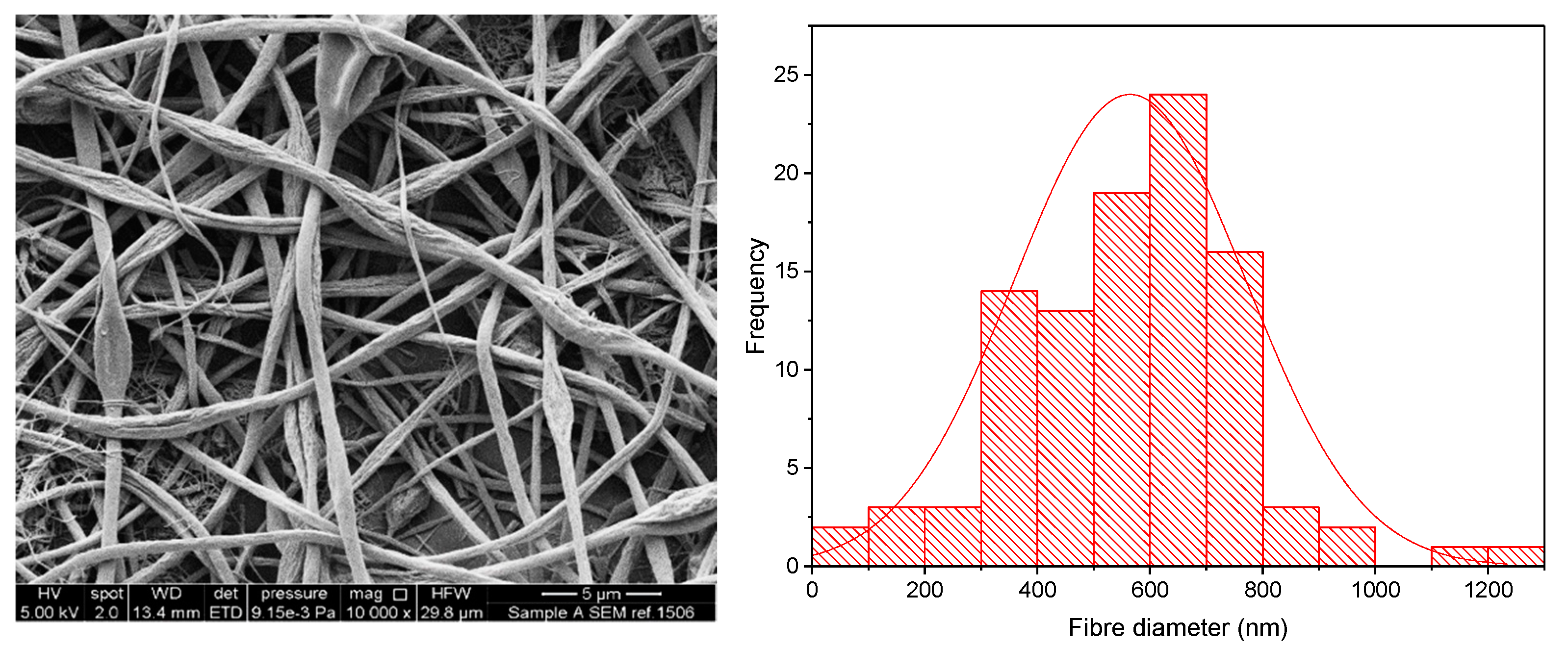
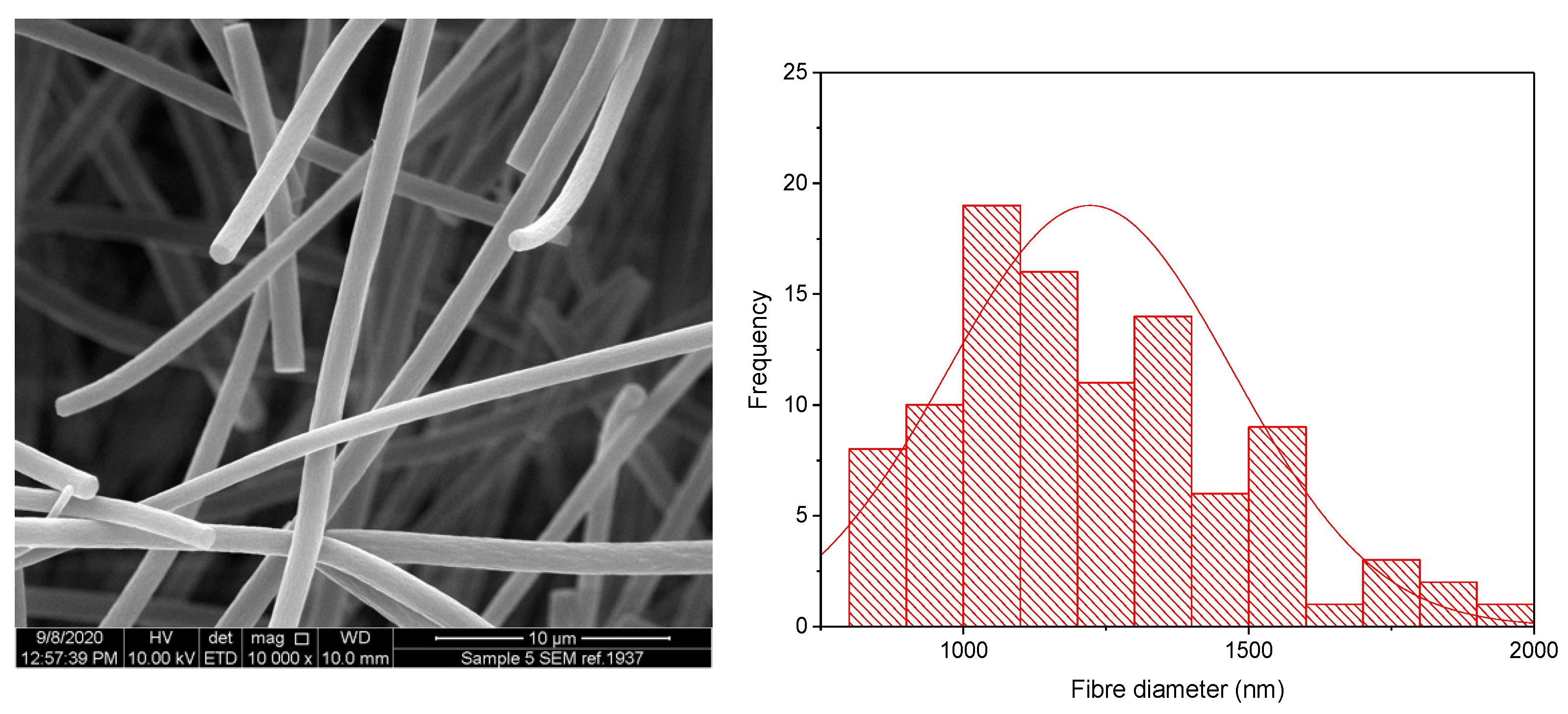
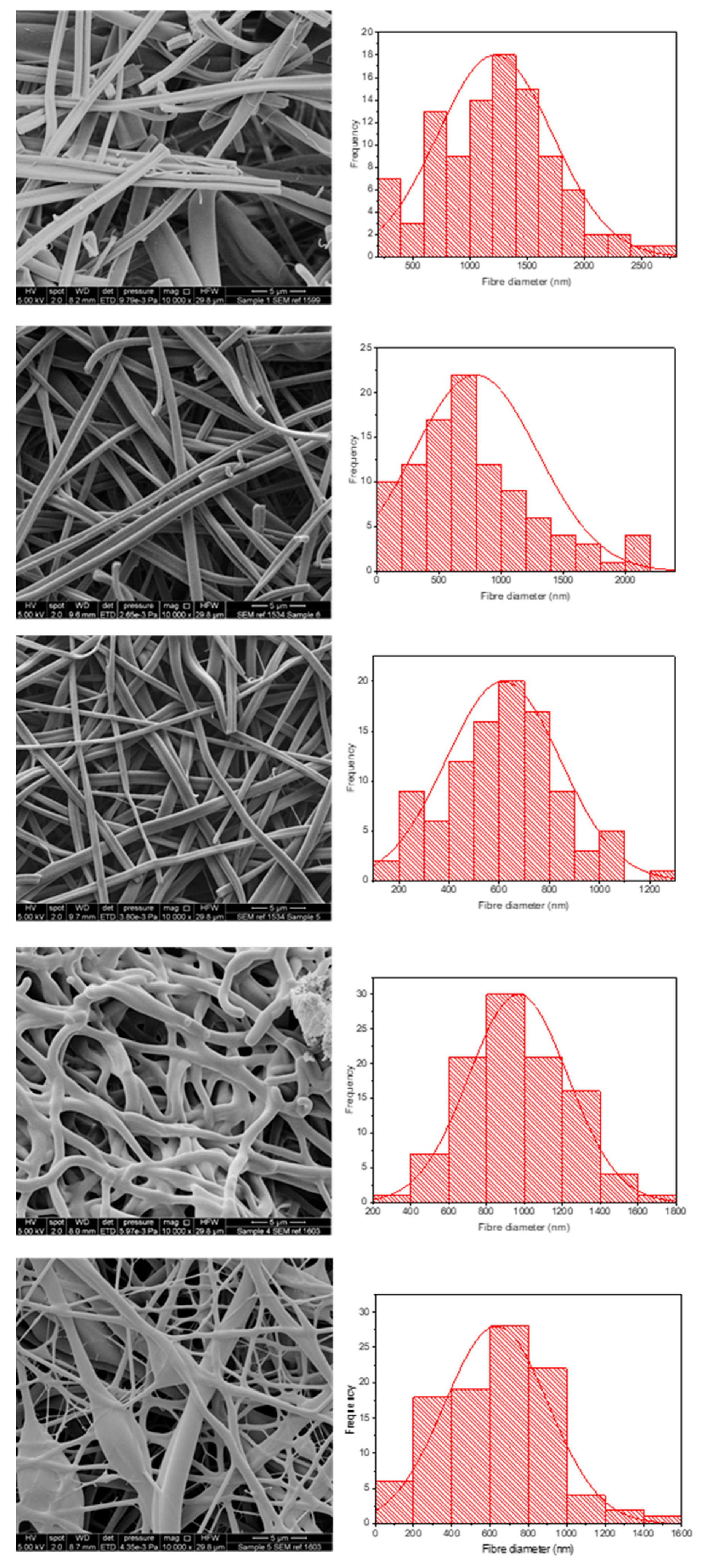
3.1. KCT Electrospinning
3.2. Co-Axial Electrospinning
3.3. Solid State Characterisation
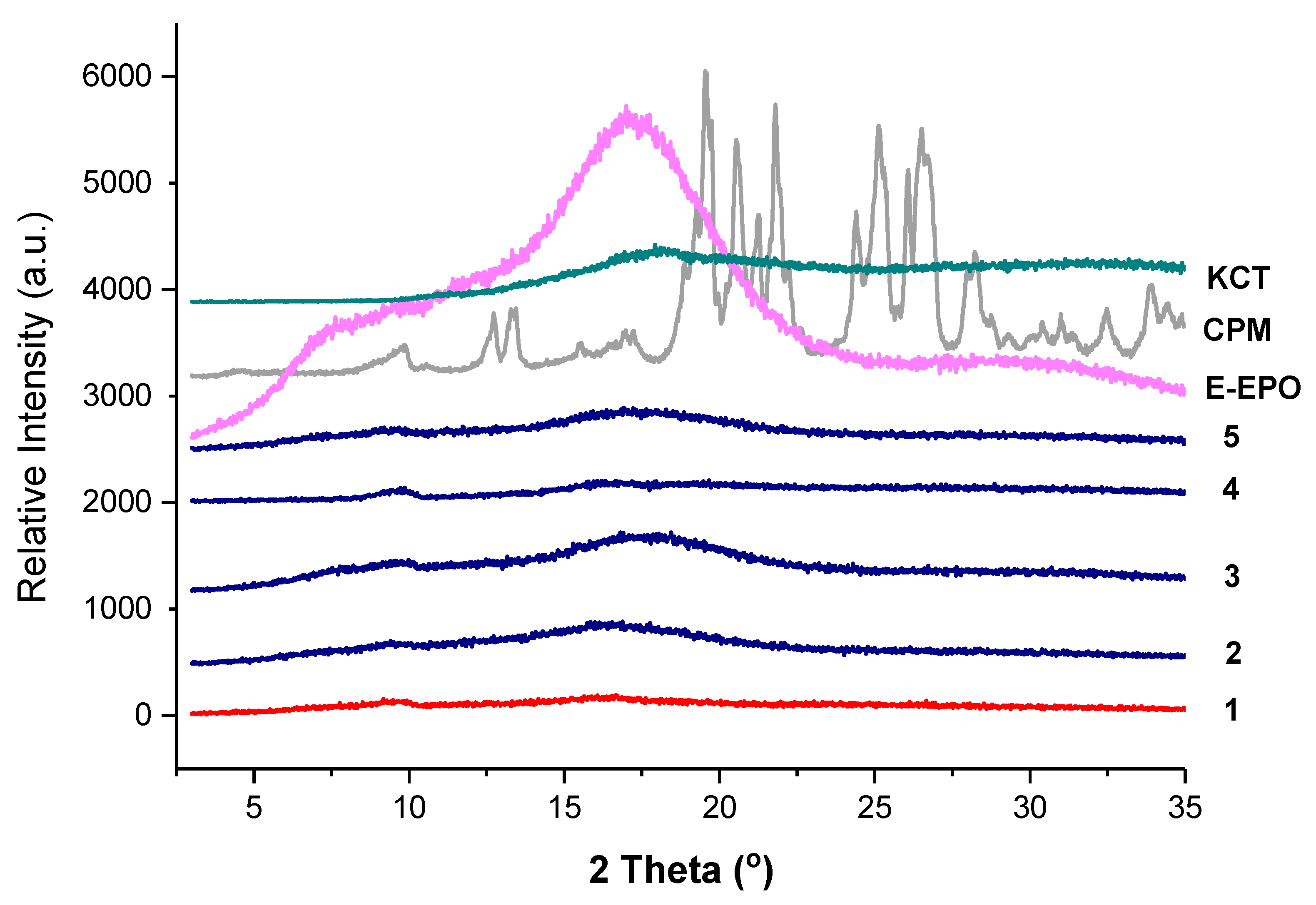
3.3.1. XRD
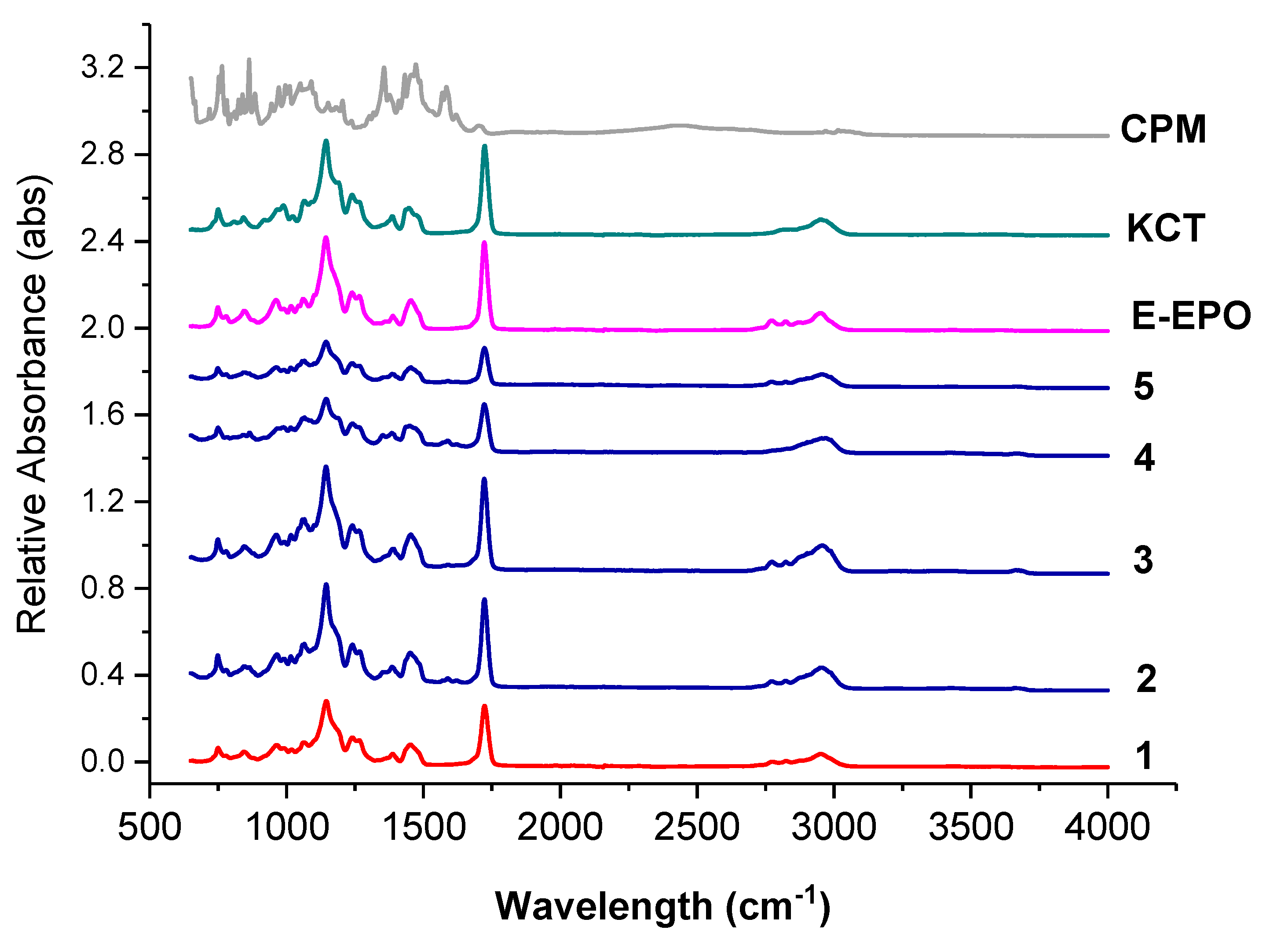
3.3.2. FTIR
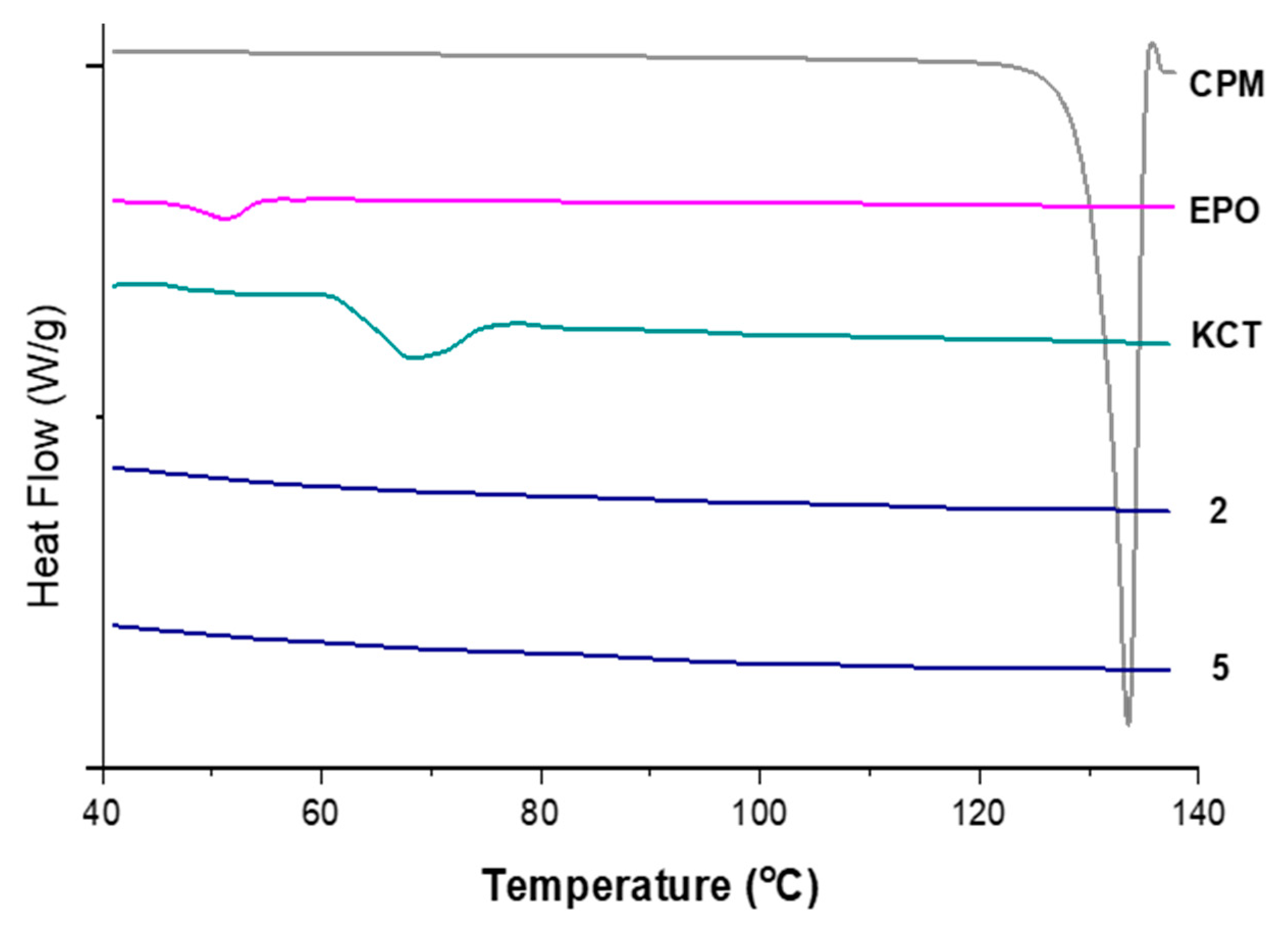
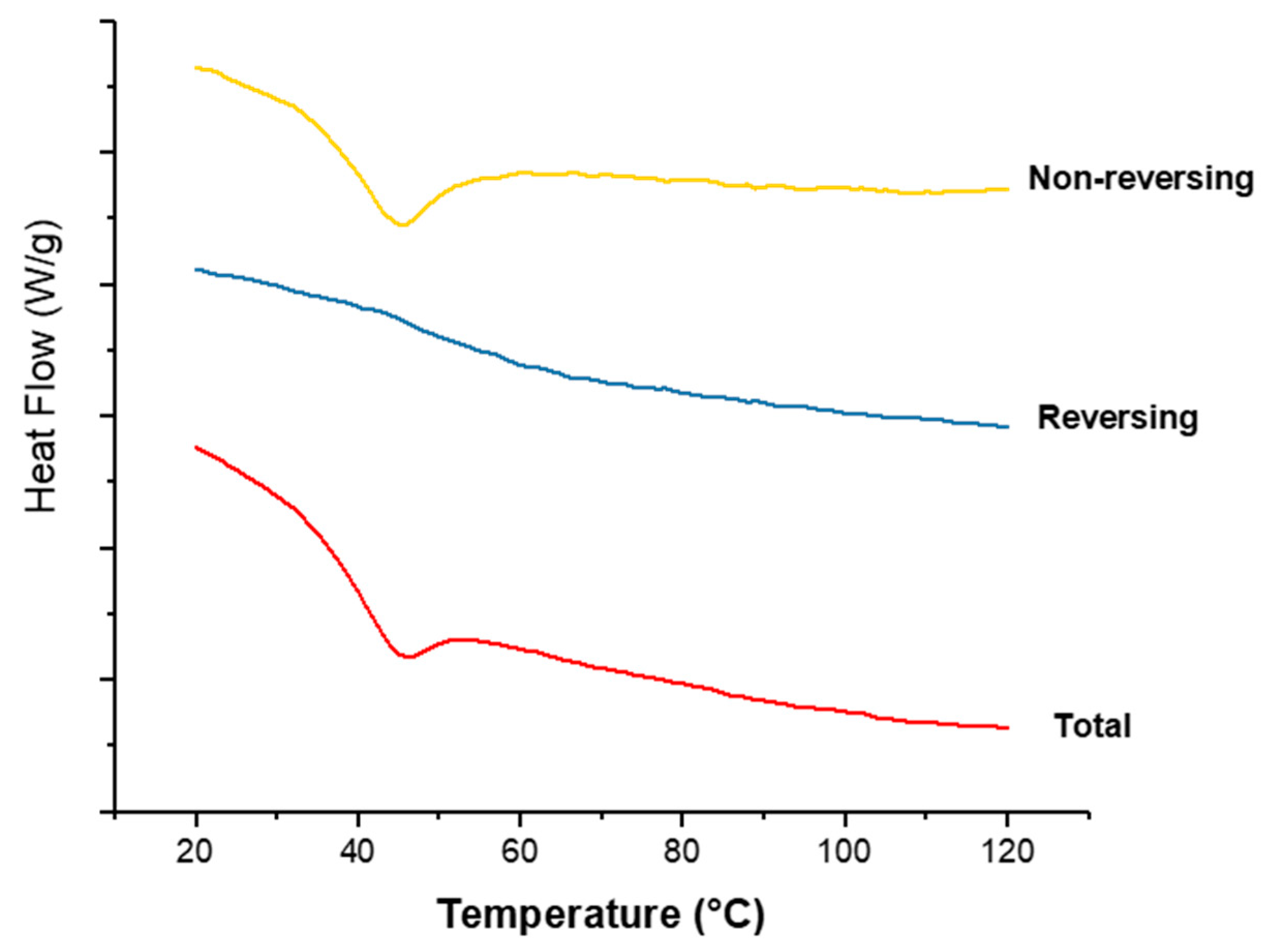
3.3.3. DSC
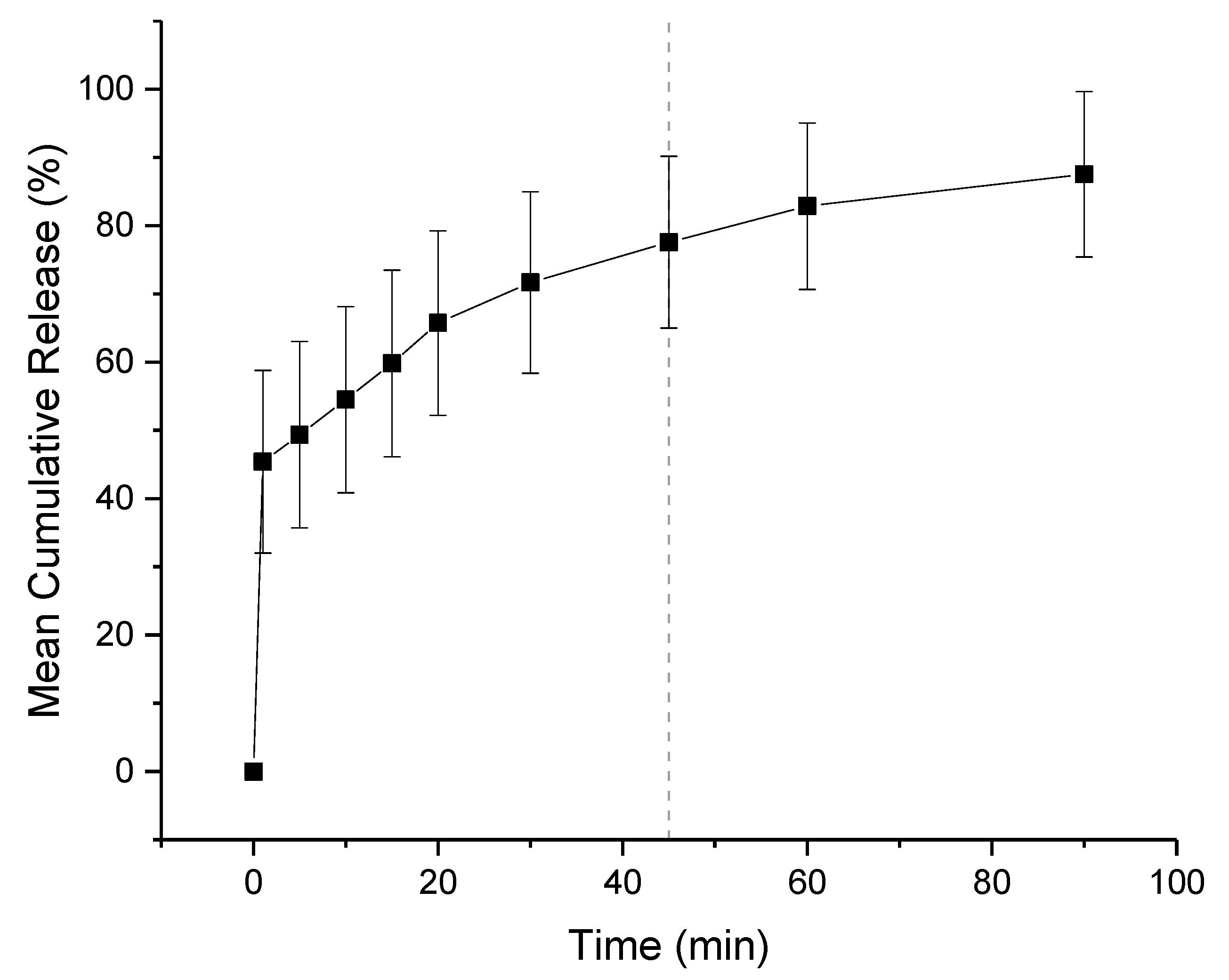
3.4. Dissolution Study
3.5. Film Thickness, pH, and Folding Endurance
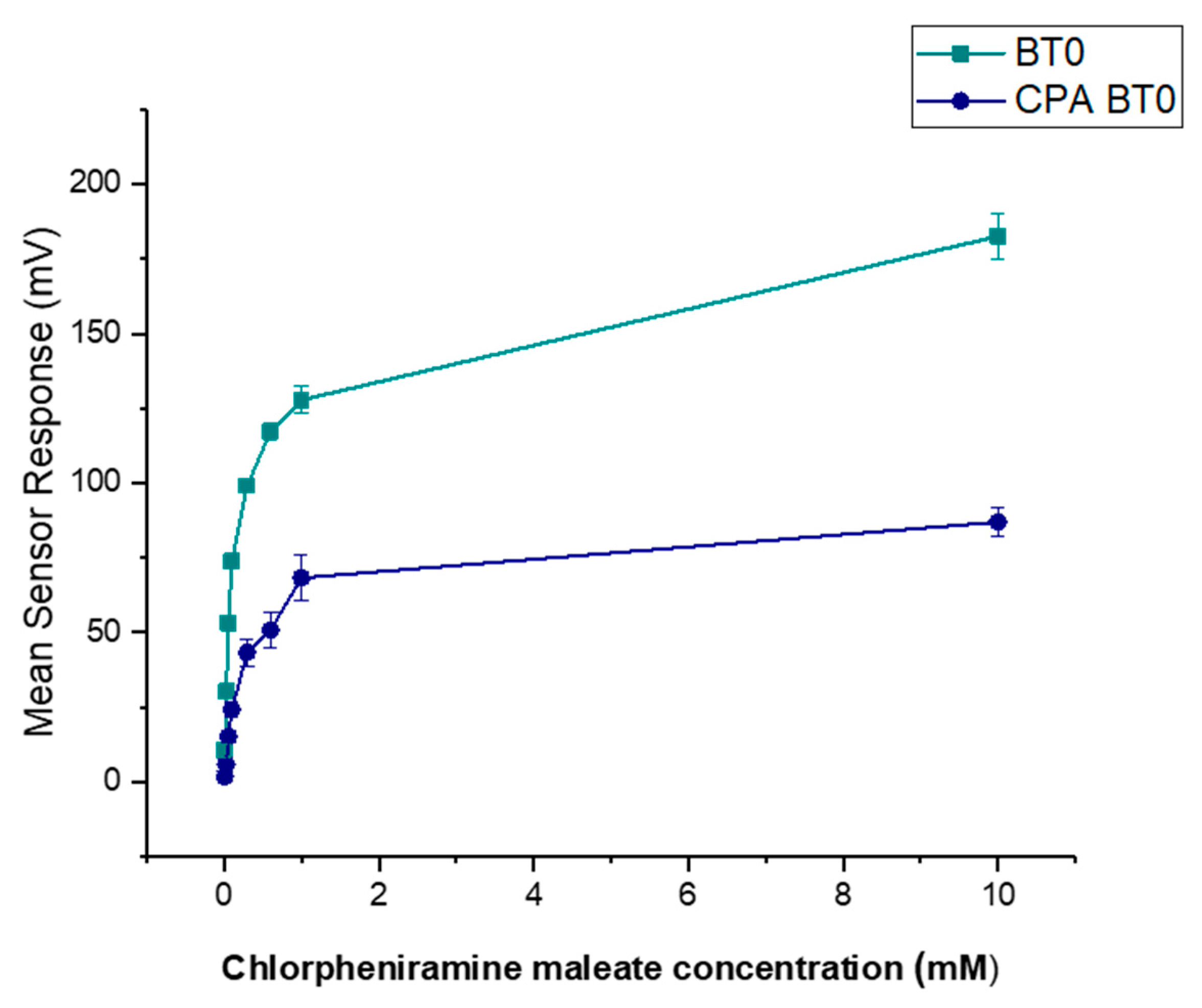
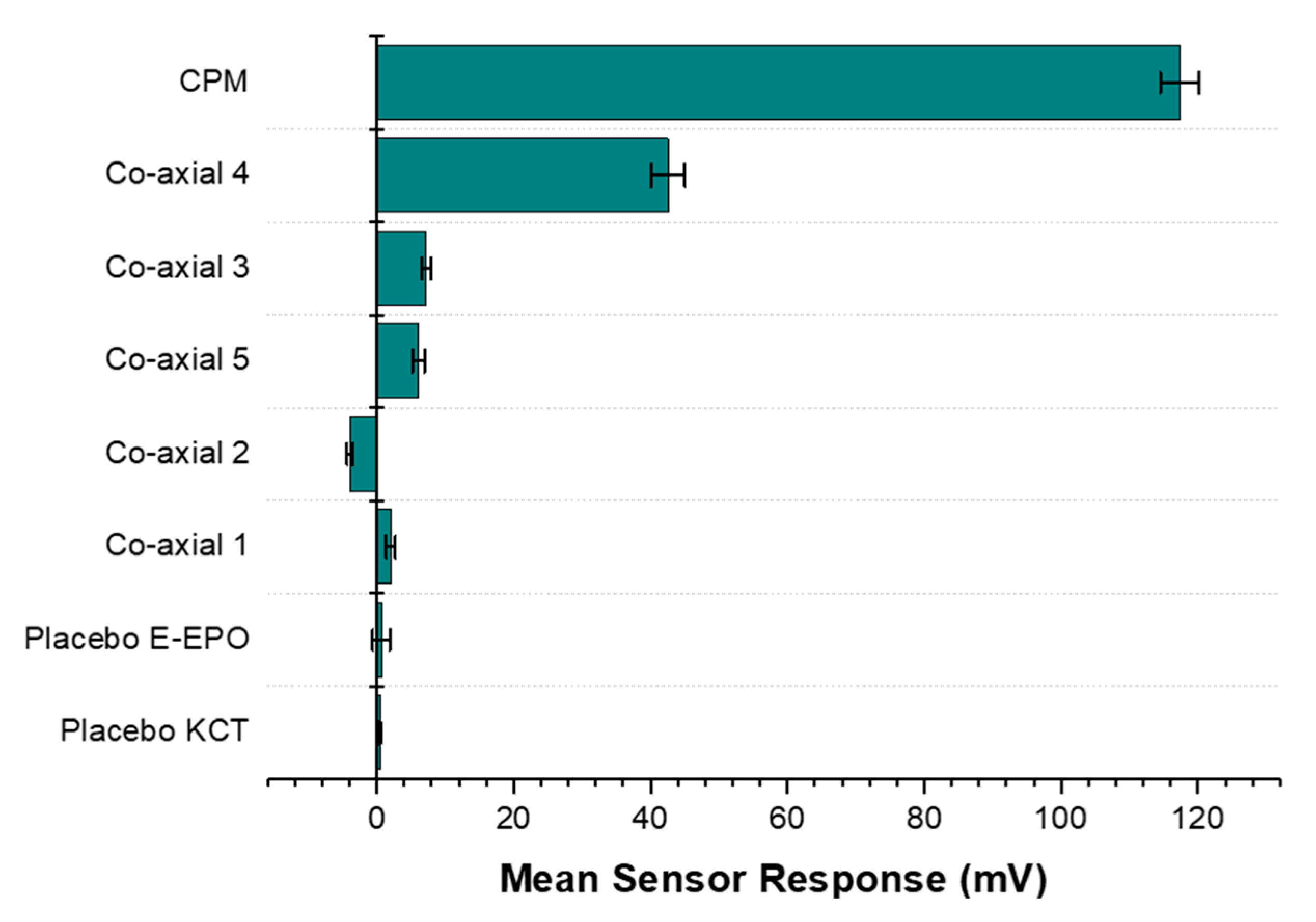
3.6. E-Tongue Taste-Assessment
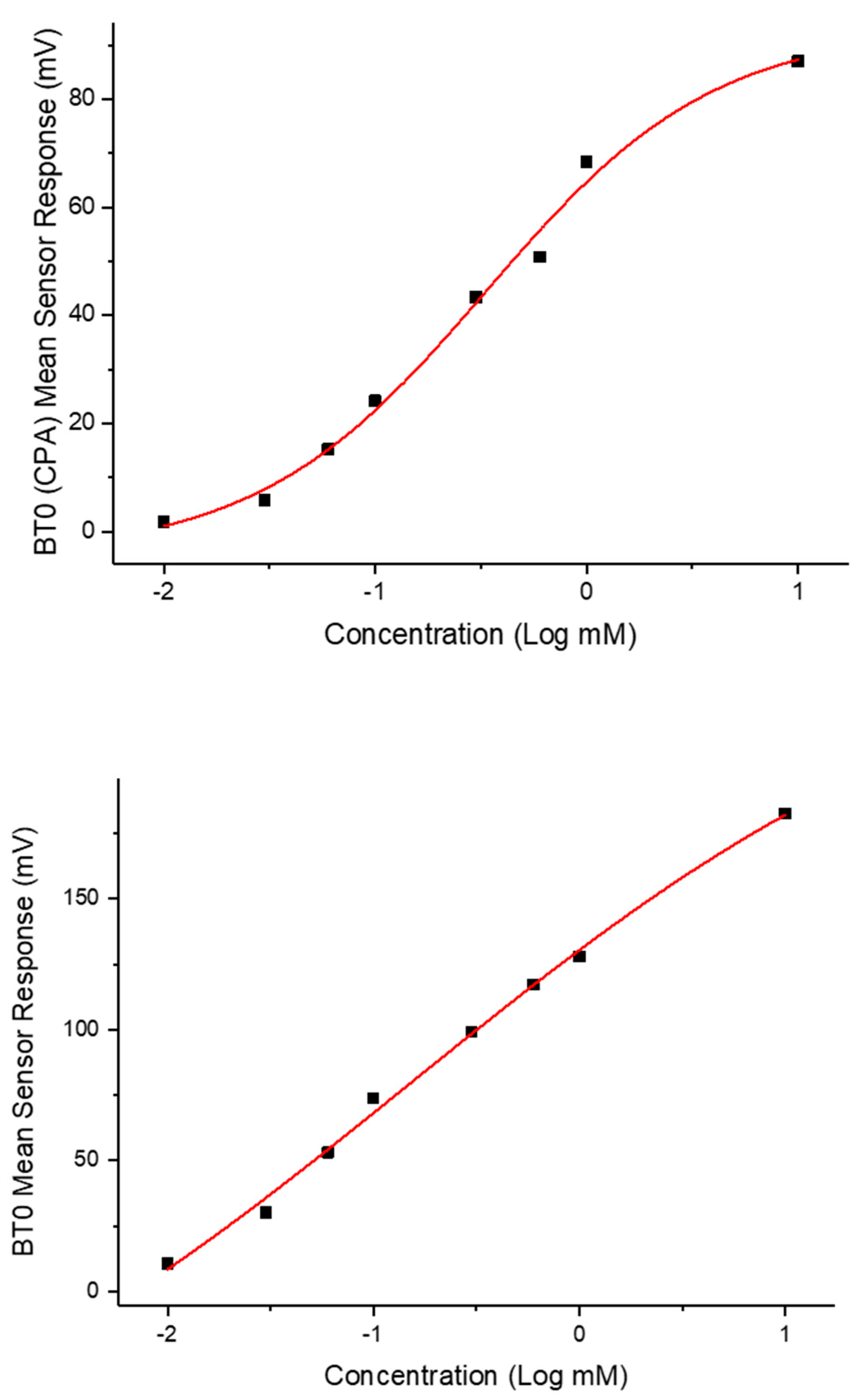
3.6.1. Dose–Response Curve
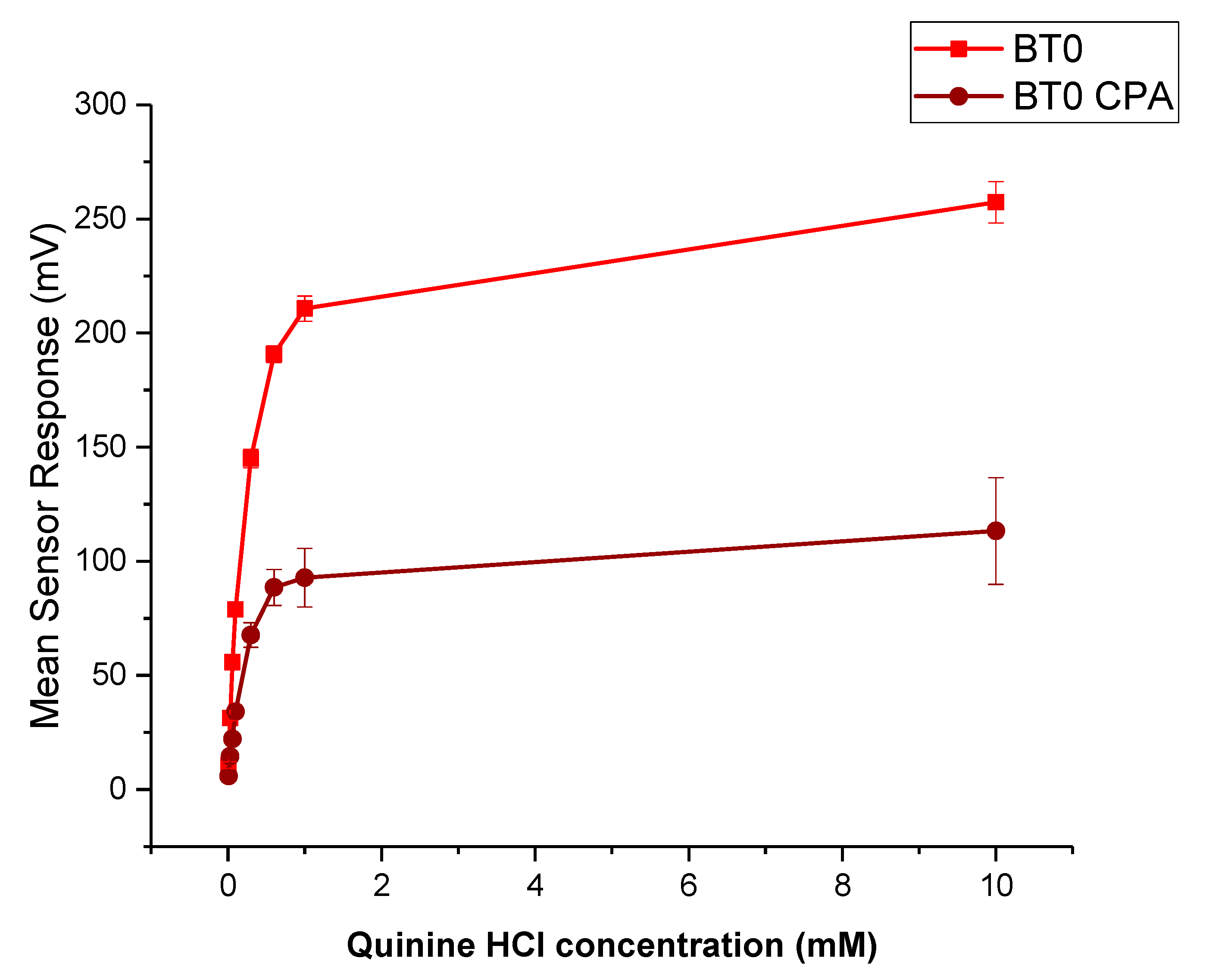
3.6.2. Bitterness Threshold
3.6.3. Formulations Taste-Assessment
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mennella, J.A.; Spector, A.C.; Reed, D.; Coldwell, S.E. The Bad Taste of Medicines: Overview of Basic Research on Bitter Taste. Clin. Ther. 2013, 35, 1225–1246. [Google Scholar] [CrossRef] [Green Version]
- European Parliament; The Council of Europe. The EU Paediatric Regulation 2007; European Parliament: Brussels, Belgium, 2006.
- Turner, M.; Catapano, M.; Hirschfeld, S.; Giaquinto, C. Paediatric drug Development: The Impact of Evolving Regulations. Adv. Drug Deliv. Rev. 2014, 73, 2–13. [Google Scholar] [CrossRef]
- Standing, J.F.; Tuleu, C. Paediatric Formulations-Getting to the Heart of the Problem. Int. J. Pharm. 2005, 300, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Slavkova, M.; Breitkreutz, J. Orodispersible Drug Formulations for Children and Elderly. Eur. J. Pharm. Sci. 2015, 75, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Abdelhakim, H.E.; Williams, G.R.; Craig, D.Q.M.; Orlu, M.; Tuleu, C. Human Mouthfeel Panel Investigating the Acceptability of Electrospun and Solvent Cast Orodispersible Films. Int. J. Pharm. 2020, 585, 119532. [Google Scholar] [CrossRef] [PubMed]
- Samprasit, W.; Akkaramongkolporn, P.; Ngawhirunpat, T.; Rojanarata, T.; Kaomongkolgit, R.; Opanasopit, P. Fast Releasing Oral Electrospun PVP/CD Nanofiber Mats of Taste-Masked Meloxicam. Int. J. Pharm. 2015, 487, 213–222. [Google Scholar] [CrossRef] [PubMed]
- Illangakoon, U.E.; Gill, H.; Shearman, G.C.; Parhizkar, M.; Mahalingam, S.; Chatterton, N.P.; Williams, G.R. Fast Dissolving Paracetamol/Caffeine Nanofibers Prepared by Electrospinning. Int. J. Pharm. 2014, 477, 369–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vrbata, P.; Berka, P.; Stránská, D.; Doležal, P.; Musilová, M.; Čižinská, L. Electrospun Drug Loaded Membranes for Sublingual Administration of Sumatriptan and Naproxen. Int. J. Pharm. 2013, 457, 168–176. [Google Scholar] [CrossRef]
- Wu, Y.-H.; Yu, D.-G.; Li, X.-Y.; Diao, A.-H.; Illangakoon, U.E.; Williams, G.R. Fast-Dissolving Sweet Sedative Nanofiber Membranes. J. Mater. Sci. 2015, 50, 3604–3613. [Google Scholar] [CrossRef]
- Abdelhakim, H.E.; Coupe, A.; Tuleu, C.; Edirisinghe, M.; Craig, D.Q.M. Electrospinning Optimization of Eudragit E PO with and without Chlorpheniramine Maleate Using a Design of Experiment Approach. Mol. Pharm. 2019, 16, 2557–2568. [Google Scholar] [CrossRef] [Green Version]
- Vass, P.; Szabó, E.; Domokos, A.; Marosi, G. Scale-Up of Electrospinning Technology: Applications in the Pharmaceutical Industry. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnology 2020, 12, e1611. [Google Scholar] [CrossRef] [Green Version]
- Shahriar, S.M.S.; Mondal, J.; Hasan, M.N.; Revuri, V.; Lee, D.Y.; Lee, Y.-K. Electrospinning Nanofibers for Therapeutics Delivery. Nanomaterials 2019, 9, 532. [Google Scholar] [CrossRef] [Green Version]
- Jia, D.; Gao, Y.; Williams, G.R. Core/Shell Poly(Ethylene Oxide)/Eudragit Fibers for Site-Specific Release. Int. J. Pharm. 2017, 523, 376–385. [Google Scholar] [CrossRef]
- Tawfik, E.A.; Craig, D.Q.; Barker, S.A. Dual Drug-Loaded Coaxial Nanofibers for the Treatment of Corneal Abrasion. Int. J. Pharm. 2020, 581, 119296. [Google Scholar] [CrossRef]
- Williams, G.R.; Raimi-Abraham, B.T.; Lou, C.J. Nanofibres in Drug Delivery; UCL Press: London, UK, 2018. [Google Scholar]
- BASF. Kollicoat Smartseal 30 D.; BASF Publications: San Diego, CA, USA, 2012; pp. 1–8. [Google Scholar]
- Krampe, R.; Visser, J.C.; Frijlink, H.W.; Breitkreutz, J.; Woerdenbag, H.J.; Preis, M. Oromucosal Film Preparations: Points to Consider for Patient Centricity and Manufacturing Processes. Expert Opin. Drug Deliv. 2015, 13, 493–506. [Google Scholar] [CrossRef] [PubMed]
- EMA. Guideline on Pharmaceutical Development of Medicines for Paediatric Use Guideline on Pharmaceutical Development of Medicines for Paediatric Use; EMA: Amsterdam, The Netherlands, 2013; Volume 44, pp. 1–23.
- Lopez, F.L.; Soto, J.; Ernest, T.B.; Gul, M.O.; Tuleu, C. Could Rodents Perceive Oral Grittiness? A Pilot Study. Int. J. Pharm. 2016, 511, 1144. [Google Scholar] [CrossRef]
- Mohamed-Ahmed, A.H.; Soto, J.; Ernest, T.; Tuleu, C. Non-Human Tools for the Evaluation of Bitter Taste in the Design and Development of Medicines: A Systematic Review. Drug Discov. Today 2016, 21, 1170–1180. [Google Scholar] [CrossRef] [PubMed]
- Pein, M.; Preis, M.; Eckert, C.; Kiene, F.E. Taste-Masking Assessment of Solid Oral Dosage Forms-A Critical Review. Int. J. Pharm. 2014, 465, 239–254. [Google Scholar] [CrossRef]
- Woertz, K.; Tissen, C.; Kleinebudde, P.; Breitkreutz, J. Taste Sensing Systems (Electronic Tongues) for Pharmaceutical Applications. Int. J. Pharm. 2011, 417, 256–271. [Google Scholar] [CrossRef] [PubMed]
- Evonik Industries. EUDRAGIT® E PO EUDRAGIT® E 12,5 EUDRAGIT® E 100, EUDRAGIT® E PO and EUDRAGIT® E 12,5 Specification and Test Methods EUDRAGIT® E PO EUDRAGIT® E 12,5; Evonik Industries: Essen, Germany, 2015; pp. 1–6. [Google Scholar]
- OriginLab Corporation. Origin Pro; OriginLab Corporation: Northampton, MA, USA, 2019. [Google Scholar]
- Kobayashi, Y.; Ikezaki, H. Biochemical Sensors: Mimicking Gustatory and Olfactory Senses; Pan Stanford Publishing: Singapore, 2013. [Google Scholar]
- Woertz, K.; Tissen, C.; Kleinebudde, P.; Breitkreutz, J. Performance Qualification of an Electronic Tongue Based on ICH Guideline Q2. J. Pharm. Biomed. Anal. 2010, 51, 497–506. [Google Scholar] [CrossRef]
- Keating, A.V.; Soto, J.; Tuleu, C.; Forbes, C.; Zhao, M.; Craig, D.Q. Solid State Characterisation and Taste Masking Efficiency Evaluation of Polymer Based Extrudates of Isoniazid for Paediatric Administration. Int. J. Pharm. 2017, 536, 536–546. [Google Scholar] [CrossRef] [PubMed]
- Bala, R.; Khanna, S.; Pawar, P.; Arora, S. Orally Dissolving Strips: A New Approach to Oral Drug Delivery System. Int. J. Pharm. Investig. 2013, 3, 67–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bin Liew, K.; Tan, Y.T.F.; Peh, K.K. Characterization of Oral Disintegrating Film Containing Donepezil for Alzheimer Disease. AAPS PharmSciTech 2011, 13, 134–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vuddanda, P.R.; Mathew, A.P.; Velaga, S. Electrospun Nanofiber Mats for Ultrafast Release of Ondansetron. React. Funct. Polym. 2016, 99, 65–72. [Google Scholar] [CrossRef]
- Hoffmann, E.M.; Breitenbach, A.; Breitkreutz, J. Advances in Orodispersible Films for Drug Delivery. Expert Opin. Drug Deliv. 2011, 8, 299–316. [Google Scholar] [CrossRef] [PubMed]
- Mahalingam, S.; Matharu, R.; Homer-Vanniasinkam, S.; Edirisinghe, M. Current Methodologies and Approaches for the Formation of Core-Sheath Polymer Fibers for Biomedical Applications. Appl. Phys. Rev. 2020, 7, 1–32. [Google Scholar] [CrossRef]
- Chang, S.; Wang, M.; Zhang, F.; Liu, Y.; Liu, X.; Yu, D.-G.; Shen, H. Sheath-Separate-Core Nanocomposites Fabricated Using a Trifluid Electrospinning. Mater. Des. 2020, 192, 108782. [Google Scholar] [CrossRef]
- Zuo, W.; Zhu, M.; Yang, W.; Yu, H.; Chen, Y.; Zhang, Y. Experimental Study on Relationship between Jet Instability and Formation of Beaded Fibers during Electrospinning. Polym. Eng. Sci. 2005, 45, 704–709. [Google Scholar] [CrossRef]
- Lopez, F.L.; Shearman, G.C.; Gaisford, S.; Williams, G.R. Amorphous Formulations of Indomethacin and Griseofulvin Prepared by Electrospinning. Mol. Pharm. 2014, 11, 4327–4338. [Google Scholar] [CrossRef]
- Sill, T.J.; von Recum, H.A. Electrospinning: Applications in Drug Delivery and Tissue Engineering. Biomaterials 2008, 29, 1989–2006. [Google Scholar] [CrossRef]
- Reneker, D.H.; Yarin, A.L. Electrospinning Jets and Polymer Nanofibers. Polymer 2008, 49, 2387–2425. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Hai, T.; Feng, Z.; Yu, D.-G.; Yang, Y.; Bligh, S.W.A. The Relationships between the Working Fluids, Process Characteristics and Products from the Modified Coaxial Electrospinning of Zein. Polymers 2019, 11, 1287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haru, Y.; Tomioka, A. Luminescent Electrospun Nanofibers Doped with Organic Dye: Toward a Disentangled Deposition. Phys. Status Solidi Basic Res. 2017, 254, 1600721. [Google Scholar] [CrossRef]
- Sigma Aldrich. FTIR Table. Available online: https://www.sigmaaldrich.com/technical-documents/articles/biology/ir-spectrum-table.html (accessed on 1 April 2021).
- PubChem. Chlopheniramine. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/chlorpheniramine#section=Solubility (accessed on 15 August 2017).
- Gupta, S.S.; Solanki, N.; Serajuddin, A.T.M. Investigation of Thermal and Viscoelastic Properties of Polymers Relevant to Hot Melt Extrusion, IV: AffinisolTM HPMC HME Polymers. AAPS PharmSciTech 2016, 17, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Saharan, V.A. Current Advances in Drug Delivery Through Fast Dissolving/Disintegrating Dosage Forms; Bentham Science Publishers: Sharjah, UAE, 2017. [Google Scholar]
- Coleman, N.J.; Craig, D.Q.M. Modulated Temperature Differential Scanning Calorimetry: A Novel Approach to Pharmaceutical Thermal Analysis. Int. J. Pharm. 1996, 135, 13–29. [Google Scholar] [CrossRef]
- Immergut, E.H.; Mark, H.F. Principles of Plasticization; American Chemical Society: Washington, DC, USA, 1965; pp. 1–26. [Google Scholar]
- Ignatious, F.; Sun, L.; Lee, C.-P.; Baldoni, J. Electrospun Nanofibers in Oral Drug Delivery. Pharm. Res. 2010, 27, 576–588. [Google Scholar] [CrossRef]
- EMA. Reflection Paper on the Dissolution Specification for Generic Solid Oral Immediate Release Products with Systemic Action; EMA: Amsterdam, The Netherlands, 2017; Volume 44.
- Liu, T.; Wan, X.; Luo, Z.; Liu, C.; Quan, P.; Cun, D.; Fang, L. A Donepezil/Cyclodextrin Complexation Orodispersible Film: Effect of Cyclodextrin on Taste-Masking Based on Dynamic Process and In Vivo Drug Absorption. Asian J. Pharm. Sci. 2019, 14, 183–192. [Google Scholar] [CrossRef]
- Prajapati, V.D.; Chaudhari, A.M.; Gandhi, A.K.; Maheriya, P. Pullulan Based Oral Thin Film Formulation of Zolmitriptan: Development and Optimization Using Factorial Design. Int. J. Biol. Macromol. 2018, 107, 2075–2085. [Google Scholar] [CrossRef]
- Tian, Y.; Orlu, M.; Woerdenbag, H.J.; Scarpa, M.; Kiefer, O.; Kottke, D.; Sjöholm, E.; Öblom, H.; Sandler, N.; Hinrichs, W.L.J.; et al. Oromucosal Films: From Patient Centricity to Production by Printing Techniques. Expert Opin. Drug Deliv. 2019, 16, 981–993. [Google Scholar] [CrossRef] [Green Version]
- Irfan, M.; Rabel, S.; Bukhtar, Q.; Qadir, M.I.; Jabeen, F.; Khan, A. Orally Disintegrating Films: A Modern Expansion in Drug Delivery System. Saudi Pharm. J. 2015, 24, 537–546. [Google Scholar] [CrossRef] [Green Version]
- Keating, A.V.; Soto, J.; Forbes, C.; Zhao, M.; Craig, D.Q.M.; Tuleu, C. Multi-Methodological Quantitative Taste Assessment of Anti-Tuberculosis Drugs to Support the Development of Palatable Paediatric Dosage Forms. Pharmaceutics 2020, 12, 369. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, Y.; Usui, R.; Ikezaki, H.; Tahara, K.; Takeuchi, H. An Advanced Technique Using an Electronic Taste-Sensing System to Evaluate the Bitterness of Orally Disintegrating Films and the Evaluation of Model Films. Int. J. Pharm. 2017, 531, 179–190. [Google Scholar] [CrossRef] [PubMed]
- Izawa, K.; Amino, Y.; Kohmura, M.; Ueda, Y.; Kuroda, M. 4.16-Human-Environment Interactions-Taste. Compr. Nat. Prod. II 2010, 4, 631–671. [Google Scholar]
- Soto, J.; Keeley, A.; Keating, A.V.; Mohamed-Ahmed, A.H.; Sheng, Y.; Winzenburg, G.; Turner, R.; Desset-Brèthes, S.; Orlu, M.; Tuleu, C. Rats Can Predict Aversiveness of Active Pharmaceutical Ingredients. Eur. J. Pharm. Biopharm. 2018, 133, 77–84. [Google Scholar] [CrossRef] [PubMed]
- BNFC. Chlorpheniramine maleate. Available online: https://www.medicinescomplete.com/mc/bnfc/current/PHP1934-chlorphenamine-maleate.htm?q=clorpheniramine maleate&t=search&ss=text&tot=33&p=1#_hit (accessed on 15 August 2017).
- Ali, J.; Zgair, A.; Hameed, G.; Garnett, M.C.; Roberts, C.; Burley, J.C.; Gershkovich, P. Application of Biorelevant Saliva-Based Dissolution for Optimisation of Orally Disintegrating Formulations of Felodipine. Int. J. Pharm. 2018, 555, 228–236. [Google Scholar] [CrossRef] [PubMed]
- Keeley, A.; Teo, M.; Ali, Z.; Frost, J.; Ghimire, M.; Rajabi-Siahboomi, A.; Orlu, M.; Tuleu, C. In Vitro Dissolution Model Can Predict the in Vivo Taste Masking Performance of Coated Multiparticulates. Mol. Pharm. 2019, 16, 2095–2105. [Google Scholar] [CrossRef]
- Melis, M.; Aragoni, M.C.; Arca, M.; Cabras, T.; Caltagirone, C.; Castagnola, M.; Crnjar, R.; Messana, I.; Tepper, B.J.; Barbarossa, I.T. Marked Increase in PROP Taste Responsiveness Following Oral Supplementation with Selected Salivary Proteins or Their Related Free Amino Acids. PLoS ONE 2013, 8, 1–8. [Google Scholar]
- Chay, S.K.; Keating, A.V.; James, C.; Aliev, A.E.; Haider, S.; Craig, D.Q.M. Evaluation of the Taste-Masking Effects of (2-Hydroxypropyl)-b-Cyclodextrin on Ranitidine Hydrochloride; A Combined Biosensor, Spectroscopic and Molecular Modelling Assessment. RSC Adv. 2018, 8, 3564–3573. [Google Scholar] [CrossRef] [Green Version]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Co-Axial Sample | Core | Shell | Theoretical DL (% w/w) | Actual DL (% w/w) | DL Efficiency (%) | Mean ± SD Diameter (nm) |
|---|---|---|---|---|---|---|
| 1 | KCT | E-EPO | N/A | N/A | Placebo | 1220 ± 501 |
| 2 | KCT | E-EPO | 7.6 | 7.4 ± 0.4 | 97.5 ± 5.3 | 795 ± 505 |
| 3 | E-EPO | E-EPO | 4.8 | 5.3 ± 0.2 | 111.3 ± 4.2 | 616 ± 228 |
| 4 | KCT | KCT | 18.9 | 17.8 ± 3.6 | 94.2 ± 19.0 | 967 ± 262 |
| 5 | E-EPO | KCT | 7.6 | 7.0 ± 1.6 | 92.1 ± 21.1 | 633 ± 271 |
| Formulation | Euclidean Distance |
|---|---|
| Co-axial 4 | 4.43 |
| Co-axial 5 | 7.51 |
| Co-axial 3 | 7.63 |
| Co-axial 2 | 9.40 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abdelhakim, H.E.; Coupe, A.; Tuleu, C.; Edirisinghe, M.; Craig, D.Q.M. Utilising Co-Axial Electrospinning as a Taste-Masking Technology for Paediatric Drug Delivery. Pharmaceutics 2021, 13, 1665. https://doi.org/10.3390/pharmaceutics13101665
Abdelhakim HE, Coupe A, Tuleu C, Edirisinghe M, Craig DQM. Utilising Co-Axial Electrospinning as a Taste-Masking Technology for Paediatric Drug Delivery. Pharmaceutics. 2021; 13(10):1665. https://doi.org/10.3390/pharmaceutics13101665
Chicago/Turabian StyleAbdelhakim, Hend E., Alastair Coupe, Catherine Tuleu, Mohan Edirisinghe, and Duncan Q. M. Craig. 2021. "Utilising Co-Axial Electrospinning as a Taste-Masking Technology for Paediatric Drug Delivery" Pharmaceutics 13, no. 10: 1665. https://doi.org/10.3390/pharmaceutics13101665