
Ustekinumab Dosing Individualization in Crohn’s Disease Guided by a Population Pharmacokinetic–Pharmacodynamic Model

 , , , , , , and
, , , , , , and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Patients and Study Design
2.2. Pharmacokinetic and Pharmacodynamic Data
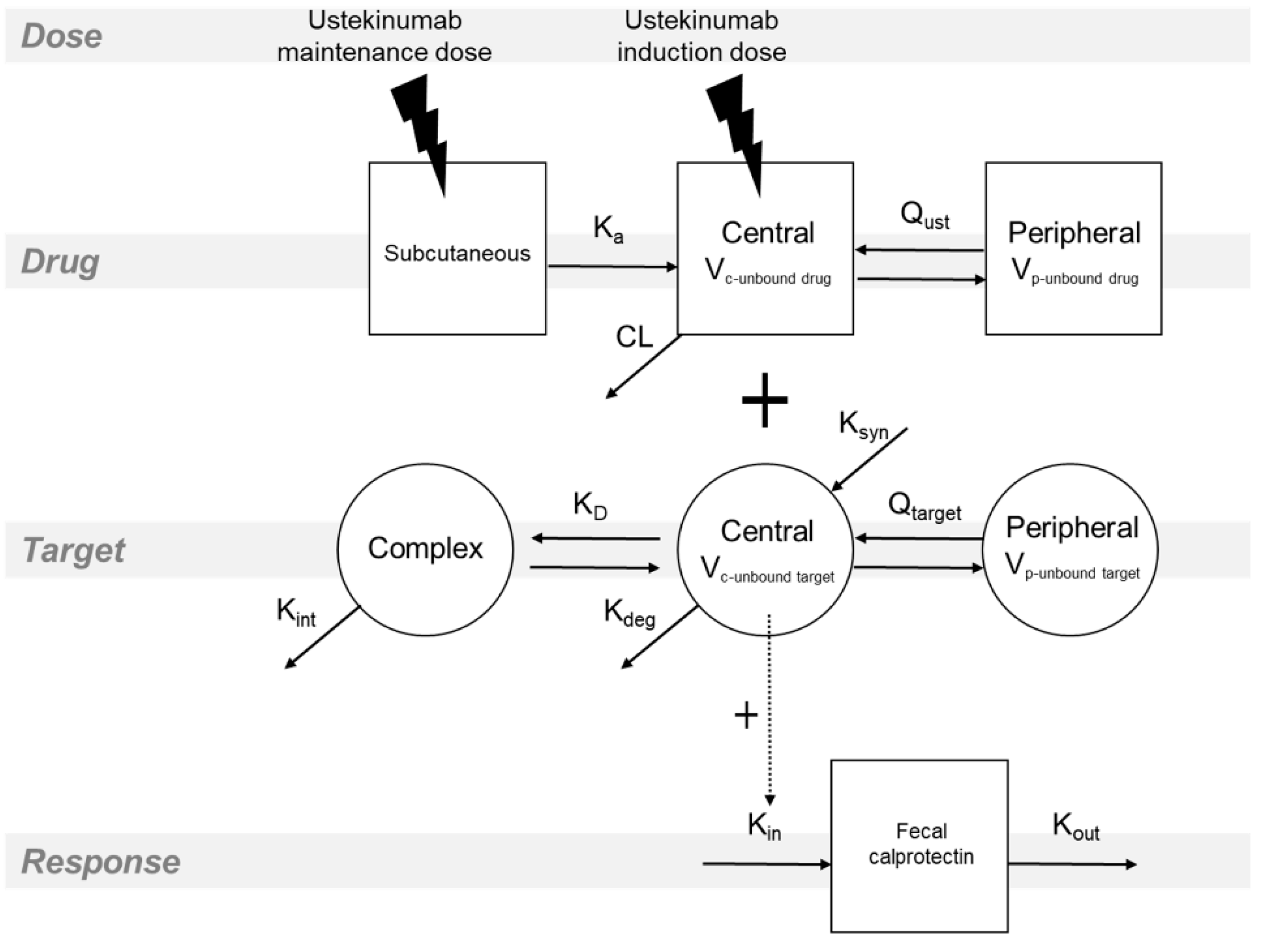
2.3. Statistical Analysis and Pharmacokinetic–Pharmacodynamic Modeling
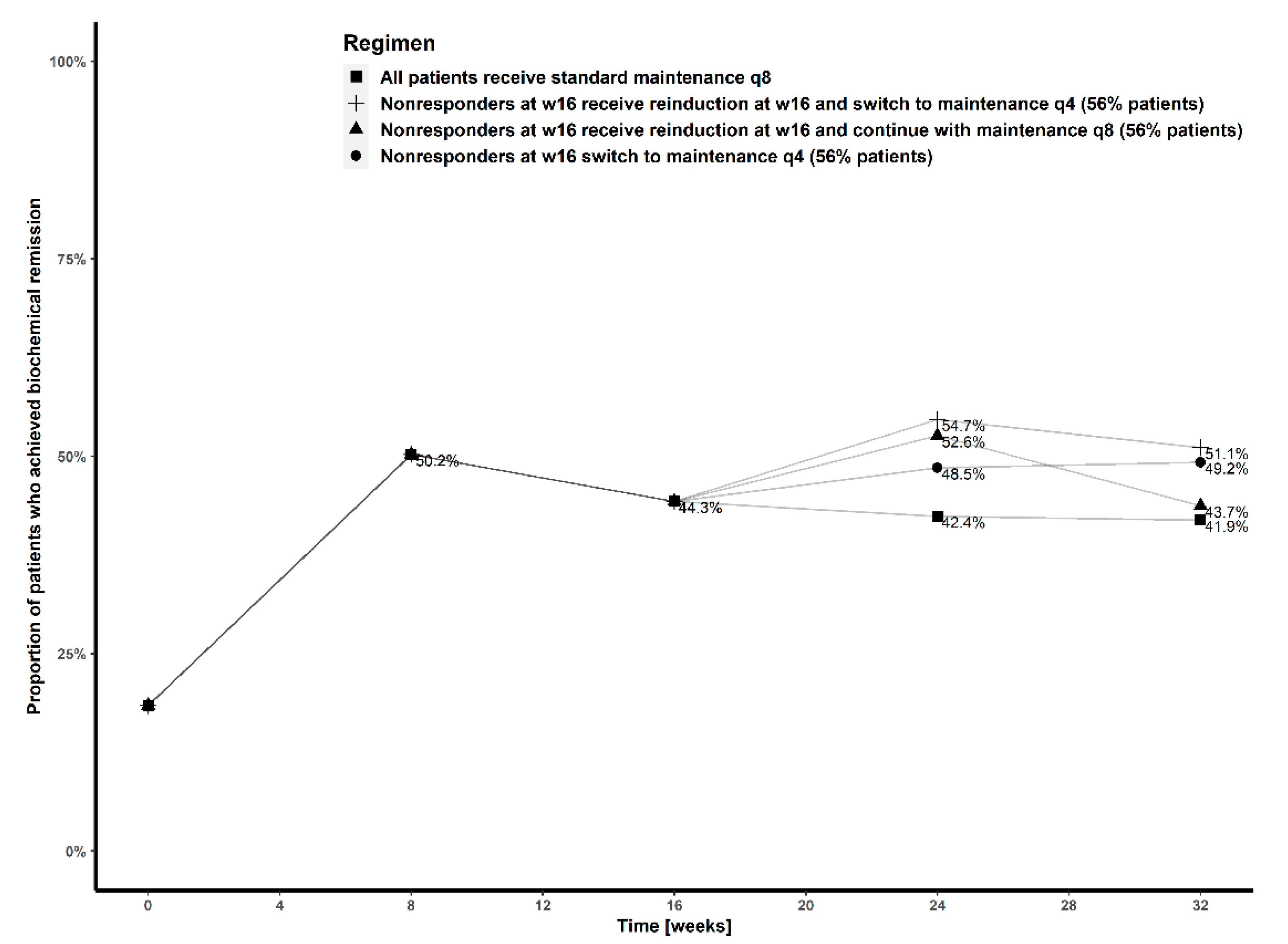
2.4. Model Based Simulations
- (a).
- All patients received standard ustekinumab treatment, with weight-based induction dose at baseline, followed by fixed 90 mg maintenance doses every eight weeks (standard treatment);
- (b).
- All patients received weight-based induction dose at baseline followed by fixed 90 mg maintenance doses every four weeks;
- (c).
- All patients received weight-based induction doses every eight weeks;
- (d).
- Patients receiving standard treatment who were not in remission at week 16, switched to maintenance doses every four weeks from week 20;
- (e).
- Patients receiving standard treatment who were not in remission at week 16 received a weight-based reinduction dose at week 16, and continued with maintenance doses every eight weeks;
- (f).
- Patients receiving standard treatment who were not in remission at week 16 received a weight-based reinduction dose at week 16 and switched to maintenance doses every four weeks from week 20.
3. Results
3.1. Baseline Patient Characteristics
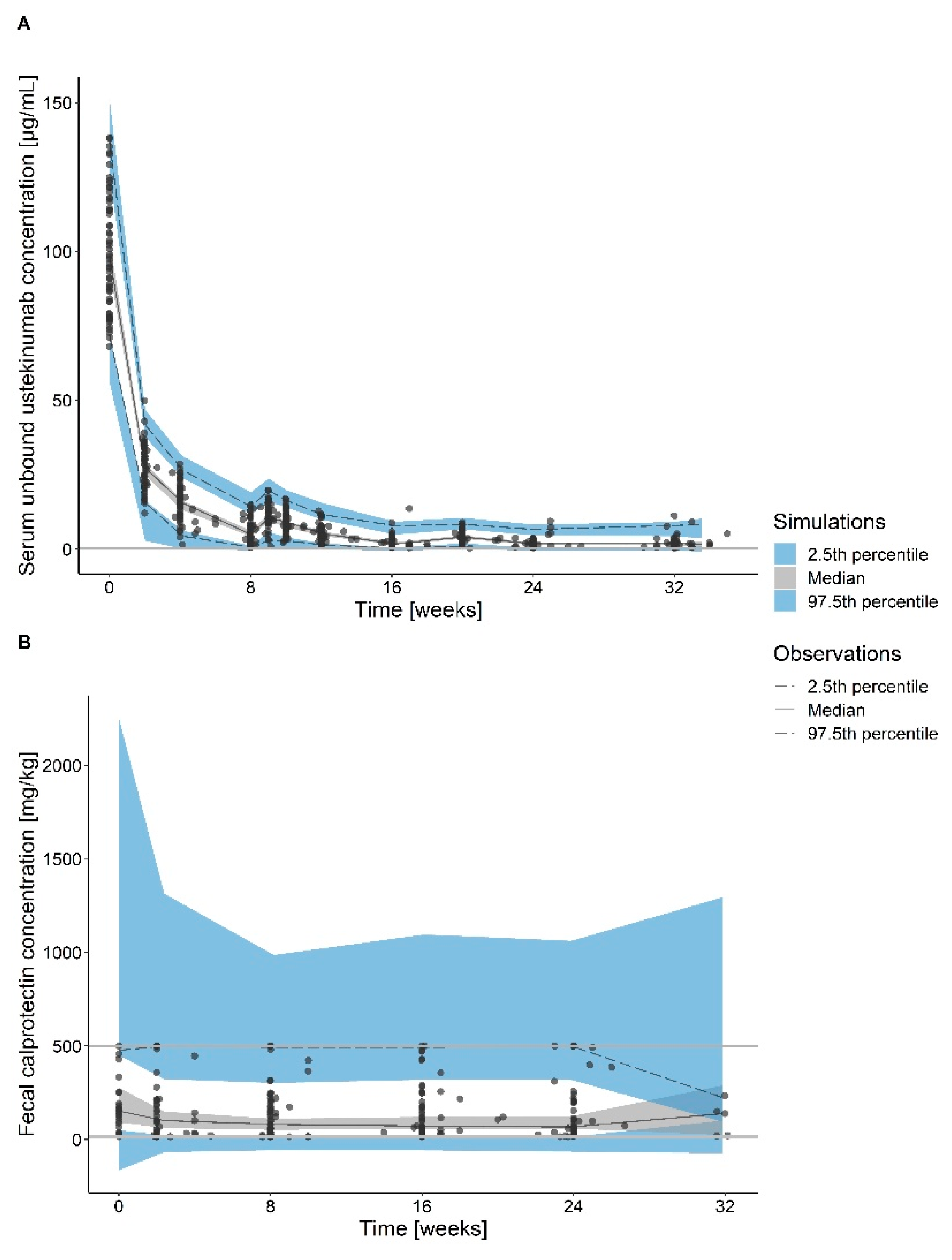
3.2. Pharmacokinetic and Pharmacodynamic Data
3.3. Pharmacokinetic–Pharmacodynamic Model
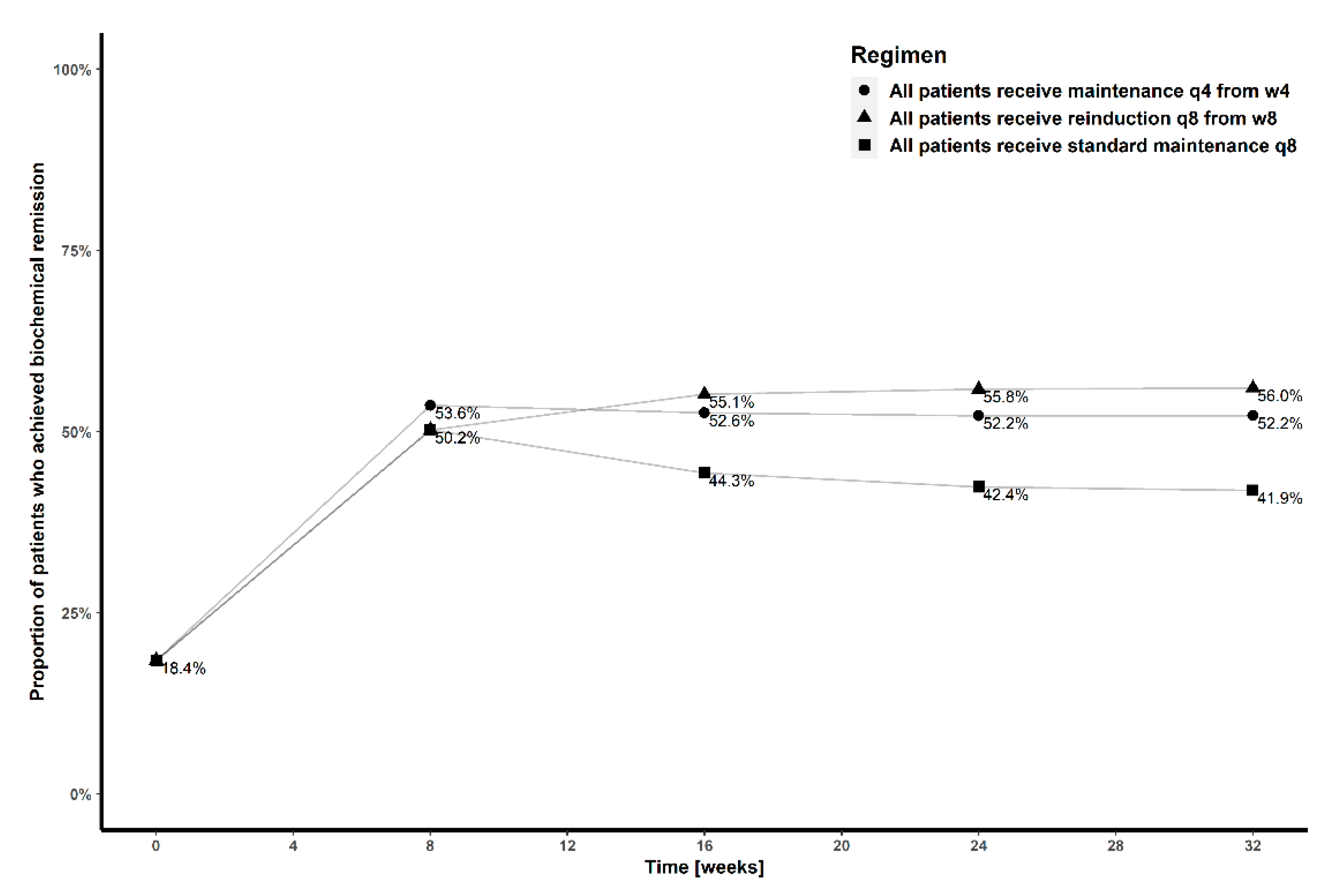
3.4. Simulations
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Torres, J.; Mehandru, S.; Colombel, J.-F.; Peyrin-Biroulet, L. Crohn’s disease. Lancet 2017, 389, 1741–1755. [Google Scholar] [CrossRef]
- Peyrin-Biroulet, L.; Sandborn, W.; Sands, B.E.; Reinisch, W.; Bemelman, W.; Bryant, R.V.; D’Haens, G.; Dotan, I.; Dubinsky, M.; Feagan, B.; et al. Selecting Therapeutic Targets in Inflammatory Bowel Disease (STRIDE): Determining Therapeutic Goals for Treat-to-Target. Am. J. Gastroenterol. 2015, 110, 1324–1338. [Google Scholar] [CrossRef]
- DʼHaens, G.; Ferrante, M.; Vermeire, S.; Baert, F.; Noman, M.; Moortgat, L.; Geens, P.; Iwens, D.; Aerden, I.; Van Assche, G.; et al. Fecal calprotectin is a surrogate marker for endoscopic lesions in inflammatory bowel disease. Inflamm. Bowel Dis. 2012, 18, 2218–2224. [Google Scholar] [CrossRef]
- Mosli, M.H.; Zou, G.; Garg, S.K.; Feagan, S.G.; MacDonald, J.K.; Chande, N.; Sandborn, W.J.; Feagan, B.G. C-Reactive Protein, Fecal Calprotectin, and Stool Lactoferrin for Detection of Endoscopic Activity in Symptomatic Inflammatory Bowel Disease Patients: A Systematic Review and Meta-Analysis. Am. J. Gastroenterol. 2015, 110, 802–819. [Google Scholar] [CrossRef]
- Moschen, A.R.; Tilg, H.; Raine, T. IL-12, IL-23 and IL-17 in IBD: Immunobiology and therapeutic targeting. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 185–196. [Google Scholar] [CrossRef] [PubMed]
- Iborra, M.; Beltrán, B.; Fernández-Clotet, A.; Gutiérrez, A.; Antolín, B.; Huguet, J.M.; De Francisco, R.; Merino, O.; Carpio, D.; García-López, S.; et al. Real-world short-term effectiveness of ustekinumab in 305 patients with Crohn’s disease: Results from the ENEIDA registry. Aliment. Pharmacol. Ther. 2019, 50, 278–288. [Google Scholar] [CrossRef] [PubMed]
- Biemans, V.B.C.; van der Meulen-de Jong, A.E.; van der Woude, C.J.; Löwenberg, M.; Dijkstra, G.; Oldenburg, B.; de Boer, N.K.H.; van der Marel, S.; Bodelier, A.G.L.; Jansen, J.M.; et al. Ustekinumab for Crohn’s Disease: Results of the ICC Registry, a Nationwide Prospective Observational Cohort Study. J. Crohn’s Colitis 2019, 14, jjz119. [Google Scholar] [CrossRef] [PubMed]
- Battat, R.; Kopylov, U.; Bessissow, T.; Bitton, A.; Cohen, A.; Jain, A.; Martel, M.; Seidman, E.; Afif, W. Association Between Ustekinumab Trough Concentrations and Clinical, Biomarker, and Endoscopic Outcomes in Patients With Crohn’s Disease. Clin. Gastroenterol. Hepatol. 2017, 15, 1427–1434.e2. [Google Scholar] [CrossRef]
- Ma, C.; Fedorak, R.N.; Kaplan, G.G.; Dieleman, L.A.; Devlin, S.M.; Stern, N.; Kroeker, K.I.; Seow, C.H.; Leung, Y.; Novak, K.L.; et al. Clinical, endoscopic and radiographic outcomes with ustekinumab in medically-refractory Crohn’s disease: Real world experience from a multicentre cohort. Aliment. Pharmacol. Ther. 2017, 45, 1232–1243. [Google Scholar] [CrossRef] [PubMed]
- Verstockt, B.; Dreesen, E.; Noman, M.; Outtier, A.; Van den Berghe, N.; Aerden, I.; Compernolle, G.; Van Assche, G.; Gils, A.; Vermeire, S.; et al. Ustekinumab Exposure-outcome Analysis in Crohn’s Disease Only in Part Explains Limited Endoscopic Remission Rates. J. Crohn’s Colitis 2019, 13, 864–872. [Google Scholar] [CrossRef]
- Hanžel, J.; Zdovc, J.; Kurent, T.; Sever, N.; Javornik, K.; Tuta, K.; Koželj, M.; Smrekar, N.; Novak, G.; Štabuc, B.; et al. Peak Concentrations of Ustekinumab After Intravenous Induction Therapy Identify Patients With Crohn’s Disease Likely to Achieve Endoscopic and Biochemical Remission. Clin. Gastroenterol. Hepatol. 2021, 19, 111–118.e10. [Google Scholar] [CrossRef] [PubMed]
- Ollech, J.E.; Normatov, I.; Peleg, N.; Wang, J.; Patel, S.A.; Rai, V.; Yi, Y.; Singer, J.; Dalal, S.R.; Sakuraba, A.; et al. Effectiveness of Ustekinumab Dose Escalation in Patients With Crohn’s Disease. Clin. Gastroenterol. Hepatol. 2020, 19, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Kopylov, U.; Hanzel, J.; Liefferinckx, C.; De Marco, D.; Imperatore, N.; Plevris, N.; Baston-Rey, I.; Harris, R.J.; Truyens, M.; Domislovic, V.; et al. Effectiveness of ustekinumab dose escalation in Crohn’s disease patients with insufficient response to standard-dose subcutaneous maintenance therapy. Aliment. Pharmacol. Ther. 2020, 52, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Fumery, M.; Peyrin-Biroulet, L.; Nancey, S.; Altwegg, R.; Gilletta, C.; Veyrard, P.; Bouguen, G.; Viennot, S.; Poullenot, F.; Filippi, J.; et al. Effectiveness And Safety Of Ustekinumab Intensification At 90 Mg Every Four Weeks In Crohn’s Disease: A Multicenter Study. J. Crohn’s Colitis 2020, 15, 222–227. [Google Scholar] [CrossRef] [PubMed]
- Hanžel, J.; Koželj, M.; Špes Hlastec, A.; Kurent, T.; Sever, N.; Zdovc, J.; Smrekar, N.; Novak, G.; Štabuc, B.; Grabnar, I.; et al. Ustekinumab concentrations shortly after escalation to monthly dosing may identify endoscopic remission in refractory Crohn’s disease. Eur. J. Gastroenterol. Hepatol. 2021. [Google Scholar] [CrossRef]
- Van den Reek, J.M.P.A.; Coenen, M.J.H.; van de L’Isle Arias, M.; Zweegers, J.; Rodijk-Olthuis, D.; Schalkwijk, J.; Vermeulen, S.H.; Joosten, I.; van de Kerkhof, P.C.M.; Seyger, M.M.B.; et al. Polymorphisms in CD84, IL12B and TNFAIP3 are associated with response to biologics in patients with psoriasis. Br. J. Dermatol. 2017, 176, 1288–1296. [Google Scholar] [CrossRef]
- Galluzzo, M.; Boca, A.N.; Botti, E.; Potenza, C.; Malara, G.; Malagoli, P.; Vesa, S.; Chimenti, S.; Buzoianu, A.D.; Talamonti, M.; et al. IL12B (p40) Gene Polymorphisms Contribute to Ustekinumab Response Prediction in Psoriasis. Dermatology 2016, 232, 230–236. [Google Scholar] [CrossRef]
- Rožman, S.; Novaković, S.; Grabnar, I.; Cerkovnik, P.; Novaković, B.J. The impact of FcγRIIa and FcγRIIIa gene polymorphisms on responses to RCHOP chemotherapy in diffuse large B-cell lymphoma patients. Oncol. Lett. 2016, 11, 3332–3336. [Google Scholar] [CrossRef] [Green Version]
- Janmahasatian, S.; Duffull, S.B.; Ash, S.; Ward, L.C.; Byrne, N.M.; Green, B. Quantification of lean bodyweight. Clin. Pharmacokinet. 2005, 44, 1051–1065. [Google Scholar] [CrossRef]
- Mager, D.E.; Jusko, W.J. General pharmacokinetic model for drugs exhibiting target-mediated drug disposition. J. Pharmacokinet. Pharmacodyn. 2001, 28, 507–532. [Google Scholar] [CrossRef]
- Gibiansky, L.; Gibiansky, E. Target-mediated drug disposition model: Approximations, identifiability of model parameters and applications to the population pharmacokinetic–pharmacodynamic modeling of biologics. Expert Opin. Drug Metab. Toxicol. 2009, 5, 803–812. [Google Scholar] [CrossRef] [PubMed]
- Ternant, D.; Monjanel, H.; Venel, Y.; Prunier-Aesch, C.; Arbion, F.; Colombat, P.; Paintaud, G.; Gyan, E. Nonlinear pharmacokinetics of rituximab in non-Hodgkin lymphomas: A pilot study. Br. J. Clin. Pharmacol. 2019, 85, 2002–2010. [Google Scholar] [CrossRef] [PubMed]
- Mager, D.E.; Krzyzanski, W. Quasi-equilibrium pharmacokinetic model for drugs exhibiting target-mediated drug disposition. Pharm. Res. 2005, 22, 1589–1596. [Google Scholar] [CrossRef] [PubMed]
- Nadeau, R.R.; Ostrowski, C.; Ni-Wu, G.; Liberato, D.J. Pharmacokinetics and pharmacodynamics of recombinant human interleukin-12 in male rhesus monkeys. J. Pharmacol. Exp. Ther. 1995, 274, 78–83. [Google Scholar]
- Zhang, T.T.; Ma, J.; Durbin, K.R.; Montavon, T.; Lacy, S.E.; Jenkins, G.J.; Doktor, S.; Kalvass, J.C. Determination of IL-23 Pharmacokinetics by Highly Sensitive Accelerator Mass Spectrometry and Subsequent Modeling to Project IL-23 Suppression in Psoriasis Patients Treated with Anti-IL-23 Antibodies. AAPS J. 2019, 21, 1–11. [Google Scholar] [CrossRef]
- Zhang, L.; Beal, S.L.; Sheiner, L.B. Simultaneous vs. Sequential Analysis for Population PK/PD Data I: Best-case Performance. Simulation 2003, 30, 387–404. [Google Scholar] [CrossRef]
- Felmlee, M.A.; Morris, M.E.; Mager, D.E. Mechanism-Based Pharmacodynamic Modeling. In Computational Toxicology; Humana Press: Totowa, NJ, USA, 2012; pp. 583–600. [Google Scholar] [CrossRef] [Green Version]
- Bergstrand, M.; Karlsson, M.O. Handling data below the limit of quantification in mixed effect models. AAPS J. 2009, 11, 371–380. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.-F.; Chen, J.-M.; Zuo, J.-H.; Yu, A.; Xiao, Z.-J.; Deng, F.-H.; Nie, B.; Jiang, B. Meta-analysis: Fecal Calprotectin for Assessment of Inflammatory Bowel Disease Activity. Inflamm. Bowel Dis. 2014, 20, 1407–1415. [Google Scholar] [CrossRef]
- Guidi, L.; Marzo, M.; Andrisani, G.; Felice, C.; Pugliese, D.; Mocci, G.; Nardone, O.; De Vitis, I.; Papa, A.; Rapaccini, G.; et al. Faecal calprotectin assay after induction with anti-Tumour Necrosis Factor α agents in inflammatory bowel disease: Prediction of clinical response and mucosal healing at one year. Dig. Liver Dis. 2014, 46, 974–979. [Google Scholar] [CrossRef]
- Adedokun, O.J.; Xu, Z.; Gasink, C.; Jacobstein, D.; Szapary, P.; Johanns, J.; Gao, L.-L.L.; Davis, H.M.; Hanauer, S.B.S.; Feagan, B.G.; et al. Pharmacokinetics and Exposure Response Relationships of Ustekinumab in Patients With Crohn’s Disease. Gastroenterology 2018, 154, 1660–1671. [Google Scholar] [CrossRef] [Green Version]
- Hudson, J.; Herfarth, H.; Barnes, E. Letter: Optimising response to ustekinumab therapy for patients with Crohn’s disease. Aliment. Pharmacol. Ther. 2020, 52, 906. [Google Scholar] [CrossRef] [PubMed]
- Sedano, R.; Guizzetti, L.; McDonald, C.; Jairath, V. Intravenous Ustekinumab Reinduction Is Effective in Prior Biologic Failure Crohn’s Disease Patients Already on Every-4-Week Dosing. Clin. Gastroenterol. Hepatol. 2020, 19, 1497–1498. [Google Scholar] [CrossRef] [PubMed]
- Danese, S.; Vermeire, S.; D’Haens, G.; Panés, J.; Dignass, A.; Magro, F.; Nazar, M.; Le Bars, M.; Sloan, S.; Lahaye, M.; et al. DOP13 Clinical and endoscopic response to ustekinumab in Crohn’s disease: Week 16 interim analysis of the STARDUST trial. J. Crohn’s Colitis 2020, 14, S049–S052. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Hu, C.; Lu, M.; Liao, S.; Marini, J.C.; Yohrling, J.; Yeilding, N.; Davis, H.M.; Zhou, H. Population pharmacokinetic modeling of ustekinumab, a human monoclonal antibody targeting IL-12/23p40, in patients with moderate to severe plaque psoriasis. J. Clin. Pharmacol. 2009, 49, 162–175. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.W.; Mendelsohn, A.; Pendley, C.; Davis, H.M.; Zhou, H. Population pharmacokinetics of ustekinumab in patients with active psoriatic arthritis. Int. J. Clin. Pharmacol. Ther. 2010, 48, 830–846. [Google Scholar] [CrossRef]
- Xu, Y.; Hu, C.; Chen, Y.; Miao, X.; Adedokun, O.J.; Xu, Z.; Sharma, A.; Zhou, H. Population Pharmacokinetics and Exposure-Response Modeling Analyses of Ustekinumab in Adults With Moderately to Severely Active Ulcerative Colitis. J. Clin. Pharmacol. 2020, 60, 889–902. [Google Scholar] [CrossRef]
- Wang, Z.; Verstockt, B.; Sabino, J.; Vermeire, S.; Ferrante, M.; Declerck, P.; Dreesen, E. Population pharmacokinetic-pharmacodynamic model-based exploration of alternative ustekinumab dosage regimens for patients with Crohn’s disease. Br. J. Clin. Pharmacol. 2021, 1–13. [Google Scholar] [CrossRef]
- Hu, C.; Adedokun, O.J.; Chen, Y.; Szapary, P.O.; Gasink, C.; Sharma, A.; Zhou, H. Challenges in longitudinal exposure-response modeling of data from complex study designs: A case study of modeling CDAI score for ustekinumab in patients with Crohn’s disease. J. Pharmacokinet. Pharmacodyn. 2017, 44, 425–436. [Google Scholar] [CrossRef]
- Berends, S.E.; van Steeg, T.J.; Ahsman, M.J.; Singh, S.; Brandse, J.F.; D’Haens, G.R.A.M.; Mathôt, R.A.A. Tumor necrosis factor-mediated disposition of infliximab in ulcerative colitis patients. J. Pharmacokinet. Pharmacodyn. 2019, 46, 543–551. [Google Scholar] [CrossRef] [Green Version]
- Dirks, N.L.; Meibohm, B. Population Pharmacokinetics of Therapeutic Monoclonal Antibodies. Clin. Pharmacokinet. 2010, 49, 633–659. [Google Scholar] [CrossRef]
- Ogawa, K.; Matsumoto, T.; Esaki, M.; Torisu, T.; Iida, M. Profiles of circulating cytokines in patients with Crohn’s disease under maintenance therapy with infliximab. J. Crohn’s Colitis 2012, 6, 529–535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lucaciu, L.; Ilies, M.; Iuga, C.; Seicean, A. P136 Serum IL-17 and IL-23 levels can distinguish between severe and non-severe inflammatory bowel disease. J. Crohn’s Colitis 2018, 12, S163. [Google Scholar] [CrossRef]
- Wong, E.C.L.; Marshall, J.K.; Reinisch, W.; Narula, N. Body Mass Index Does Not Impact Clinical Efficacy of Ustekinumab in Crohn’s Disease: A Post Hoc Analysis of the IM-UNITI Trial. Inflamm. Bowel Dis. 2020, 27, 848–854. [Google Scholar] [CrossRef] [PubMed]
- Sand, K.M.K.; Bern, M.; Nilsen, J.; Noordzij, H.T.; Sandlie, I.; Andersen, J.T. Unraveling the Interaction between FcRn and Albumin: Opportunities for Design of Albumin-Based Therapeutics. Front. Immunol. 2015, 5, 682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moroi, R.; Endo, K.; Kinouchi, Y.; Shiga, H.; Kakuta, Y.; Kuroha, M.; Kanazawa, Y.; Shimodaira, Y.; Horiuchi, T.; Takahashi, S.; et al. FCGR3A-158 polymorphism influences the biological response to infliximab in Crohn’s disease through affecting the ADCC activity. Immunogenetics 2013, 65, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Louis, E.; El Ghoul, Z.; Vermeire, S.; Dall’Ozzo, S.; Rutgeerts, P.; Paintaud, G.; Belaiche, J.; De Vos, M.; Van Gossum, A.; Colombel, J.-F.; et al. Association between polymorphism in IgG Fc receptor IIIa coding gene and biological response to infliximab in Crohn’s disease. Aliment. Pharmacol. Ther. 2004, 19, 511–519. [Google Scholar] [CrossRef]
- Castro-Dopico, T.; Clatworthy, M.R. IgG and Fcγ receptors in intestinal immunity and inflammation. Front. Immunol. 2019, 10, 805. [Google Scholar] [CrossRef]
- Dreesen, E.; Berends, S.; Laharie, D.; D’Haens, G.; Vermeire, S.; Gils, A.; Mathôt, R. Modelling of the relationship between infliximab exposure, faecal calprotectin and endoscopic remission in patients with Crohn’s disease. Br. J. Clin. Pharmacol. 2020, 1–13. [Google Scholar] [CrossRef]
- Feagan, B.G.; Sandborn, W.J.; Gasink, C.; Jacobstein, D.; Lang, Y.; Friedman, J.R.; Blank, M.A.; Johanns, J.; Gao, L.L.; Miao, Y.; et al. Ustekinumab as induction and maintenance therapy for Crohn’s disease. N. Engl. J. Med. 2016, 375, 1946–1960. [Google Scholar] [CrossRef]
- Hanauer, S.B.; Sandborn, W.J.; Feagan, B.G.; Gasink, C.; Jacobstein, D.; Zou, B.; Johanns, J.; Adedokun, O.J.; Sands, B.E.; Rutgeerts, P.; et al. IM-UNITI: Three-year efficacy, safety, and immunogenicity of ustekinumab treatment of Crohn’s disease. J. Crohn’s Colitis 2020, 14, 23–32. [Google Scholar] [CrossRef]
- Sands, B.E.; Sandborn, W.J.; Panaccione, R.; O’Brien, C.D.; Zhang, H.; Johanns, J.; Adedokun, O.J.; Li, K.; Peyrin-Biroulet, L.; Van Assche, G.; et al. Ustekinumab as Induction and Maintenance Therapy for Ulcerative Colitis. N. Engl. J. Med. 2019, 381, 1201–1214. [Google Scholar] [CrossRef]
- Labaere, D.; Smismans, A.; Van Olmen, A.; Christiaens, P.; D’Haens, G.; Moons, V.; Cuyle, P.-J.; Frans, J.; Bossuyt, P. Comparison of six different calprotectin assays for the assessment of inflammatory bowel disease. United Eur. Gastroenterol. J. 2014, 2, 30–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | Value |
|---|---|
| Women, n (%) | 32 (56) |
| Age at UST initiation, years, median (IQR) | 49 (32–56) |
| Weight, kg, median (IQR) | 70 (59–84) |
| Fat-free mass a, median (IQR) | 45 (39–62) |
| Height, cm, median (IQR) | 169 (163–179) |
| Intravenous ustekinumab dose, n (%) | |
| 260 mg | 9 (15.8) |
| 390 mg | 35 (61.4) |
| 520 mg | 13 (22.8) |
| Disease duration, years, median (IQR) | 14 (7–22) |
| Disease location, n (%) | |
| ileal (L1) | 17 (29.8) |
| colonic (L2) | 4 (7) |
| ileocolonic (L3) | 36 (63.2) |
| upper gastrointestinal involvement (L4) | 6 (10.5) |
| Fistulizing perianal disease, n (%) | 10 (17.5) |
| History of CD-related surgery, n (%) | 34 (59.6) |
| Smoking status, n (%) | |
| active smoking | 10 (17.5) |
| previously smoking | 12 (21.1) |
| never smoked | 35 (61.4) |
| Previous biological therapy, n (%) | 38 (66.7) |
| previous anti-TNF exposure | 38 (66.7) |
| previous vedolizumab exposure | 10 (17.5) |
| previous anti-TNF and vedolizumab exposure | 9 (15.8) |
| Systemic steroids at baseline, n (%) | 9 (15.8) |
| Topical steroids at baseline, n (%) | 3 (5.3) |
| Immunomodulators at baseline, n (%) | 5 (8.8) |
| azathioprine | 4 (7) |
| methotrexate | 1 (1.8) |
| Harvey–Bradshaw score, median (IQR) | 6 (3–10) |
| Fecal calprotectin, mg/kg, median (IQR) | 134 (53–213) |
| C-reactive protein, mg/L, median (IQR) | 3 (3–11) |
| Serum albumin, g/L, median (IQR) | 43 (41–44) |
| Endoscopically active disease at baseline, n (%) | 44 (77.2) |
| Samples available (ustekinumab measurement), n | 574 |
| Samples available (Fecal calprotectin), n | 224 |
| Samples with fecal calprotectin below the limit of quantification, n (%) | 15 (6.8) |
| Samples with fecal calprotectin above the limit of quantification, n (%) | 11 (5) |
| Genotype frequencies | |
| IL12B rs3212227, n (%) | |
| A/A | 37 (64.9) |
| A/C | 17 (29.8) |
| C/C | 3 (5.3) |
| IL12B rs3213094, n (%) | |
| C/C | 37 (64.9) |
| C/T | 17 (29.8) |
| T/T | 3 (5.3) |
| IL12B rs6887695, n (%) | |
| G/G | 22 (38.6) |
| C/G | 25 (43.9) |
| C/C | 10 (17.5) |
| FcGR2A rs1801274, n (%) | |
| A/A | 21 (36.8) |
| A/G | 29 (50.9) |
| G/G | 7 (12.3) |
| FcGR3A rs396991, n (%) | |
| A/A | 21 (36.8) |
| A/C | 31 (54.4) |
| C/C | 5 (8.8) |
| Parameter (Units) | Base Model | Final Model | |
|---|---|---|---|
| Estimate | Estimate | Bootstrap Median (95% CI) | |
| Ustekinumab pharmacokinetics | |||
| Ka (day−1) | 0.518 | 0.381 | 0.380 (0.341–0.422) |
| CL (L/day) a | 0.264 | 0.277 | 0.275 (0.259–0.294) |
| FFM on CL | / | 0.598 | 0.596 (0.539–0.673) |
| bio-naïve on CL | / | −0.227 | −0.232 (−0.280–−0.192) |
| Serum albumin on CL | / | −0.0165 | −0.0170 (−0.0224–−0.0127) |
| Vc (L) b | 2.18 | 3.57 | 3.56 (3.41–3.70) |
| FFM on Vc | / | 0.590 | 0.587 (0.534–0.644) |
| Q (L/day) | 20.1 | 1.89 | 1.88 (1.69–2.11) |
| Vp (L) c | 5.04 | 3.30 | 3.27 (3.01–3.52) |
| FFM on Vp | / | 0.586 | 0.581 (0.512–0.660) |
| Fraction absorbed, F (%) | 71.7 | / | / |
| FCGR3A-158 V/V | / | 88.8 | 88.8 (86.1–92.4) |
| FCGR3A-158 V/F, F/F | / | 71.0 | 70.8 (65.3–75.8) |
| Target pharmacokinetics | |||
| Ksyn (nmol/L × day−1) d | 1.65 × 10−8 | 9.86 × 10−9 | 9.85 × 10−9 (8.75 × 10−9–1.09 × 10−8) |
| Serum CRP on Ksyn | / | 0.0846 | 0.0843 (0.0772–0.0883) |
| Kdeg (day−1) | 1.85 × 10−10 | 9.26 × 10−10 | 9.26 × 10−10 (8.53 × 10−10–1.06 × 10−9) |
| Vc–target (L) | 18.8 | 2.44 | 2.44 (2.27–2.81) |
| Qtarget (L/d) | 0.752 | 0.493 | 0.488 (0.440–0.539) |
| Vp–target (L) | 22.6 | 11.0 | 10.9 (9.87–12.0) |
| Binding | |||
| Kint (day−1) | 1.71 × 10−7 | 2.83 × 10−6 | 2.82 × 10−6 (2.56 × 10−6–3.15 × 10−6) |
| Kd (nmol/L) | 0.350 | 0.168 | 0.168 (0.154–0.196) |
| Interindividual variability | |||
| IIV CL (%, CV) e | 27.9 (13) | 18.0 (16) | 18.0 (16.0–20.1) |
| IIV Vc (%, CV) e | 32.9 (16) | 9.79 (22) | 9.79 (8.88–10.8) |
| IIV Vp (%, CV) e | 21.5 (23) | 24.1 (24) | 23.6 (19.7–26.0) |
| IIV F (%, SD) e | 16.6 (21) | 17.3 (22) | 17.4 (15.8–20.2) |
| IIV Ksyn (%, CV) e | 105 (25) | 99.2 (27) | 98.4 (83.4–110) |
| Residual variability | |||
| Additive RUV (nmol/L) e | 4.46 (18) | 4.55 (17) | 4.58 (4.09–5.86) |
| Proportional RUV (%) e | 7.84 (18) | 7.77 (17) | 7.74 (6.94–8.55) |
| Parameter (Units) | Estimate | Bootstrap Median (95% CI) |
|---|---|---|
| Kout (day−1) | 0.0581 | 0.0641 (0.0249–0.110) |
| FC0 (mg/kg) | ||
| Patients without ulcers at baseline | 102 | 105 (54.4–188) |
| Patients with ulcers at baseline | 213 | 214 (157–287) |
| Emax (%) | 219 | 227 (128–442) |
| C50 (nmol/L) | 2.46 | 2.56 (0.413–13.9) |
| Interindividual variability | ||
| IIV FC0 (%) b | 99.0 (2) | 98.0 (75.0–128) |
| Residual variability | ||
| Proportional RUV (%) b | 57.3 (17) | 56.4 (48.8–65.0) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aguiar Zdovc, J.; Hanžel, J.; Kurent, T.; Sever, N.; Koželj, M.; Smrekar, N.; Novak, G.; Štabuc, B.; Dreesen, E.; Thomas, D.; et al. Ustekinumab Dosing Individualization in Crohn’s Disease Guided by a Population Pharmacokinetic–Pharmacodynamic Model. Pharmaceutics 2021, 13, 1587. https://doi.org/10.3390/pharmaceutics13101587
Aguiar Zdovc J, Hanžel J, Kurent T, Sever N, Koželj M, Smrekar N, Novak G, Štabuc B, Dreesen E, Thomas D, et al. Ustekinumab Dosing Individualization in Crohn’s Disease Guided by a Population Pharmacokinetic–Pharmacodynamic Model. Pharmaceutics. 2021; 13(10):1587. https://doi.org/10.3390/pharmaceutics13101587
Chicago/Turabian StyleAguiar Zdovc, Jurij, Jurij Hanžel, Tina Kurent, Nejc Sever, Matic Koželj, Nataša Smrekar, Gregor Novak, Borut Štabuc, Erwin Dreesen, Debby Thomas, and et al. 2021. "Ustekinumab Dosing Individualization in Crohn’s Disease Guided by a Population Pharmacokinetic–Pharmacodynamic Model" Pharmaceutics 13, no. 10: 1587. https://doi.org/10.3390/pharmaceutics13101587