An Update on Antimicrobial Peptides (AMPs) and Their Delivery Strategies for Wound Infections


Abstract
:
1. Introduction
2. Antimicrobial Peptides (AMPs) and AMP Dendrimers (AMPDs)
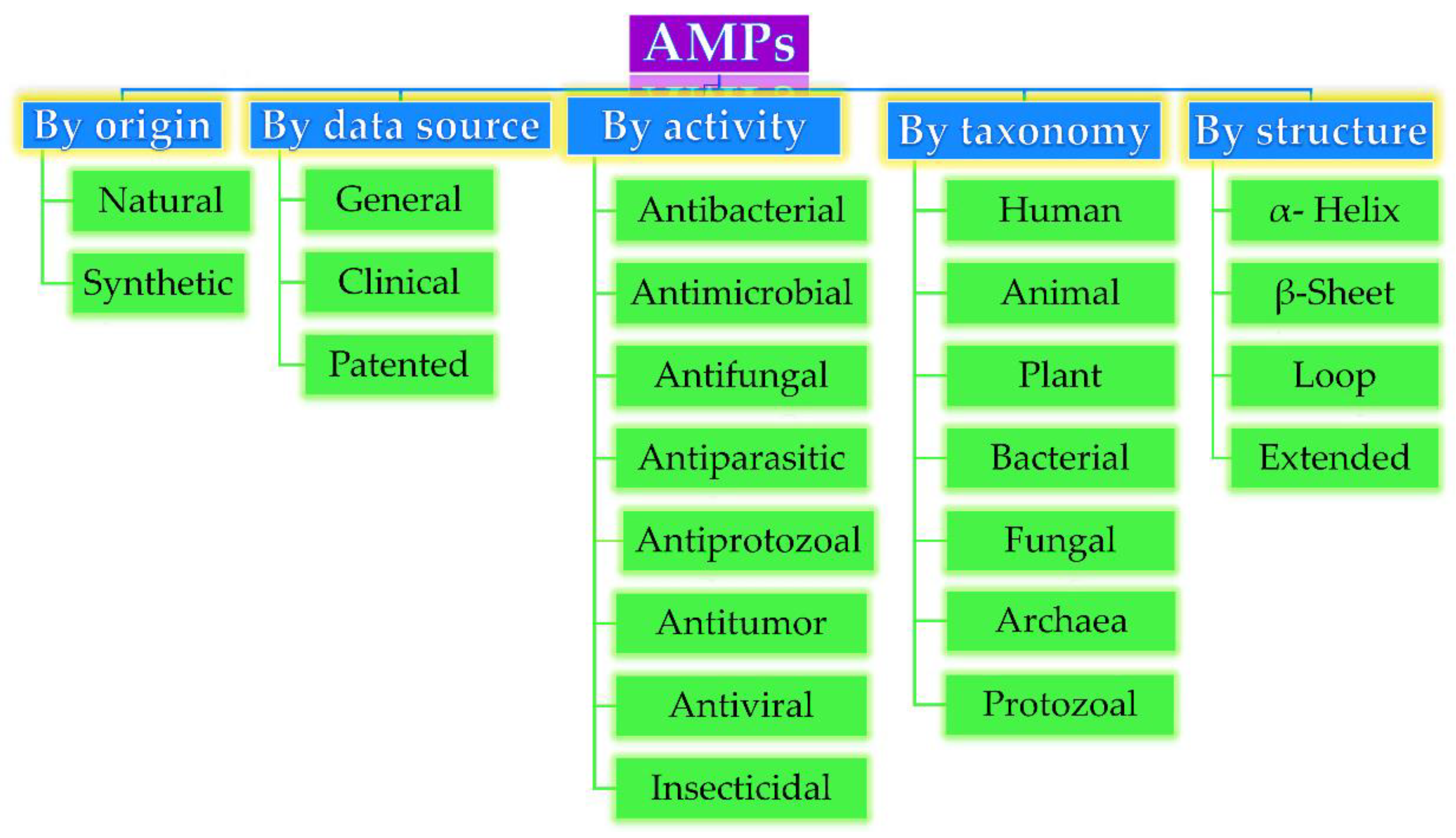
2.1. AMPs: Classification, Mechanism of Action
2.1.1. Wound-Healing Promoting AMPs
2.1.2. The Challenge of Resistance Development
2.1.3. AMP’s Activity against Biofilm Formation
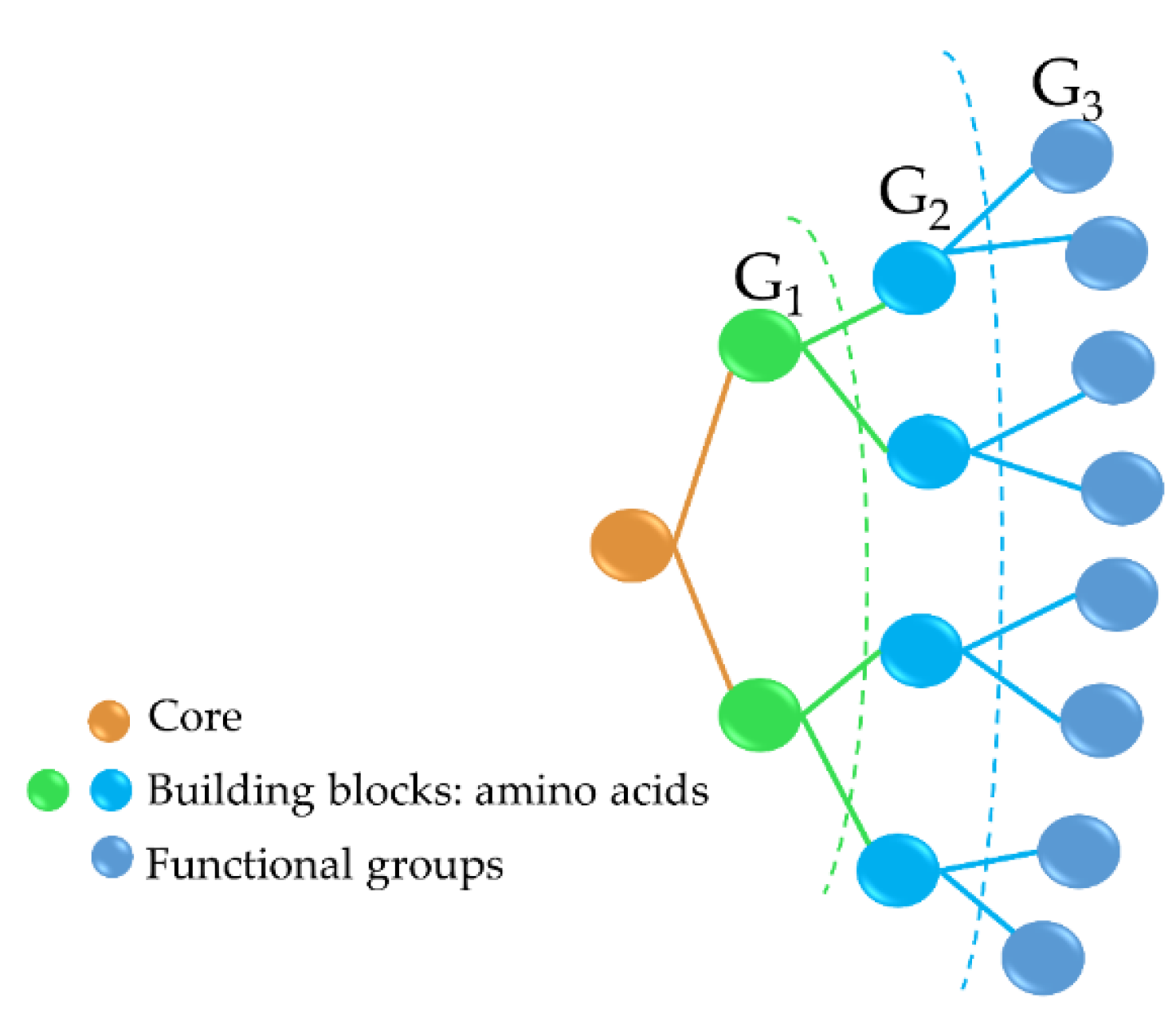
2.2. Antimicrobial Dendrimer Peptides
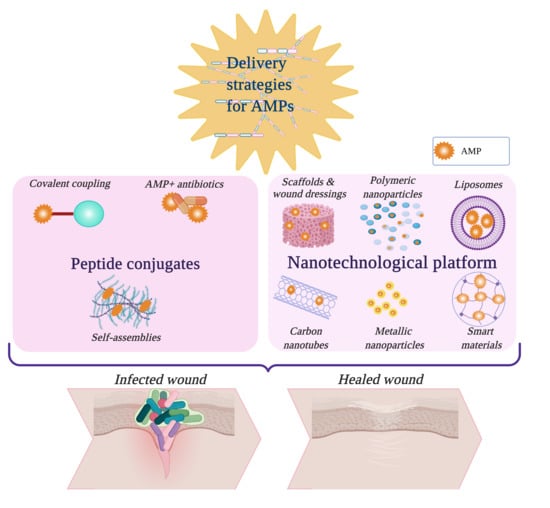
3. Peptide Conjugates
3.1. Covalent Coupling to Polymers
3.2. Self-Assembled AMPs
3.3. Combination of AMPs with Antibiotics
4. Nanotechnological Platforms and Scaffolds for Peptide Delivery
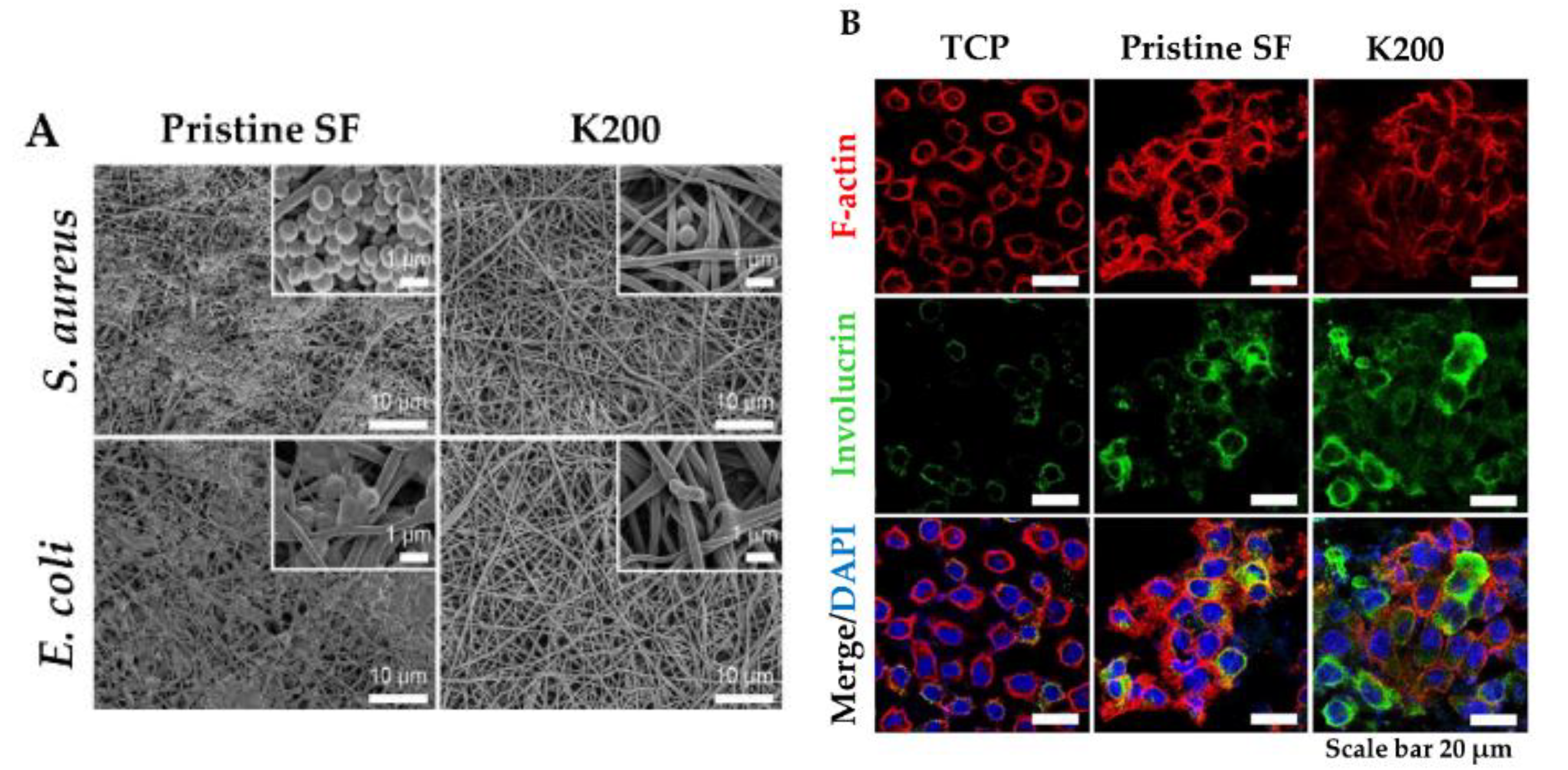
4.1. Polymeric Scaffolds and Wound Dressings
4.2. Organic Nanoparticles
4.2.1. Polymeric Nanoparticles, Nanoemulsions, and Micelles
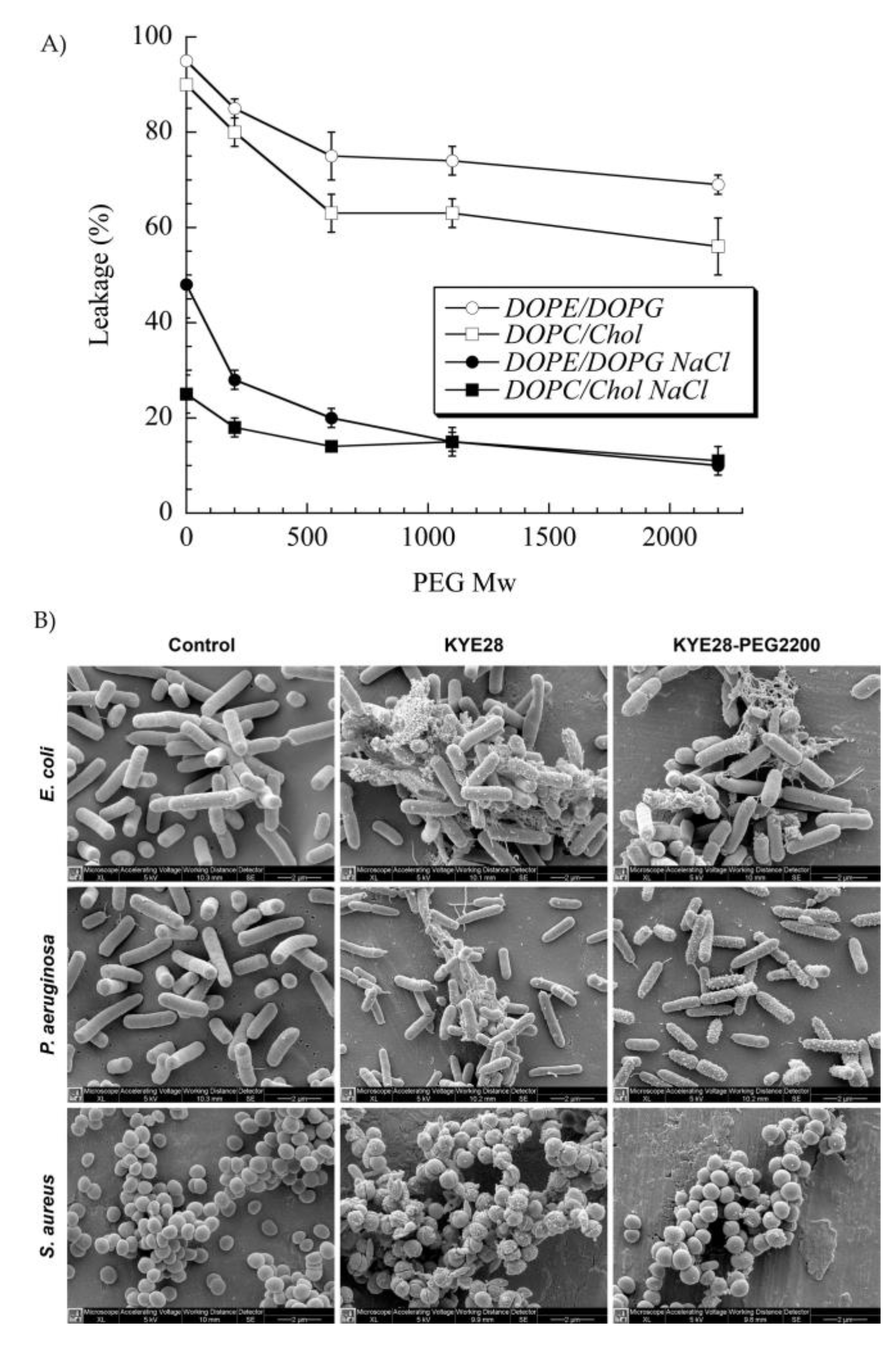
4.2.2. Liposomes
4.3. Inorganic NPs
4.3.1. Metallic NPs
4.3.2. Carbon Nanotubes, Graphene, Fullerenes
4.4. Smart Nanomaterials
5. Discussion and Further Insights
Author Contributions
Funding
Conflicts of Interest
References
- Patrulea, V.; Ostafe, V.; Borchard, G.; Jordan, O. Chitosan as a starting material for wound healing applications. Eur. J. Pharm. Biopharm. 2015, 97, 417–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, M.; Jonnalagadda, S. Advances in bioprinting using additive manufacturing. Eur. J. Pharm. Sci. 2020, 143, 105167. [Google Scholar] [CrossRef] [PubMed]
- Thapa, R.K.; Diep, D.B.; Tønnesen, H.H. Topical antimicrobial peptide formulations for wound healing: Current developments and future prospects. Acta Biomater. 2020, 103, 52–67. [Google Scholar] [CrossRef] [PubMed]
- Gieringer, M.; Gosepath, J.; Naim, R. Radiotherapy and wound healing: Principles, management and prospects (review). Oncol. Rep. 2011, 26, 299–307. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Sayed, P.; Kaeppeli, A.; Siriwardena, T.; Darbre, T.; Perron, K.; Jafari, P.; Reymond, J.-L.; Pioletti, D.P.; Applegate, L.A. Anti-Microbial Dendrimers against Multidrug-Resistant P. aeruginosa Enhance the Angiogenic Effect of Biological Burn-wound Bandages. Sci. Rep. 2016, 6, 22020. [Google Scholar] [CrossRef] [PubMed]
- Patrulea, V.; Younes, I.; Jordan, O.; Borchard, G. Chitosan-based systems for controlled delivery of antimicrobial peptides for biomedical application. In Functional Chitosan: Drug Delivery and Biomedical Applications; Jana, S., Jana, S., Eds.; Springer: Singapore, 2019; pp. 415–455. [Google Scholar]
- Omar, A.; Wright, J.B.; Schultz, G.; Burrell, R.; Nadworny, P. Microbial Biofilms and Chronic Wounds. Microorganisms 2017, 5, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lombardi, L.; Falanga, A.; Del Genio, V.; Galdiero, S. A New Hope: Self-Assembling Peptides with Antimicrobial Activity. Pharmaceutics 2019, 11, 166. [Google Scholar] [CrossRef] [Green Version]
- Seppanen, E.J.; Thornton, R.B.; Corscadden, K.J.; Granland, C.M.; Hibbert, J.; Fuery, A.; Wiertsema, S.P.; Vijayasekaran, S.; Coates, H.L.; Jacoby, P.; et al. High concentrations of middle ear antimicrobial peptides and proteins and proinflammatory cytokines are associated with detection of middle ear pathogens in children with recurrent acute otitis media. PLoS ONE 2019, 14, e0227080. [Google Scholar] [CrossRef] [Green Version]
- Rios, A.C.; Moutinho, C.G.; Pinto, F.C.; Del Fiol, F.S.; Jozala, A.; Chaud, M.V.; Vila, M.M.D.C.; Teixeira, J.A.; Balcão, V.M. Alternatives to overcoming bacterial resistances: State-of-the-art. Microbiol. Res. 2016, 191, 51–80. [Google Scholar] [CrossRef]
- Hincapié, O.; Giraldo, P.; Orduz, S. In silico design of polycationic antimicrobial peptides active against Pseudomonas aeruginosa and Staphylococcus aureus. Antonie Van Leeuwenhoek 2018, 111, 1871–1882. [Google Scholar] [CrossRef]
- Gull, S.; Shamim, N.; Minhas, F. AMAP: Hierarchical multi-label prediction of biologically active and antimicrobial peptides. Comput. Biol. Med. 2019, 107, 172–181. [Google Scholar] [CrossRef] [PubMed]
- Haney, E.F.; Hancock, R.E.W. Peptide design for antimicrobial and immunomodulatory applications. Biopolymers 2013, 100, 572–583. [Google Scholar] [CrossRef] [PubMed]
- Meher, P.K.; Sahu, T.K.; Saini, V.; Rao, A.R. Predicting antimicrobial peptides with improved accuracy by incorporating the compositional, physico-chemical and structural features into Chou’s general PseAAC. Sci. Rep. 2017, 7, 42362. [Google Scholar] [CrossRef] [PubMed]
- Magana, M.; Pushpanathan, M.; Santos, A.L.; Leanse, L.; Fernandez, M.; Ioannidis, A.; Giulianotti, M.A.; Apidianakis, Y.; Bradfute, S.; Ferguson, A.L.; et al. The value of antimicrobial peptides in the age of resistance. Lancet Infect. Dis. 2020, 20, e216–e230. [Google Scholar] [CrossRef]
- Porto, W.F.; Pires, A.S.; Franco, O.L. Computational tools for exploring sequence databases as a resource for antimicrobial peptides. Biotechnol. Adv. 2017, 35, 337–349. [Google Scholar] [CrossRef]
- Torres, M.D.T.; Sothiselvam, S.; Lu, T.K.; de la Fuente-Nunez, C. Peptide Design Principles for Antimicrobial Applications. J. Mol. Biol. 2019, 431, 3547–3567. [Google Scholar] [CrossRef]
- Fleming, A.; Wright, A.E. On a remarkable bacteriolytic element found in tissues and secretions. Proc. R. Soc. B 1922, 93, 306–317. [Google Scholar] [CrossRef] [Green Version]
- Fleming, A. On the Antibacterial Action of Cultures of a Penicillium, with Special Reference to their Use in the Isolation of B. influenzæ. Br. J. Exp. Pathol. 1929, 10, 226–236. [Google Scholar] [CrossRef]
- APD (The Antimicrobial Peptide Database). Available online: http://aps.unmc.edu/AP/main.php (accessed on 5 March 2020).
- DRAMP (Data Repository of Antimicrobial Peptides). Available online: http://dramp.cpu-bioinfor.org/browse/ (accessed on 5 March 2020).
- Kang, X.; Dong, F.; Shi, C.; Liu, S.; Sun, J.; Chen, J.; Li, H.; Xu, H.; Lao, X.; Zheng, H. DRAMP 2.0, an updated data repository of antimicrobial peptides. Sci. Data 2019, 6, 148. [Google Scholar] [CrossRef] [Green Version]
- Koo, H.B.; Seo, J. Antimicrobial peptides under clinical investigation. Pept. Sci. 2019, 111, e24122. [Google Scholar] [CrossRef]
- Nuti, R.; Goud, N.S.; Saraswati, A.P.; Alvala, R.; Alvala, M. Antimicrobial Peptides: A Promising Therapeutic Strategy in Tackling Antimicrobial Resistance. Curr. Med. Chem. 2017, 24, 4303–4314. [Google Scholar] [CrossRef] [PubMed]
- Ho, Y.-H.; Shah, P.; Chen, Y.-W.; Chen, C.-S. Systematic Analysis of Intracellular-targeting Antimicrobial Peptides, Bactenecin 7, Hybrid of Pleurocidin and Dermaseptin, Proline-Arginine-rich Peptide, and Lactoferricin B, by Using Escherichia coli Proteome Microarrays. Mol. Cell. Proteom. 2016, 15, 1837–1847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rowe-Magnus, D.A.; Kao, A.Y.; Prieto, A.C.; Pu, M.; Kao, C. Cathelicidin Peptides Restrict Bacterial Growth via Membrane Perturbation and Induction of Reactive Oxygen Species. mBio 2019, 10, e02021-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, J.; Hwang, I.-s.; Choi, H.J.; Ji Hong, H.; Hwang, J.-S.; Lee, D.G. The novel biological action of antimicrobial peptides via apoptosis induction. J. Microbiol. Biotechnol. 2012, 22, 1457–1466. [Google Scholar] [CrossRef] [Green Version]
- Mangoni, M.L.; McDermott, A.M.; Zasloff, M. Antimicrobial peptides and wound healing: Biological and therapeutic considerations. Exp. Dermatol. 2016, 25, 167–173. [Google Scholar] [CrossRef]
- Steinstraesser, L.; Hirsch, T.; Schulte, M.; Kueckelhaus, M.; Jacobsen, F.; Mersch, E.A.; Stricker, I.; Afacan, N.; Jenssen, H.; Hancock, R.E.; et al. Innate defense regulator peptide 1018 in wound healing and wound infection. PLoS ONE 2012, 7, e39373. [Google Scholar] [CrossRef] [Green Version]
- Hirsch, T.; Spielmann, M.; Zuhaili, B.; Fossum, M.; Metzig, M.; Koehler, T.; Steinau, H.U.; Yao, F.; Onderdonk, A.B.; Steinstraesser, L.; et al. Human beta-defensin-3 promotes wound healing in infected diabetic wounds. J. Gene Med. 2009, 11, 220–228. [Google Scholar] [CrossRef]
- Kolar, S.S.; McDermott, A.M. Role of host-defence peptides in eye diseases. Cell. Mol. Life Sci. 2011, 68, 2201. [Google Scholar] [CrossRef] [Green Version]
- Shaykhiev, R.; Beisswenger, C.; Kandler, K.; Senske, J.; Puchner, A.; Damm, T.; Behr, J.; Bals, R. Human endogenous antibiotic LL-37 stimulates airway epithelial cell proliferation and wound closure. Am. J. Physiol. Lung Cell. Mol. Physiol. 2005, 289, L842–L848. [Google Scholar] [CrossRef]
- Gomes, A.; Teixeira, C.; Ferraz, R.; Prudencio, C.; Gomes, P. Wound-Healing Peptides for Treatment of Chronic Diabetic Foot Ulcers and Other Infected Skin Injuries. Molecules 2017, 22, 1743. [Google Scholar] [CrossRef] [Green Version]
- Teirlinck, E.; Samal, S.K.; Coenye, T.; Braeckmans, K. Chapter 3-Penetrating the Bacterial Biofilm: Challenges for Antimicrobial Treatment. In Functionalized Nanomaterials for the Management of Microbial Infection; Boukherroub, R., Szunerits, S., Drider, D., Eds.; Elsevier: Boston, MA, USA, 2017; pp. 49–76. [Google Scholar]
- Nishikawa, T.; Nakagami, H.; Maeda, A.; Morishita, R.; Miyazaki, N.; Ogawa, T.; Tabata, Y.; Kikuchi, Y.; Hayashi, H.; Tatsu, Y.; et al. Development of a novel antimicrobial peptide, AG-30, with angiogenic properties. J. Cell. Mol. Med. 2009, 13, 535–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakagami, H.; Nishikawa, T.; Tamura, N.; Maeda, A.; Hibino, H.; Mochizuki, M.; Shimosato, T.; Moriya, T.; Morishita, R.; Tamai, K.; et al. Modification of a novel angiogenic peptide, AG30, for the development of novel therapeutic agents. J. Cell. Mol. Med. 2012, 16, 1629–1639. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Mu, L.; Tang, J.; Shen, C.; Gao, C.; Rong, M.; Zhang, Z.; Liu, J.; Wu, X.; Yu, H.; et al. A potential wound healing-promoting peptide from frog skin. Int. J. Biochem. Cell Biol. 2014, 49, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Mu, L.; Tang, J.; Liu, H.; Shen, C.; Rong, M.; Zhang, Z.; Lai, R. A potential wound-healing-promoting peptide from salamander skin. FASEB J. 2014, 28, 3919–3929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, J.; Liu, H.; Gao, C.; Mu, L.; Yang, S.; Rong, M.; Zhang, Z.; Liu, J.; Ding, Q.; Lai, R. A small peptide with potential ability to promote wound healing. PLoS ONE 2014, 9, e92082. [Google Scholar] [CrossRef]
- Ma, Z.; Han, J.; Chang, B.; Gao, L.; Lu, Z.; Lu, F.; Zhao, H.; Zhang, C.; Bie, X. Membrane-Active Amphipathic Peptide WRL3 with in Vitro Antibiofilm Capability and in Vivo Efficacy in Treating Methicillin-Resistant Staphylococcus aureus Burn Wound Infections. ACS Infect. Dis. 2017, 3, 820–832. [Google Scholar] [CrossRef]
- Huang, H.-N.; Pan, C.-Y.; Wu, H.-Y.; Chen, J.-Y. Antimicrobial peptide Epinecidin-1 promotes complete skin regeneration of methicillin-resistant Staphylococcus aureus-infected burn wounds in a swine model. Oncotarget 2017, 8, 21067–21080. [Google Scholar] [CrossRef] [Green Version]
- Nordström, R.; Malmsten, M. Delivery systems for antimicrobial peptides. Adv. Colloid Interface Sci. 2017, 242, 17–34. [Google Scholar] [CrossRef]
- Chen, C.H.; Lu, T.K. Development and Challenges of Antimicrobial Peptides for Therapeutic Applications. Antibiotics 2020, 9, 24. [Google Scholar] [CrossRef] [Green Version]
- Song, D.W.; Kim, S.H.; Kim, H.H.; Lee, K.H.; Ki, C.S.; Park, Y.H. Multi-biofunction of antimicrobial peptide-immobilized silk fibroin nanofiber membrane: Implications for wound healing. Acta Biomater. 2016, 39, 146–155. [Google Scholar] [CrossRef]
- Gunasekera, S.; Muhammad, T.; Strömstedt, A.A.; Rosengren, K.J.; Göransson, U. Backbone Cyclization and Dimerization of LL-37-Derived Peptides Enhance Antimicrobial Activity and Proteolytic Stability. Front. Microbiol. 2020, 11, 168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paranjape, S.M.; Lauer, T.W.; Montelaro, R.C.; Mietzner, T.A.; Vij, N. Modulation of proinflammatory activity by the engineered cationic antimicrobial peptide WLBU-2. F1000Research 2 2013, 2, 36. [Google Scholar] [CrossRef] [PubMed]
- Lin, Q.; Deslouches, B.; Montelaro, R.C.; Di, Y.P. Prevention of ESKAPE pathogen biofilm formation by antimicrobial peptides WLBU2 and LL37. Int. J. Antimicrob. Agents 2018, 52, 667–672. [Google Scholar] [CrossRef] [PubMed]
- Orlov, D.S.; Shamova, O.V.; Eliseev, I.E.; Zharkova, M.S.; Chakchir, O.B.; Antcheva, N.; Zachariev, S.; Panteleev, P.V.; Kokryakov, V.N.; Ovchinnikova, T.V.; et al. Redesigning Arenicin-1, an Antimicrobial Peptide from the Marine Polychaeta Arenicola marina, by Strand Rearrangement or Branching, Substitution of Specific Residues, and Backbone Linearization or Cyclization. Mar. Drugs 2019, 17, 376. [Google Scholar] [CrossRef] [Green Version]
- Elliott, A.G.; Huang, J.X.; Neve, S.; Zuegg, J.; Edwards, I.A.; Cain, A.K.; Boinett, C.J.; Barquist, L.; Lundberg, C.V.; Steen, J.; et al. An amphipathic peptide with antibiotic activity against multidrug-resistant Gram-negative bacteria. Nat. Commun. 2020, 11, 3184. [Google Scholar] [CrossRef] [PubMed]
- Kunda, N.K. Antimicrobial peptides as novel therapeutics for non-small cell lung cancer. Drug Discov. Today 2020, 25, 238–247. [Google Scholar] [CrossRef]
- Hammami, R.; Fernandez, B.; Lacroix, C.; Fliss, I. Anti-infective properties of bacteriocins: An update. Cell. Mol. Life Sci. 2013, 70, 2947–2967. [Google Scholar] [CrossRef]
- Dover, S.E.; Aroutcheva, A.A.; Faro, S.; Chikindas, M.L. Safety study of an antimicrobial peptide lactocin 160, produced by the vaginal Lactobacillus rhamnosus. Infect Dis Obs. Gynecol 2007, 2007, 78248. [Google Scholar] [CrossRef] [Green Version]
- Meade, E.; Slattery, M.A.; Garvey, M. Bacteriocins, Potent Antimicrobial Peptides and the Fight against Multi Drug Resistant Species: Resistance Is Futile? Antibiotics 2020, 9, 32. [Google Scholar] [CrossRef] [Green Version]
- Sherwood, E.J.; Bibb, M.J. The antibiotic planosporicin coordinates its own production in the actinomycete Planomonospora alba. Proc. Natl. Acad. Sci. USA 2013, 110, E2500–E2509. [Google Scholar] [CrossRef] [Green Version]
- Castiglione, F.; Lazzarini, A.; Carrano, L.; Corti, E.; Ciciliato, I.; Gastaldo, L.; Candiani, P.; Losi, D.; Marinelli, F.; Selva, E.; et al. Determining the Structure and Mode of Action of Microbisporicin, a Potent Lantibiotic Active Against Multiresistant Pathogens. Chem. Biol. 2008, 15, 22–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, C.; Sarkar, P.; Issa, R.; Haldar, J. Alternatives to Conventional Antibiotics in the Era of Antimicrobial Resistance. Trends Microbiol. 2019, 27, 323–338. [Google Scholar] [CrossRef] [PubMed]
- Hara, S.; Mukae, H.; Sakamoto, N.; Ishimoto, H.; Amenomori, M.; Fujita, H.; Ishimatsu, Y.; Yanagihara, K.; Kohno, S. Plectasin has antibacterial activity and no affect on cell viability or IL-8 production. Biochem. Biophys. Res. Commun. 2008, 374, 709–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahlapuu, M.; Håkansson, J.; Ringstad, L.; Björn, C. Antimicrobial Peptides: An Emerging Category of Therapeutic Agents. Front. Cell. Infect. Microbiol. 2016, 6, 194. [Google Scholar] [CrossRef] [Green Version]
- Muchintala, D.; Suresh, V.; Raju, D.; Sashidhar, R.B. Synthesis and characterization of cecropin peptide-based silver nanocomposites: Its antibacterial activity and mode of action. Mater. Sci. Eng. C 2020, 110, 110712. [Google Scholar] [CrossRef]
- Van Groenendael, R.; Kox, M.; Leijte, G.; Koeneman, B.; Gerretsen, J.; van Eijk, L.; Pickkers, P. A randomized double-blind, placebo-controlled clinical phase IIa trial on safety, immunomodulatory effects and pharmacokinetics of EA-230 during experimental human endotoxaemia. Br. J. Clin. Pharm. 2019, 85, 1559–1571. [Google Scholar] [CrossRef]
- Hazam, P.K.; Goyal, R.; Ramakrishnan, V. Peptide based antimicrobials: Design strategies and therapeutic potential. Prog. Biophys. Mol. Biol. 2019, 142, 10–22. [Google Scholar] [CrossRef]
- Han, J.; Jyoti, M.A.; Song, H.-Y.; Jang, W.S. Antifungal Activity and Action Mechanism of Histatin 5-Halocidin Hybrid Peptides against Candida ssp. PLoS ONE 2016, 11, e0150196. [Google Scholar] [CrossRef] [Green Version]
- Costa, F.; Maia, S.; Gomes, J.; Gomes, P.; Martins, M.C.L. Characterization of hLF1–11 immobilization onto chitosan ultrathin films, and its effects on antimicrobial activity. Acta Biomater. 2014, 10, 3513–3521. [Google Scholar] [CrossRef] [Green Version]
- Bruni, N.; Capucchio, M.T.; Biasibetti, E.; Pessione, E.; Cirrincione, S.; Giraudo, L.; Corona, A.; Dosio, F. Antimicrobial Activity of Lactoferrin-Related Peptides and Applications in Human and Veterinary Medicine. Molecules 2016, 21, 752. [Google Scholar] [CrossRef]
- Boto, A.; de Pérez la Lastra, J.M.; González, C.C. The Road from Host-Defense Peptides to a New Generation of Antimicrobial Drugs. Molecules 2018, 23, 311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Overhage, J.; Campisano, A.; Bains, M.; Torfs, E.C.W.; Rehm, B.H.A.; Hancock, R.E.W. Human Host Defense Peptide LL-37 Prevents Bacterial Biofilm Formation. Infect. Immun. 2008, 76, 4176–4182. [Google Scholar] [CrossRef] [Green Version]
- US National Library of Medicine. A Phase II Study to Evaluate the Efficacy and Safety of Two Doses of LTX-109 in Impetigo. Available online: https://clinicaltrials.gov/ct2/show/NCT01803035 (accessed on 26 March 2020).
- Saravolatz, L.D.; Pawlak, J.; Johnson, L.; Bonilla, H.; Saravolatz, L.D., 2nd; Fakih, M.G.; Fugelli, A.; Olsen, W.M. In vitro activities of LTX-109, a synthetic antimicrobial peptide, against methicillin-resistant, vancomycin-intermediate, vancomycin-resistant, daptomycin-nonsusceptible, and linezolid-nonsusceptible Staphylococcus aureus. Antimicrob. Agents Chemother. 2012, 56, 4478–4482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yasir, M.; Dutta, D.; Hossain, K.R.; Chen, R.; Ho, K.K.K.; Kuppusamy, R.; Clarke, R.J.; Kumar, N.; Willcox, M.D.P. Mechanism of Action of Surface Immobilized Antimicrobial Peptides Against Pseudomonas aeruginosa. Front. Microbiol. 2020, 10, 3053. [Google Scholar] [CrossRef] [PubMed]
- Yasir, M.; Dutta, D.; Willcox, M.D.P. Comparative mode of action of the antimicrobial peptide melimine and its derivative Mel4 against Pseudomonas aeruginosa. Sci. Rep. 2019, 9, 7063. [Google Scholar] [CrossRef] [Green Version]
- Czyzewski, A.M.; Jenssen, H.; Fjell, C.D.; Waldbrook, M.; Chongsiriwatana, N.P.; Yuen, E.; Hancock, R.E.; Barron, A.E. In Vivo, In Vitro, and In Silico Characterization of Peptoids as Antimicrobial Agents. PLoS ONE 2016, 11, e0135961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vila, J.; Moreno-Morales, J.; Ballesté-Delpierre, C. Current landscape in the discovery of novel antibacterial agents. Clin. Microbiol. Infect. 2019, 26, 569–603. [Google Scholar] [CrossRef]
- Du, H.; Puri, S.; McCall, A.; Norris, H.L.; Russo, T.; Edgerton, M. Human Salivary Protein Histatin 5 Has Potent Bactericidal Activity against ESKAPE Pathogens. Front. Cell. Infect. Microbiol. 2017, 7, 41. [Google Scholar] [CrossRef] [Green Version]
- US National Library of Medicine. Phase III Efficacy and Safety Study of AB103 in the Treatment of Patients with Necrotizing Soft Tissue Infections (ACCUTE). Available online: https://clinicaltrials.gov/ct2/show/NCT02469857 (accessed on 26 March 2020).
- de la Cruz, M.; González, I.; Parish, C.A.; Onishi, R.; Tormo, J.R.; Martín, J.; Peláez, F.; Zink, D.; El Aouad, N.; Reyes, F.; et al. Production of Ramoplanin and Ramoplanin Analogs by Actinomycetes. Front. Microbiol. 2017, 8, 343. [Google Scholar] [CrossRef] [Green Version]
- Cheng, M.; Huang, J.X.; Ramu, S.; Butler, M.S.; Cooper, M.A. Ramoplanin at bactericidal concentrations induces bacterial membrane depolarization in Staphylococcus aureus. Antimicrob. Agents Chemother. 2014, 58, 6819–6827. [Google Scholar] [CrossRef] [Green Version]
- Bienvenu, A.-L.; Pradat, P.; Guerin, C.; Aubrun, F.; Fellahi, J.-L.; Friggeri, A.; Guichon, C.; Hernu, R.; Menotti, J.; Monard, C.; et al. Evaluation of first-line therapies for the treatment of candidemia in ICU patients: A propensity score analysis. Int. J. Infect. Dis. 2020, 93, 15–21. [Google Scholar] [CrossRef]
- Pfaller, M.A.; Diekema, D.J.; Ostrosky-Zeichner, L.; Rex, J.H.; Alexander, B.D.; Andes, D.; Brown, S.D.; Chaturvedi, V.; Ghannoum, M.A.; Knapp, C.C.; et al. Correlation of MIC with Outcome for Candida Species Tested against Caspofungin, Anidulafungin, and Micafungin: Analysis and Proposal for Interpretive MIC Breakpoints. J. Clin. Microbiol. 2008, 46, 2620–2629. [Google Scholar] [CrossRef] [Green Version]
- Kabir, V.; Maertens, J.; Kuypers, D. Fungal infections in solid organ transplantation: An update on diagnosis and treatment. Transpl. Rev. 2019, 33, 77–86. [Google Scholar] [CrossRef]
- Hakim, A.; Braun, H.; Thornton, D.; Strymish, J. Successful treatment of methicillin-sensitive Staphylococcus aureus tricuspid-valve endocarditis with dalbavancin as an outpatient in a person who injects drugs: A case report. Int. J. Infect. Dis. 2020, 91, 202–205. [Google Scholar] [CrossRef] [Green Version]
- Chen, A.Y.; Zervos, M.J.; Vazquez, J.A. Dalbavancin: A novel antimicrobial. Int. J. Clin. Pr. 2007, 61, 853–863. [Google Scholar] [CrossRef] [Green Version]
- Giannella, M.; Bartoletti, M.; Gatti, M.; Viale, P. Advances in the therapy of bacterial bloodstream infections. Clin. Microbiol. Infect. 2020, 26, 158–167. [Google Scholar] [CrossRef]
- Berditsch, M.; Lux, H.; Babii, O.; Afonin, S.; Ulrich, A.S. Therapeutic Potential of Gramicidin S in the Treatment of Root Canal Infections. Pharmaceuticals 2016, 9, 56. [Google Scholar] [CrossRef] [Green Version]
- Mirski, T.; Niemcewicz, M.; Bartoszcze, M.; Gryko, R.; Michalski, A. Utilisation of peptides against microbial infections—A review. Ann. Agric. Environ. Med. 2018, 25, 205–210. [Google Scholar] [CrossRef]
- Loutet, S.A.; Valvano, M.A. Extreme antimicrobial Peptide and polymyxin B resistance in the genus burkholderia. Front. Microbiol. 2011, 1, 159. [Google Scholar] [CrossRef]
- Gomes, B.; Augusto, M.T.; Felício, M.R.; Hollmann, A.; Franco, O.L.; Gonçalves, S.; Santos, N.C. Designing improved active peptides for therapeutic approaches against infectious diseases. Biotechnol. Adv. 2018, 36, 415–429. [Google Scholar] [CrossRef]
- Brade, K.D.; Rybak, J.M.; Rybak, M.J. Oritavancin: A New Lipoglycopeptide Antibiotic in the Treatment of Gram-Positive Infections. Infect. Dis. 2016, 5, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kosowska-Shick, K.; Clark, C.; Pankuch, G.A.; McGhee, P.; Dewasse, B.; Beachel, L.; Appelbaum, P.C. Activity of Telavancin against Staphylococci and Enterococci Determined by MIC and Resistance Selection Studies. Antimicrob. Agents Chemother. 2009, 53, 4217–4224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leshem, A.; Liwinski, T.; Elinav, E. Immune-Microbiota Interplay and Colonization Resistance in Infection. Mol. Cell 2020, 78, 597–613. [Google Scholar] [CrossRef] [PubMed]
- Bauer, M.E.; Shafer, W.M. On the in vivo significance of bacterial resistance to antimicrobial peptides. Biochim. Biophys. Acta (BBA)-Biomembr. 2015, 1848, 3101–3111. [Google Scholar] [CrossRef] [Green Version]
- Lewies, A.; Du Plessis, L.H.; Wentzel, J.F. Antimicrobial Peptides: The Achilles’ Heel of Antibiotic Resistance? Probiotics Antimicrob. Proteins 2019, 11, 370–381. [Google Scholar] [CrossRef]
- Kawano, Y.; Jordan, O.; Hanawa, T.; Borchard, G.; Patrulea, V. Are Antimicrobial Peptide Dendrimers an Escape from ESKAPE? Adv. Wound Care 2020, 9, 378–395. [Google Scholar] [CrossRef]
- Park, S.-C.; Park, Y.; Hahm, K.-S. The role of antimicrobial peptides in preventing multidrug-resistant bacterial infections and biofilm formation. Int. J. Mol. Sci. 2011, 12, 5971–5992. [Google Scholar] [CrossRef] [Green Version]
- Kumar, P.; Pletzer, D.; Haney, E.F.; Rahanjam, N.; Cheng, J.T.J.; Yue, M.; Aljehani, W.; Hancock, R.E.W.; Kizhakkedathu, J.N.; Straus, S.K. Aurein-Derived Antimicrobial Peptides Formulated with Pegylated Phospholipid Micelles to Target Methicillin-Resistant Staphylococcus aureus Skin Infections. ACS Infect. Dis. 2019, 5, 443–453. [Google Scholar] [CrossRef]
- Mirakabad, F.S.T.; Khoramgah, M.S.; Keshavarz, K.; Tabarzad, M.; Ranjbari, J. Peptide dendrimers as valuable biomaterials in medical sciences. Life Sci. 2019, 233, 116754. [Google Scholar] [CrossRef]
- Siriwardena, T.N.; Lüscher, A.; Köhler, T.; van Delden, C.; Javor, S.; Reymond, J.-L. Antimicrobial Peptide Dendrimer Chimera. Helv. Chim. Acta 2019, 102, e1900034. [Google Scholar] [CrossRef]
- Tam, J.P.; Lu, Y.-A.; Yang, J.-L. Antimicrobial dendrimeric peptides. Eur. J. Biochem. 2002, 269, 923–932. [Google Scholar] [CrossRef] [PubMed]
- Sowińska, M.; Laskowska, A.; Guśpiel, A.; Solecka, J.; Bochynska-Czyż, M.; Lipkowski, A.W.; Trzeciak, K.; Urbanczyk-Lipkowska, Z. Bioinspired Amphiphilic Peptide Dendrimers as Specific and Effective Compounds against Drug Resistant Clinical Isolates of E. coli. Bioconjugate Chem. 2018, 29, 3571–3585. [Google Scholar] [CrossRef] [PubMed]
- Pires, J.; Siriwardena, T.N.; Stach, M.; Tinguely, R.; Kasraian, S.; Luzzaro, F.; Leib, S.L.; Darbre, T.; Reymond, J.L.; Endimiani, A. In Vitro Activity of the Novel Antimicrobial Peptide Dendrimer G3KL against Multidrug-Resistant Acinetobacter baumannii and Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2015, 59, 7915–7918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, X.; Liu, Y.; Ma, Y.; Zhang, M.; He, Z.; Siriwardena, T.N.; Xu, H.; Bai, Y.; Zhang, X.; Reymond, J.-L.; et al. Peptide dendrimers G3KL and TNS18 inhibit Pseudomonas aeruginosa biofilms. Appl. Microbiol. Biotechnol. 2019, 103, 5821–5830. [Google Scholar] [CrossRef] [PubMed]
- Gan, B.-H.; Siriwardena, T.N.; Javor, S.; Darbre, T.; Reymond, J.-L. Fluorescence Imaging of Bacterial Killing by Antimicrobial Peptide Dendrimer G3KL. ACS Infect. Dis. 2019, 5, 2164–2173. [Google Scholar] [CrossRef]
- Pompilio, A.; Geminiani, C.; Mantini, P.; Siriwardena, T.N.; Di Bonaventura, I.; Reymond, J.L.; Di Bonaventura, G. Peptide dendrimers as “lead compounds” for the treatment of chronic lung infections by Pseudomonas aeruginosa in cystic fibrosis patients: In vitro and in vivo studies. Infect. Drug Resist. 2018, 11, 1767–1782. [Google Scholar] [CrossRef] [Green Version]
- Scorciapino, M.A.; Serra, I.; Manzo, G.; Rinaldi, A.C. Antimicrobial Dendrimeric Peptides: Structure, Activity and New Therapeutic Applications. Int. J. Mol. Sci. 2017, 18, 542. [Google Scholar] [CrossRef] [Green Version]
- Fosgerau, K.; Hoffmann, T. Peptide therapeutics: Current status and future directions. Drug Discov. Today 2015, 20, 122–128. [Google Scholar] [CrossRef] [Green Version]
- da Cunha, N.B.; Cobacho, N.B.; Viana, J.F.C.; Lima, L.A.; Sampaio, K.B.O.; Dohms, S.S.M.; Ferreira, A.C.R.; de la Fuente-Núñez, C.; Costa, F.F.; Franco, O.L.; et al. The next generation of antimicrobial peptides (AMPs) as molecular therapeutic tools for the treatment of diseases with social and economic impacts. Drug Discov. Today 2017, 22, 234–248. [Google Scholar] [CrossRef]
- Laws, M.; Shaaban, A.; Rahman, K.M. Antibiotic resistance breakers: Current approaches and future directions. Fems Microbiol. Rev. 2019, 43, 490–516. [Google Scholar] [CrossRef] [Green Version]
- González-Bello, C. Antibiotic adjuvants—A strategy to unlock bacterial resistance to antibiotics. Bioorganic Med. Chem. Lett. 2017, 27, 4221–4228. [Google Scholar] [CrossRef] [PubMed]
- Brown, D. Antibiotic resistance breakers: Can repurposed drugs fill the antibiotic discovery void? Nat. Rev. Drug Discov. 2015, 14, 821–832. [Google Scholar] [CrossRef] [PubMed]
- Vasile, C.; Pamfil, D.; Stoleru, E.; Baican, M. New Developments in Medical Applications of Hybrid Hydrogels Containing Natural Polymers. Molecules 2020, 25, 1539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Townsend, L.; Williams, R.L.; Anuforom, O.; Berwick, M.R.; Halstead, F.; Hughes, E.; Stamboulis, A.; Oppenheim, B.; Gough, J.; Grover, L.; et al. Antimicrobial peptide coatings for hydroxyapatite: Electrostatic and covalent attachment of antimicrobial peptides to surfaces. J. R. Soc. Interface 2017, 14, 20160657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patrulea, V.; Hirt-Burri, N.; Jeannerat, A.; Applegate, L.A.; Ostafe, V.; Jordan, O.; Borchard, G. Peptide-decorated chitosan derivatives enhance fibroblast adhesion and proliferation in wound healing. Carbohydr. Polym. 2016, 142, 114–123. [Google Scholar] [CrossRef]
- Patrulea, V.; Laurent-Applegate, L.A.; Ostafe, V.; Borchard, G.; Jordan, O. Polyelectrolyte nanocomplexes based on chitosan derivatives for wound healing application. Eur. J. Pharm. Biopharm. 2019, 140, 100–108. [Google Scholar] [CrossRef]
- Barbosa, M.; Costa, F.; Monteiro, C.; Duarte, F.; Martins, M.C.L.; Gomes, P. Antimicrobial coatings prepared from Dhvar-5-click-grafted chitosan powders. Acta Biomater. 2019, 84, 242–256. [Google Scholar] [CrossRef]
- Monteiro, C.; Fernandes, H.; Oliveira, D.; Vale, N.; Barbosa, M.; Gomes, P.; MC, L.M. AMP-Chitosan Coating with Bactericidal Activity in the Presence of Human Plasma Proteins. Molecules 2020, 25, 3046. [Google Scholar] [CrossRef]
- Veronese, F.M.; Mero, A. The Impact of PEGylation on Biological Therapies. BioDrugs 2008, 22, 315–329. [Google Scholar] [CrossRef]
- Imura, Y.; Nishida, M.; Ogawa, Y.; Takakura, Y.; Matsuzaki, K. Action mechanism of tachyplesin I and effects of PEGylation. Biochim. Biophys. Acta (BBA)-Biomembr. 2007, 1768, 1160–1169. [Google Scholar] [CrossRef] [Green Version]
- Imura, Y.; Nishida, M.; Matsuzaki, K. Action mechanism of PEGylated magainin 2 analogue peptide. Biochim. Biophys. Acta (BBA)-Biomembr. 2007, 1768, 2578–2585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, S.; Papareddy, P.; Mörgelin, M.; Schmidtchen, A.; Malmsten, M. Effects of PEGylation on Membrane and Lipopolysaccharide Interactions of Host Defense Peptides. Biomacromolecules 2014, 15, 1337–1345. [Google Scholar] [CrossRef] [PubMed]
- Papathanasiou, E.; Finkelman, M.; Hanley, J.; Parashis, A.O. Prevalence, Etiology and Treatment of Peri-Implant Mucositis and Peri-Implantitis: A Survey of Periodontists in the United States. J. Periodontol. 2016, 87, 493–501. [Google Scholar] [CrossRef] [PubMed]
- Dutta, D.; Ozkan, J.; Willcox, M.D. Biocompatibility of antimicrobial melimine lenses: Rabbit and human studies. Optom. Vis. Sci. 2014, 91, 570–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dutta, D.; Zhao, T.; Cheah, K.B.; Holmlund, L.; Willcox, M.D.P. Activity of a melimine derived peptide Mel4 against Stenotrophomonas, Delftia, Elizabethkingia, Burkholderia and biocompatibility as a contact lens coating. Contact Lens Anterio. 2017, 40, 175–183. [Google Scholar] [CrossRef]
- Taotao, F.; Xiaoyan, Y.; Bing, S.; Leming, S. Peptide self-assembled nanostructures for drug delivery applications. J. Nanomater. 2017, 2017, 16. [Google Scholar] [CrossRef] [Green Version]
- Itzhaki, R.F.; Lathe, R.; Balin, B.J.; Ball, M.J.; Bearer, E.L.; Braak, H.; Bullido, M.J.; Carter, C.; Clerici, M.; Cosby, S.L.; et al. Microbes and Alzheimer’s Disease. J. Alzheimer’s Dis. 2016, 51, 979–984. [Google Scholar] [CrossRef] [Green Version]
- Bourdenx, M.; Koulakiotis, N.S.; Sanoudou, D.; Bezard, E.; Dehay, B.; Tsarbopoulos, A. Protein aggregation and neurodegeneration in prototypical neurodegenerative diseases: Examples of amyloidopathies, tauopathies and synucleinopathies. Prog. Neurobiol. 2017, 155, 171–193. [Google Scholar] [CrossRef]
- Liu, Y.; Ren, B.; Zhang, Y.; Sun, Y.; Chang, Y.; Liang, G.; Xu, L.; Zheng, J. Molecular simulation aspects of amyloid peptides at membrane interface. Biochim. Biophys. Acta (BBA)-Biomembr. 2018, 1860, 1906–1916. [Google Scholar] [CrossRef]
- Malekkhaiat Häffner, S.; Malmsten, M. Influence of self-assembly on the performance of antimicrobial peptides. Curr. Opin. Colloid Interface Sci. 2018, 38, 56–79. [Google Scholar] [CrossRef]
- Görbitz, C.H. The structure of nanotubes formed by diphenylalanine, the core recognition motif of Alzheimer’s β-amyloid polypeptide. Chem. Commun. 2006, 22, 2332–2334. [Google Scholar] [CrossRef] [PubMed]
- Krysmann, M.J.; Castelletto, V.; Kelarakis, A.; Hamley, I.W.; Hule, R.A.; Pochan, D.J. Self-Assembly and Hydrogelation of an Amyloid Peptide Fragment. Biochemistry 2008, 47, 4597–4605. [Google Scholar] [CrossRef]
- Last, N.B.; Miranker, A.D. Common mechanism unites membrane poration by amyloid and antimicrobial peptides. Proc. Natl. Acad. Sci. USA 2013, 110, 6382–6387. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Yang, S.; Li, S.; Lang, J.C.; Mao, C.; Kroll, P.; Tang, L.; Dong, H. Self-Assembled Peptide Nanofibers Display Natural Antimicrobial Peptides to Selectively Kill Bacteria without Compromising Cytocompatibility. ACS Appl. Mater. Interfaces 2019, 11, 28681–28689. [Google Scholar] [CrossRef] [PubMed]
- Yazici, H.; Habib, G.; Boone, K.; Urgen, M.; Utku, F.S.; Tamerler, C. Self-assembling antimicrobial peptides on nanotubular titanium surfaces coated with calcium phosphate for local therapy. Mater. Sci. Eng. C 2019, 94, 333–343. [Google Scholar] [CrossRef]
- Hong, W.; Zhao, Y.; Guo, Y.; Huang, C.; Qiu, P.; Zhu, J.; Chu, C.; Shi, H.; Liu, M. PEGylated Self-Assembled Nano-Bacitracin A: Probing the Antibacterial Mechanism and Real-Time Tracing of Target Delivery in Vivo. ACS Appl. Mater. Interfaces 2018, 10, 10688–10705. [Google Scholar] [CrossRef] [PubMed]
- Cirioni, O.; Silvestri, C.; Ghiselli, R.; Orlando, F.; Riva, A.; Mocchegiani, F.; Chiodi, L.; Castelletti, S.; Gabrielli, E.; Saba, V.; et al. Protective effects of the combination of α-helical antimicrobial peptides and rifampicin in three rat models of Pseudomonas aeruginosa infection. J. Antimicrob. Chemother. 2008, 62, 1332–1338. [Google Scholar] [CrossRef] [Green Version]
- Cirioni, O.; Ghiselli, R.; Silvestri, C.; Kamysz, W.; Orlando, F.; Mocchegiani, F.; Matteo, D.F.; Riva, A.; Łukasiak, J.; Scalise, G.; et al. Efficacy of tachyplesin III, colistin, and imipenem against a multiresistant Pseudomonas aeruginosa strain. Antimicrob. Agents Chemother. 2007, 51, 2005. [Google Scholar] [CrossRef] [Green Version]
- Jeong, N.; Kim, J.-Y.; Park, S.-C.; Lee, J.-K.; Gopal, R.; Yoo, S.; Son, B.K.; Hahm, J.S.; Park, Y.; Hahm, K.-S. Antibiotic and synergistic effect of Leu–Lys rich peptide against antibiotic resistant microorganisms isolated from patients with cholelithiasis. Biochem. Biophys. Res. Commun. 2010, 399, 581–586. [Google Scholar] [CrossRef]
- Lakshminarayanan, R.; Tan, W.X.; Aung, T.T.; Goh, E.T.L.; Muruganantham, N.; Li, J.; Chang, J.Y.T.; Dikshit, N.; Saraswathi, P.; Lim, R.R.; et al. Branched Peptide, B2088, Disrupts the Supramolecular Organization of Lipopolysaccharides and Sensitizes the Gram-negative Bacteria. Sci. Rep. 2016, 6, 25905. [Google Scholar] [CrossRef] [Green Version]
- Dosler, S.; Karaaslan, E. Inhibition and destruction of Pseudomonas aeruginosa biofilms by antibiotics and antimicrobial peptides. Peptides 2014, 62, 32–37. [Google Scholar] [CrossRef]
- Chen, H.; Wubbolts, R.W.; Haagsman, H.P.; Veldhuizen, E.J.A. Inhibition and Eradication of Pseudomonas aeruginosa Biofilms by Host Defence Peptides. Sci. Rep. 2018, 8, 10446. [Google Scholar] [CrossRef] [PubMed]
- Tyers, M.; Wright, G.D. Drug combinations: A strategy to extend the life of antibiotics in the 21st century. Nat. Rev. Microbiol. 2019, 17, 141–155. [Google Scholar] [CrossRef] [PubMed]
- Pal, I.; Bhattacharyya, D.; Kar, R.K.; Zarena, D.; Bhunia, A.; Atreya, H.S. A Peptide-Nanoparticle System with Improved Efficacy against Multidrug Resistant Bacteria. Sci. Rep. 2019, 9, 4485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bahrami, A.; Delshadi, R.; Jafari, S.M. Active delivery of antimicrobial nanoparticles into microbial cells through surface functionalization strategies. Trends Food Sci. Technol. 2020, 99, 217–228. [Google Scholar] [CrossRef]
- Borro, B.C.; Malmsten, M. Complexation between antimicrobial peptides and polyelectrolytes. Adv. Colloid Interface Sci. 2019, 270, 251–260. [Google Scholar] [CrossRef]
- Nikolova, M.P.; Chavali, M.S. Recent advances in biomaterials for 3D scaffolds: A review. Bioact. Mater. 2019, 4, 271–292. [Google Scholar] [CrossRef]
- Calori, I.R.; Braga, G.; Carvalho de Jesus, P.D.C.; Bi, H.; Tedesco, A.C. Polymer Scaffolds as Drug Delivery Systems. Eur. Polym. J. 2020, 106921. [Google Scholar] [CrossRef]
- Liu, C.; Zhang, Q.; Zhu, S.; Liu, H.; Chen, J. Preparation and applications of peptide-based injectable hydrogels. RSC Adv. 2019, 9, 28299–28311. [Google Scholar] [CrossRef] [Green Version]
- Lozeau, L.D.; Grosha, J.; Kole, D.; Prifti, F.; Dominko, T.; Camesano, T.A.; Rolle, M.W. Collagen tethering of synthetic human antimicrobial peptides cathelicidin LL37 and its effects on antimicrobial activity and cytotoxicity. Acta Biomater. 2017, 52, 9–20. [Google Scholar] [CrossRef] [Green Version]
- Cassin, M.E.; Ford, A.J.; Orbach, S.M.; Saverot, S.E.; Rajagopalan, P. The design of antimicrobial LL37-modified collagen-hyaluronic acid detachable multilayers. Acta Biomater. 2016, 40, 119–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- US National Library of Medicine. A Study of Granexin Gel in the Treatment of Diabetic Foot Ulcer. Available online: https://clinicaltrials.gov/ct2/show/NCT02667327 (accessed on 23 March 2020).
- Grek, C.L.; Prasad, G.M.; Viswanathan, V.; Armstrong, D.G.; Gourdie, R.G.; Ghatnekar, G.S. Topical administration of a connexin43-based peptide augments healing of chronic neuropathic diabetic foot ulcers: A multicenter, randomized trial. Wound Repair Regen. 2015, 23, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.; Zhan, B.; Zhang, W.; Qin, D.; Xia, G.; Zhang, H.; Peng, M.; Li, S.-A.; Zhang, Y.; Gao, Y.; et al. Carboxymethyl chitosan nanoparticles loaded with bioactive peptide OH-CATH30 benefit nonscar wound healing. Int. J. Nanomed. 2018, 13, 5771–5786. [Google Scholar] [CrossRef] [Green Version]
- Almaaytah, A.; Mohammed, G.K.; Abualhaijaa, A.; Al-Balas, Q. Development of novel ultrashort antimicrobial peptide nanoparticles with potent antimicrobial and antibiofilm activities against multidrug-resistant bacteria. Drug Des. Dev. Ther. 2017, 11, 3159–3170. [Google Scholar] [CrossRef] [Green Version]
- Nordström, R.; Nyström, L.; Andrén, O.C.J.; Malkoch, M.; Umerska, A.; Davoudi, M.; Schmidtchen, A.; Malmsten, M. Membrane interactions of microgels as carriers of antimicrobial peptides. J. Colloid Interface Sci. 2018, 513, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Nordström, R.; Browning, K.L.; Parra-Ortiz, E.; Damgaard, L.S.E.; Häffner, S.M.; Maestro, A.; Campbell, R.A.; Cooper, J.F.K.; Malmsten, M. Membrane interactions of antimicrobial peptide-loaded microgels. J. Colloid Interface Sci. 2020, 562, 322–332. [Google Scholar] [CrossRef] [PubMed]
- Boge, L.; Hallstensson, K.; Ringstad, L.; Johansson, J.; Andersson, T.; Davoudi, M.; Larsson, P.T.; Mahlapuu, M.; Håkansson, J.; Andersson, M. Cubosomes for topical delivery of the antimicrobial peptide LL-37. Eur. J. Pharm. Biopharm. 2019, 134, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Innocenti Malini, R.; Zabara, M.; Gontsarik, M.; Maniura-Weber, K.; Rossi, R.M.; Spano, F.; Salentinig, S. Self-assembly of glycerol monooleate with the antimicrobial peptide LL-37: A molecular dynamics study. RSC Adv. 2020, 10, 8291–8302. [Google Scholar] [CrossRef]
- Silva, J.P.; Gonçalves, C.; Costa, C.; Sousa, J.; Silva-Gomes, R.; Castro, A.G.; Pedrosa, J.; Appelberg, R.; Gama, F.M. Delivery of LLKKK18 loaded into self-assembling hyaluronic acid nanogel for tuberculosis treatment. J. Control. Release 2016, 235, 112–124. [Google Scholar] [CrossRef] [Green Version]
- Lin, Z.; Wu, T.; Wang, W.; Li, B.; Wang, M.; Chen, L.; Xia, H.; Zhang, T. Biofunctions of antimicrobial peptide-conjugated alginate/hyaluronic acid/collagen wound dressings promote wound healing of a mixed-bacteria-infected wound. Int. J. Biol. Macromol. 2019, 140, 330–342. [Google Scholar] [CrossRef]
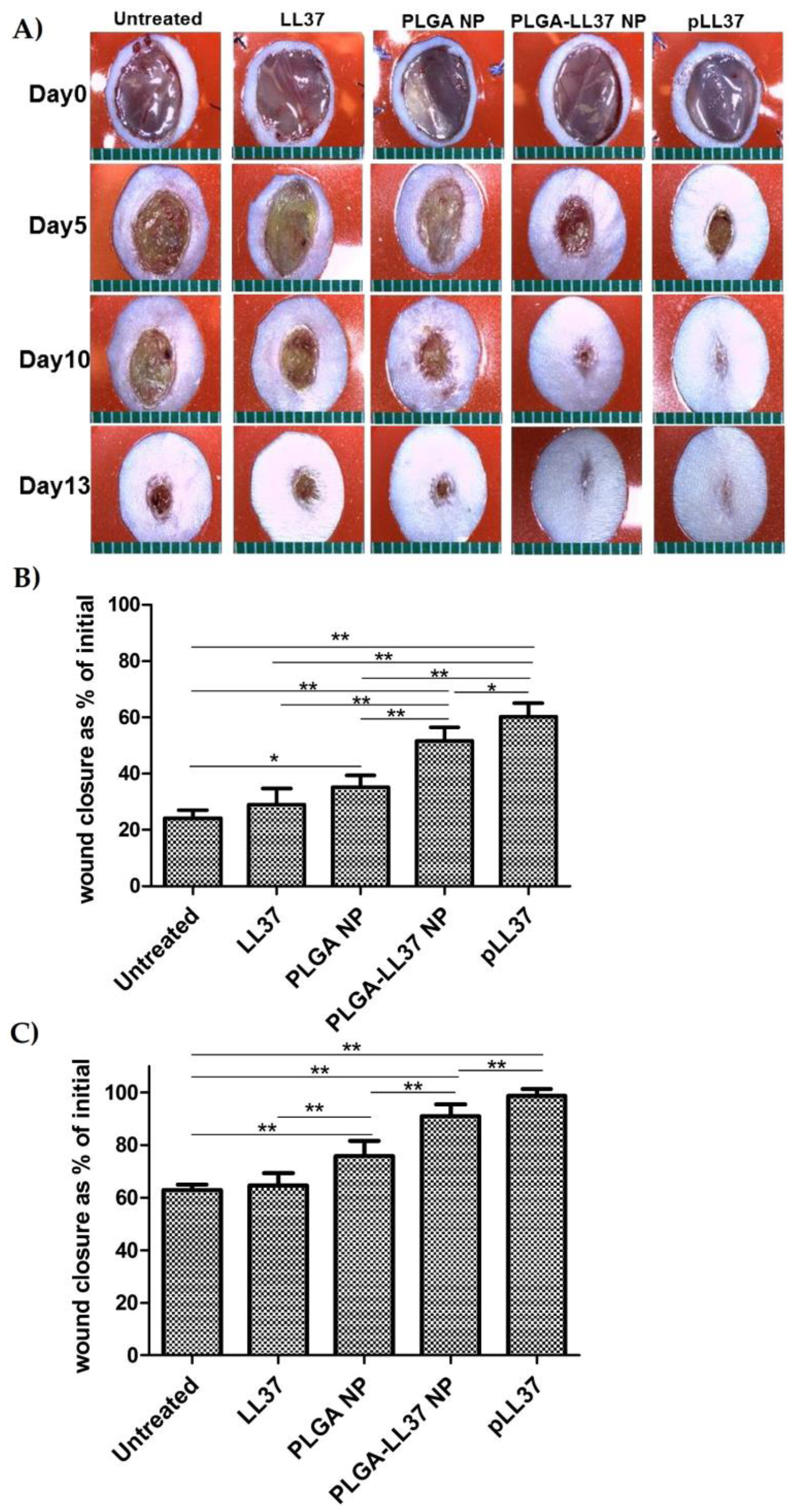
- Chereddy, K.K.; Her, C.-H.; Comune, M.; Moia, C.; Lopes, A.; Porporato, P.E.; Vanacker, J.; Lam, M.C.; Steinstraesser, L.; Sonveaux, P.; et al. PLGA nanoparticles loaded with host defense peptide LL37 promote wound healing. J. Control. Release 2014, 194, 138–147. [Google Scholar] [CrossRef] [PubMed]
- Casciaro, B.; d’Angelo, I.; Zhang, X.; Loffredo, M.R.; Conte, G.; Cappiello, F.; Quaglia, F.; Di, Y.-P.P.; Ungaro, F.; Mangoni, M.L. Poly(lactide-co-glycolide) Nanoparticles for Prolonged Therapeutic Efficacy of Esculentin-1a-Derived Antimicrobial Peptides against Pseudomonas aeruginosa Lung Infection: In Vitro and in Vivo Studies. Biomacromolecules 2019, 20, 1876–1888. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Feng, S.; Qie, J.; Wei, X.; Yan, H.; Liu, K. Polyion complexes of a cationic antimicrobial peptide as a potential systemically administered antibiotic. Int. J. Pharm. 2019, 554, 284–291. [Google Scholar] [CrossRef]
- Garcia-Orue, I.; Gainza, G.; Girbau, C.; Alonso, R.; Aguirre, J.J.; Pedraz, J.L.; Igartua, M.; Hernandez, R.M. LL37 loaded nanostructured lipid carriers (NLC): A new strategy for the topical treatment of chronic wounds. Eur. J. Pharm. Biopharm. 2016, 108, 310–316. [Google Scholar] [CrossRef] [PubMed]
- Cheng, R.; Liu, L.; Xiang, Y.; Lu, Y.; Deng, L.; Zhang, H.; Santos, H.A.; Cui, W. Advanced liposome-loaded scaffolds for therapeutic and tissue engineering applications. Biomaterials 2020, 232, 119706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wijnant, G.-J.; Van Bocxlaer, K.; Yardley, V.; Harris, A.; Alavijeh, M.; Silva-Pedrosa, R.; Antunes, S.; Mauricio, I.; Murdan, S.; Croft, S.L. Comparative efficacy, toxicity and biodistribution of the liposomal amphotericin B formulations Fungisome® and AmBisome® in murine cutaneous leishmaniasis. Int. J. Parasitol. Drugs Drug Resist. 2018, 8, 223–228. [Google Scholar] [CrossRef]
- Yang, Z.; Liu, J.; Gao, J.; Chen, S.; Huang, G. Chitosan coated vancomycin hydrochloride liposomes: Characterizations and evaluation. Int. J. Pharm. 2015, 495, 508–515. [Google Scholar] [CrossRef]
- Yamakami, K.; Tsumori, H.; Shimizu, Y.; Sakurai, Y.; Nagatoshi, K.; Sonomoto, K. Cationic Lipid Content in Liposome-Encapsulated Nisin Improves Sustainable Bactericidal Activity against Streptococcus mutans. Open Dent. J. 2016, 10, 360–366. [Google Scholar] [CrossRef]
- Alipour, M.; Suntres, Z.E.; Halwani, M.; Azghani, A.O.; Omri, A. Activity and interactions of liposomal antibiotics in presence of polyanions and sputum of patients with cystic fibrosis. PLoS ONE 2009, 4, e5724. [Google Scholar] [CrossRef]
- He, J.; Abdelraouf, K.; Ledesma, K.R.; Chow, D.S.L.; Tam, V.H. Pharmacokinetics and efficacy of liposomal polymyxin B in a murine pneumonia model. Int. J. Antimicrob. Agents 2013, 42, 559–564. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Zhang, X.; Huang, X.; Wang, X.; Liao, G.; Chen, Z. Preparation and characterization of flexible nanoliposomes loaded with daptomycin, a novel antibiotic, for topical skin therapy. Int. J. Nanomed. 2013, 8, 1285–1292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prasher, P.; Sharma, M.; Mudila, H.; Gupta, G.; Sharma, A.K.; Kumar, D.; Bakshi, H.A.; Negi, P.; Kapoor, D.N.; Chellappan, D.K.; et al. Emerging trends in clinical implications of bio-conjugated silver nanoparticles in drug delivery. Colloid Interface Sci. Commun. 2020, 35, 100244. [Google Scholar] [CrossRef]
- Casciaro, B.; Moros, M.; Rivera-Fernández, S.; Bellelli, A.; de la Fuente, J.M.; Mangoni, M.L. Gold-nanoparticles coated with the antimicrobial peptide esculentin-1a(1-21)NH2 as a reliable strategy for antipseudomonal drugs. Acta Biomater. 2017, 47, 170–181. [Google Scholar] [CrossRef] [Green Version]
- de Alteriis, E.; Maselli, V.; Falanga, A.; Galdiero, S.; Di Lella, F.M.; Gesuele, R.; Guida, M.; Galdiero, E. Efficiency of gold nanoparticles coated with the antimicrobial peptide indolicidin against biofilm formation and development of Candida spp. clinical isolates. Infect. Drug Resist. 2018, 11, 915–925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rai, A.; Pinto, S.; Velho, T.R.; Ferreira, A.F.; Moita, C.; Trivedi, U.; Evangelista, M.; Comune, M.; Rumbaugh, K.P.; Simões, P.N.; et al. One-step synthesis of high-density peptide-conjugated gold nanoparticles with antimicrobial efficacy in a systemic infection model. Biomaterials 2016, 85, 99–110. [Google Scholar] [CrossRef]
- Pal, I.; Brahmkhatri, V.P.; Bera, S.; Bhattacharyya, D.; Quirishi, Y.; Bhunia, A.; Atreya, H.S. Enhanced stability and activity of an antimicrobial peptide in conjugation with silver nanoparticle. J. Colloid Interface Sci. 2016, 483, 385–393. [Google Scholar] [CrossRef]
- Lambadi, P.R.; Sharma, T.K.; Kumar, P.; Vasnani, P.; Thalluri, S.M.; Bisht, N.; Pathania, R.; Navani, N.K. Facile biofunctionalization of silver nanoparticles for enhanced antibacterial properties, endotoxin removal, and biofilm control. Int. J. Nanomed. 2015, 10, 2155–2171. [Google Scholar] [CrossRef] [Green Version]
- Zheng, K.; Setyawati, M.I.; Lim, T.-P.; Leong, D.T.; Xie, J. Antimicrobial Cluster Bombs: Silver Nanoclusters Packed with Daptomycin. ACS Nano 2016, 10, 7934–7942. [Google Scholar] [CrossRef]
- Arvizo, R.; Bhattacharya, R.; Mukherjee, P. Gold nanoparticles: Opportunities and challenges in nanomedicine. Expert Opin. Drug Deliv. 2010, 7, 753–763. [Google Scholar] [CrossRef] [Green Version]
- Balfourier, A.; Luciani, N.; Wang, G.; Lelong, G.; Ersen, O.; Khelfa, A.; Alloyeau, D.; Gazeau, F.; Carn, F. Unexpected intracellular biodegradation and recrystallization of gold nanoparticles. Proc. Natl. Acad. Sci. USA 2020, 117, 103–113. [Google Scholar] [CrossRef]
- Li, P.; Han, F.; Cao, W.; Zhang, G.; Li, J.; Zhou, J.; Gong, X.; Turnbull, G.; Shu, W.; Xia, L.; et al. Carbon quantum dots derived from lysine and arginine simultaneously scavenge bacteria and promote tissue repair. Appl. Mater. Today 2020, 19, 100601. [Google Scholar] [CrossRef]
- Maleki Dizaj, S.; Mennati, A.; Jafari, S.; Khezri, K.; Adibkia, K. Antimicrobial Activity of Carbon-Based Nanoparticles. Adv Pharm. Bull. 2015, 5, 19–23. [Google Scholar] [CrossRef] [PubMed]
- Chaudhari, A.A.; Ashmore, D.A.; Nath, S.d.; Kate, K.; Dennis, V.; Singh, S.R.; Owen, D.R.; Palazzo, C.; Arnold, R.D.; Miller, M.E.; et al. A novel covalent approach to bio-conjugate silver coated single walled carbon nanotubes with antimicrobial peptide. J. Nanobiotechnol. 2016, 14, 58. [Google Scholar] [CrossRef] [Green Version]
- Sur, A.; Pradhan, B.; Banerjee, A.; Aich, P. Immune activation efficacy of indolicidin is enhanced upon conjugation with carbon nanotubes and gold nanoparticles. PLoS ONE 2015, 10, e0123905. [Google Scholar] [CrossRef] [PubMed]
- Kanchanapally, R.; Viraka Nellore, B.P.; Sinha, S.S.; Pedraza, F.; Jones, S.J.; Pramanik, A.; Chavva, S.R.; Tchounwou, C.; Shi, Y.; Vangara, A.; et al. Antimicrobial peptide-conjugated graphene oxide membrane for efficient removal and effective killing of multiple drug resistant bacteria. RSC Adv. 2015, 5, 18881–18887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viraka Nellore, B.P.; Kanchanapally, R.; Pedraza, F.; Sinha, S.S.; Pramanik, A.; Hamme, A.T.; Arslan, Z.; Sardar, D.; Ray, P.C. Bio-Conjugated CNT-Bridged 3D Porous Graphene Oxide Membrane for Highly Efficient Disinfection of Pathogenic Bacteria and Removal of Toxic Metals from Water. ACS Appl. Mater. Interfaces 2015, 7, 19210–19218. [Google Scholar] [CrossRef] [Green Version]
- Dostalova, S.; Moulick, A.; Milosavljevic, V.; Guran, R.; Kominkova, M.; Cihalova, K.; Heger, Z.; Blazkova, L.; Kopel, P.; Hynek, D.; et al. Antiviral activity of fullerene C60 nanocrystals modified with derivatives of anionic antimicrobial peptide maximin H5. Mon. Für Chem.-Chem. Mon. 2016, 147, 905–918. [Google Scholar] [CrossRef]
- Barron, A.R. [60]Fullerene-peptides: Bio-nano conjugates with structural and chemical diversity. J. Enzym. Inhib. Med. Chem. 2016, 31, 164–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pellarini, F.; Pantarotto, D.; Da Ros, T.; Giangaspero, A.; Tossi, A.; Prato, M. A Novel [60]Fullerene Amino Acid for Use in Solid-Phase Peptide Synthesis. Org. Lett. 2001, 3, 1845–1848. [Google Scholar] [CrossRef]
- Pantarotto, D.; Bianco, A.; Pellarini, F.; Tossi, A.; Giangaspero, A.; Zelezetsky, I.; Briand, J.-P.; Prato, M. Solid-Phase Synthesis of Fullerene-peptides. J. Am. Chem. Soc. 2002, 124, 12543–12549. [Google Scholar] [CrossRef]
- Ashraf, M.U.; Hussain, M.A.; Muhammad, G.; Haseeb, M.T.; Bashir, S.; Hussain, S.Z.; Hussain, I. A superporous and superabsorbent glucuronoxylan hydrogel from quince (Cydonia oblanga): Stimuli responsive swelling, on-off switching and drug release. Int. J. Biol. Macromol. 2017, 95, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Rivero, G.; Meuter, M.; Pepe, A.; Guevara, M.G.; Boccaccini, A.R.; Abraham, G.A. Nanofibrous membranes as smart wound dressings that release antibiotics when an injury is infected. Colloids Surf. A: Physicochem. Eng. Asp. 2020, 587, 124313. [Google Scholar] [CrossRef]
- Muhammad, G.; Hussain, M.A.; Amin, M.; Hussain, S.Z.; Hussain, I.; Abbas Bukhari, S.N.; Naeem-ul-Hassan, M. Glucuronoxylan-mediated silver nanoparticles: Green synthesis, antimicrobial and wound healing applications. RSC Adv. 2017, 7, 42900–42908. [Google Scholar] [CrossRef] [Green Version]
- Wu, K.; Wu, X.; Chen, M.; Wu, H.; Jiao, Y.; Zhou, C. H2O2-responsive smart dressing for visible H2O2 monitoring and accelerating wound healing. Chem. Eng. J. 2020, 387, 124127. [Google Scholar] [CrossRef]
- Hu, M.; Korschelt, K.; Daniel, P.; Landfester, K.; Tremel, W.; Bannwarth, M.B. Fibrous Nanozyme Dressings with Catalase-Like Activity for H2O2 Reduction To Promote Wound Healing. ACS Appl. Mater. Interfaces 2017, 9, 38024–38031. [Google Scholar] [CrossRef]
- Rezaei, N.; Hamidabadi, H.G.; Khosravimelal, S.; Zahiri, M.; Ahovan, Z.A.; Bojnordi, M.N.; Eftekhari, B.S.; Hashemi, A.; Ganji, F.; Darabi, S.; et al. Antimicrobial peptides-loaded smart chitosan hydrogel: Release behavior and antibacterial potential against antibiotic resistant clinical isolates. Int. J. Biol. Macromol. 2020, 164, 855–862. [Google Scholar] [CrossRef]
- Panzarasa, G.; Osypova, A.; Toncelli, C.; Buhmann, M.T.; Rottmar, M.; Ren, Q.; Maniura-Weber, K.; Rossi, R.M.; Boesel, L.F. The pyranine-benzalkonium ion pair: A promising fluorescent system for the ratiometric detection of wound pH. Sens. Actuators B Chem. 2017, 249, 156–160. [Google Scholar] [CrossRef]
- Lombardi, L.; Shi, Y.; Falanga, A.; Galdiero, E.; de Alteriis, E.; Franci, G.; Chourpa, I.; Azevedo, H.S.; Galdiero, S. Enhancing the Potency of Antimicrobial Peptides through Molecular Engineering and Self-Assembly. Biomacromolecules 2019, 20, 1362–1374. [Google Scholar] [CrossRef]
- Dimatteo, R.; Darling, N.J.; Segura, T. In situ forming injectable hydrogels for drug delivery and wound repair. Adv. Drug Deliv. Rev. 2018, 127, 167–184. [Google Scholar] [CrossRef]
- Bradshaw, M.; Ho, D.; Fear, M.W.; Gelain, F.; Wood, F.M.; Iyer, K.S. Designer self-assembling hydrogel scaffolds can impact skin cell proliferation and migration. Sci. Rep. 2014, 4, 6903. [Google Scholar] [CrossRef] [Green Version]
- Zhou, A.; Chen, S.; He, B.; Zhao, W.; Chen, X.; Jiang, D. Controlled release of TGF-beta 1 from RADA self-assembling peptide hydrogel scaffolds. Drug Des. Dev. Ther. 2016, 10, 3043–3051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Håkansson, J.; Ringstad, L.; Umerska, A.; Johansson, J.; Andersson, T.; Boge, L.; Rozenbaum, R.T.; Sharma, P.K.; Tollbäck, P.; Björn, C.; et al. Characterization of the in vitro, ex vivo, and in vivo Efficacy of the Antimicrobial Peptide DPK-060 Used for Topical Treatment. Front. Cell. Infect. Microbiol. 2019, 9, 174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vlieghe, P.; Lisowski, V.; Martinez, J.; Khrestchatisky, M. Synthetic therapeutic peptides: Science and market. Drug Discov. Today 2010, 15, 40–56. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, S.; Suzuki, T.; Suzuki, T.; Takatsuka, H.; Ishikawa, M.; Hattori, N.; Fujishiro, T.; Miyauchi, H.; Oami, T.; Ariyoshi, N.; et al. An extremely high bioavailability of orally administered vancomycin in a patient with severe colitis and renal insufficiency. J. Infect. Chemother. 2017, 23, 848–851. [Google Scholar] [CrossRef]
- Cimolai, N. Does oral vancomycin use necessitate therapeutic drug monitoring? Infection 2020, 48, 173–182. [Google Scholar] [CrossRef]
- Davis, C.A.; Janssen, E.M.L. Environmental fate processes of antimicrobial peptides daptomycin, bacitracins, and polymyxins. Environ. Int. 2020, 134, 105271. [Google Scholar] [CrossRef]
- Saravolatz, L.D.; Cleveland, K.O.; Rikabi, K.; Hassoun, A.; Reilly, J.; Johnson, L.B.; Spak, C.; Valenti, S.; Szpunar, S. Real-world use of telavancin in the treatment of osteomyelitis. Diagn. Microbiol. Infect. Dis. 2019, 95, 185–190. [Google Scholar] [CrossRef]
- Chastain, D.B.; Davis, A. Treatment of chronic osteomyelitis with multidose oritavancin: A case series and literature review. Int. J. Antimicrob. Agents 2019, 53, 429–434. [Google Scholar] [CrossRef]
- Sinha, R.; Shukla, P. Antimicrobial Peptides: Recent Insights on Biotechnological Interventions and Future Perspectives. Protein Pept. Lett. 2019, 26, 79–87. [Google Scholar] [CrossRef]
- Pursey, E.; Sünderhauf, D.; Gaze, W.H.; Westra, E.R.; van Houte, S. CRISPR-Cas antimicrobials: Challenges and future prospects. PLoS Pathog. 2018, 14, e1006990. [Google Scholar] [CrossRef] [Green Version]
- Hanson, M.A.; Dostálová, A.; Ceroni, C.; Poidevin, M.; Kondo, S.; Lemaître, B. Correction: Synergy and remarkable specificity of antimicrobial peptides in vivo using a systematic knockout approach. eLife 2019, 8, e48778. [Google Scholar] [CrossRef] [PubMed]
- Unckless, R.L.; Howick, V.M.; Lazzaro, B.P. Convergent Balancing Selection on an Antimicrobial Peptide in Drosophila. Curr. Biol. 2016, 26, 257–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choudhary, E.; Sharma, R.; Kumar, Y.; Agarwal, N. Conditional Silencing by CRISPRi Reveals the Role of DNA Gyrase in Formation of Drug-Tolerant Persister Population in Mycobacterium tuberculosis. Front. Cell. Infect. Microbiol. 2019, 9, 70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, W.; Zhang, S.; Fleming, J.; Chen, Y.; Li, Z.; Fan, S.; Liu, Y.; Wang, W.; Wang, T.; Liu, Y.; et al. Mycobacterium tuberculosis type III-A CRISPR/Cas system crRNA and its maturation have atypical features. FASEB J. 2019, 33, 1496–1509. [Google Scholar] [CrossRef] [PubMed]
- Citorik, R.J.; Mimee, M.; Lu, T.K. Sequence-specific antimicrobials using efficiently delivered RNA-guided nucleases. Nat. Biotechnol. 2014, 32, 1141–1145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bikard, D.; Euler, C.W.; Jiang, W.; Nussenzweig, P.M.; Goldberg, G.W.; Duportet, X.; Fischetti, V.A.; Marraffini, L.A. Exploiting CRISPR-Cas nucleases to produce sequence-specific antimicrobials. Nat. Biotechnol. 2014, 32, 1146–1150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Peng, N. Endogenous CRISPR-Cas System-Based Genome Editing and Antimicrobials: Review and Prospects. Front. Microbiol. 2019, 10, 2471. [Google Scholar] [CrossRef] [Green Version]
- Kosikowska, P.; Lesner, A. Antimicrobial peptides (AMPs) as drug candidates: A patent review (2003–2015). Expert Opin. Ther. Pat. 2016, 26, 689–702. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
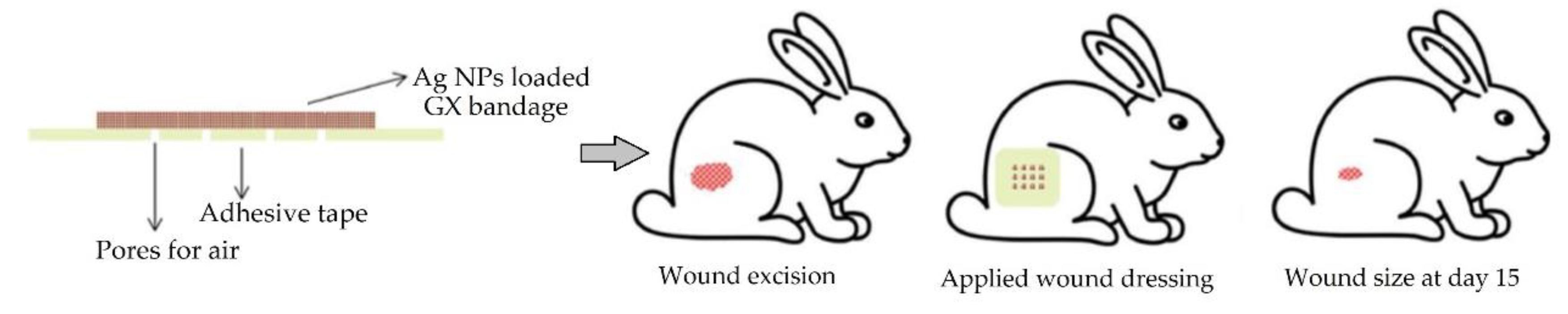{kind=link}
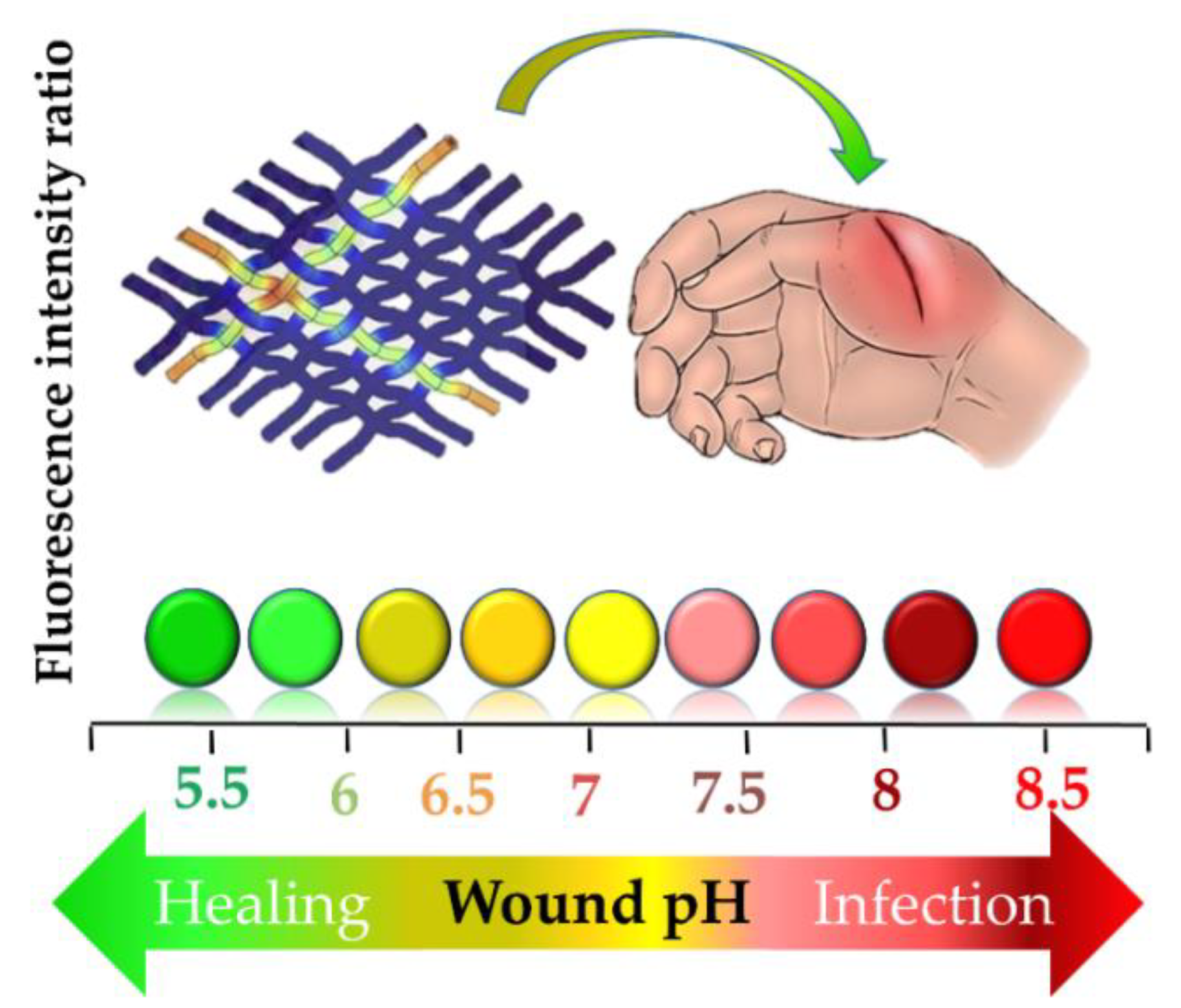{kind=link}
| Activity AMP | Selected Examples of AMPs | Total No. |
|---|---|---|
| Antibacterial peptides | Abaecin; andropin; bombinin; 1 bBD-1-13; cecropin A, B, C, D, P; cryptdin; drosocin; esculentin-1-2; dermaseptin-B2-B5, B6, S1-S4; 2 hBD-26,27; LL-37; magainin; melittin; nisin; protegrin 1; pyrrhocoricin; temporin A, B, C, E, F, G, K, L; thanatin; tritrpticin | 2678 |
| Antibiofilm peptides | BMAP-27,28; citropin 1.1; colistin A; Dhvar4; gramicidin S; hBD-3; holothuroidin 1; indolicidin; LL-37; nisin A; polymyxin B; protegrin 1; SMAP-29 (Ovispirin); tachyplesin III; temporin B; temporin-1CEb | 57 |
| Anticancer peptides | Alloferon 1,2; aurein 1–3; buforin II; gomesin; indolicidin; lactoferricin B; LL-37; magainin 2; mastoparan B; melittin; nisin A,Z; tritrpticin | 237 |
| Anti-diabetic peptides | Amolopin; brevinin-1E, 2EC; esculentin-1, 1B; magainin-AM2 | 15 |
| Antifungal peptides | Androctonin; antifungal protein; aurein 1–3; cecropin 2, A, B; dermaseptin-S1-S5; HD-2-6; HNP-1-6; indolicidin; lactoferricin B; magainin 2; melittin; protegrin 1–5; ponericin G1-G4; G7, W1-W5; thanatin; tritrpticin | 1142 |
| Anti-HIV peptides | Aurein 1.2; cecropin A; dermaseptin-S1,S4, S9; hBD-2,3; HNP-1-4; indolicidin; lactoferricin B; LL-37; melittin; protegrin 1 | 109 |
| Anti-inflammatory peptides | Allomyrinasin; cathelicidin-PY; coprisin; defensin DEFB126; lucilin; papiliocin | 20 |
| Anti-MRSA peptides | Acipensin 1,2; BMAP-27,28; CAP18, citropin 1.1; clavanin A; cryptdin-4; 4 Dhvar5; esculentin-1,2 ISa-ISb; hBD-3; hedistin; 3 HNP-1; hominicin; imcroporin; indolicidin; LL-37; micasin-1; omega76; SMAP-29; plectasin; pleurocidin; protegrin 1; ubiquicidin | 165 |
| Antiparasitic peptides | Batroxicidin; cecropin A; dermaseptin-S1-S5; kalata B2, B5-B7; LL-37; magainin 2; melittin; temporin A, B, F, L; | 116 |
| Anti-sepsis peptides | Apidaecin IA; bactenecin 7; buforin II; cathelicidin-PY; cecropin 2, P1; drosocin; 5 HD-5; HNP-1; lactoferricin B; LL-37; melittin; polymyxin B; protegrin 1; pyrrhocoricin; SMAP-29; tachyplesin I; temporin L; thanatin | 75 |
| Anti-toxin peptides | hBD-1-4; HNP-1-5; retrocyclin-1-3 | 15 |
| Anti-tuberculosis peptides | Griselimycin; hBD consensus; hBD10; human granulysin; lassomycin; laterosporulin10; LL-37; micrococcin P1; pantocin wh-1; RNase 7; Teixobactin; VpAmp1.0, 2.0 | 13 |
| Antiviral peptides | Alloferon 1,2; antiviral protein Y3; aurein 1.2; BMAP-27,28; dermaseptin-S1, S4; hBD-1-3; HNP-1-6; indolicidin; lactoferricin B; LL-37; magainin 2; melittin; mucroporin; protegrin 1–5; thanatin; temporin A, B | 189 |
| Wound-healing peptides | AH90; AG-30; AG-30/5C; bactenecin; coprisin; epinecidin-1; hBD-2, 3; HD-5; HNP-1; IDR-1018; indolicidin; LL-37; lucifensin; magainin 2; nisin A; temporin A | 22 |
| AMPs | Mechanism of Action | Activity Against | Side Effects | Application and Administration Route | Ref |
|---|---|---|---|---|---|
| Potent AMPs | |||||
| AG30 | Membrane disruption | Gram (+), (−); E. coli (MIC: 40 µg/mL) and S. aureus (MIC: 20 µg/mL) | Lack of stability in saline | Topical | [36] |
| AG30/5C | Membrane disruption | Gram (+), (−); P. aeruginosa (MIC: 5 µg/mL), S. aureus (MIC: 50 µg/mL) | More stable than AG30 | Topical | [37] |
| Cys-KR12 | Membrane disruption | Gram (+), (−); E. coli (MIC: 4 µg/mL), S. aureus (MIC: 8 µg/mL) | Not reported | Topical | [45] |
| KR12 | Membrane disruption | Gram (+), (−); E. coli (MIC: 2.5 µM), P. aeruginosa (MIC: 10 µM) | Not reported | Topical | [46] |
| WLBU2 | Binding to lipopolysaccharide and DNA inhibition | Gram (+), (−); P. aeruginosa (MIC: 3–8 µM); S. aureus (MIC: 6–16 µM) | Not reported | Eradicated lethal P. aeruginosa septicemia in mice | [47,48] |
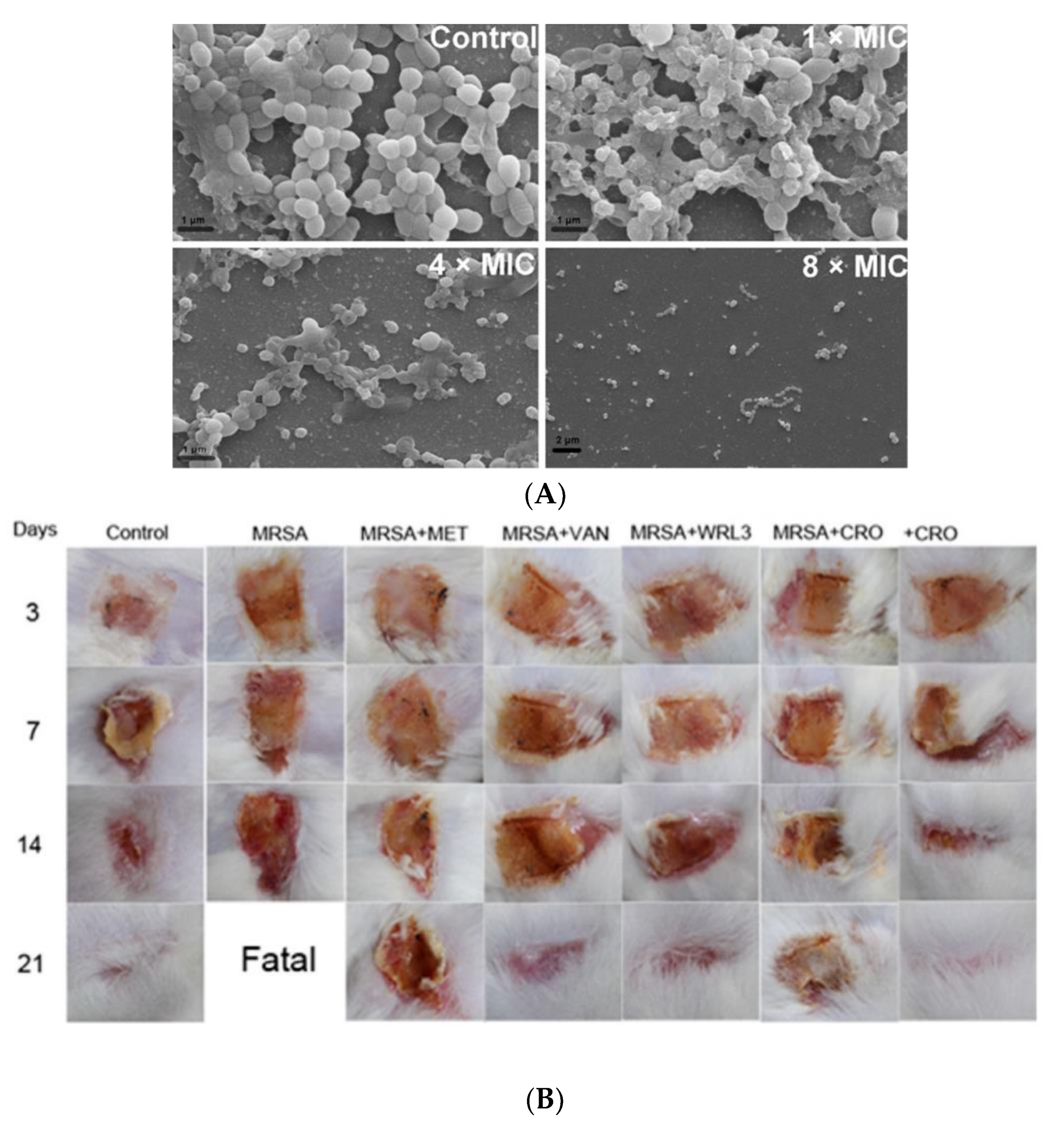
| WRL3 | Membrane lysis | Gram (+), (−); MRSA (MIC: 2 μg/mL) | Not reported | MRSA-related infections in skin burn wounds; topical | [41] |
| Preclinical Phase | |||||
| Arenicin | Membrane pore formation | Gram (+), (−); MRSA infection; E. coli (MIC: 1 µg/mL); K. pneumoniae (2 µg/mL) | Significant toxicity to mammalian cells | Urinary tract infections; hospital-acquired infections | [23,49,50] |
| Avidocin and purocin | Membrane disruption | Gram (+) and (−) | Safety reported | Treatment of C. difficile infections (colitis); topical | [23] |
| Buforin II | Inhibition of DNA/RNA synthesis | Gram (+), (−); fungi; E. coli (MIC: 1 µg/mL); K. pneumoniae (MIC: 2 µg/mL) | Safety reported | Used as bacteriostatic; bactericide; anti-sepsis | [25,51] |
| Lactocin 160 (Bacteriocin) | Membrane disruption | G. vaginalis; P. bivia; Lactobacillus spp. (MIC: > 200 mg/mL) | Safety reported | Urogenital tract infections; Bacterial vaginosis | [52,53] |
| LTX-109 (Lytixar) | Membrane disruption and cell lysis | Gram (+); MRSA; 2 VISA; 3 VRSA (MIC: 2–4 µg/mL) | Itching, pain and burning effects | Treatment of diabetic foot ulcers; topical | [23] |
| Mersacidin | Inhibition of cell wall | Gram (+), MRSA (MIC: 2–16 µg/mL); Clostridium spp. (MIC: 1–16 µg/mL) | Safety reported | Treatment of hospital-acquired infections | [54] |
| Planosporicin (Bacteriocin) | Inhibition of cell wall | Gram (+), (−); Planomonospora sp., MDR strains; S. aureus (MIC: 2–>128 μg/mL); S. epidermidis; E. faecalis (MIC: 32 μg/mL) | Not reported | Hospital-acquired infections | [55,56] |
| Plectasin (NZ2114) | Inhibition of cell wall synthesis | Gram (+), (−); MRSA (MIC: 16–32 μg/mL); P. aeruginosa (MIC >128 μg/mL) | Not reported | Pneumococcal peritonitis and pneumonia infections | [57,58] |
| Clinical Phase | |||||
| CZEN-002/(CKPV)2 (phase IIb) | Immunomodulation | C. albicans | Not reported | Vulvo-vaginal candidiasis; topical | [59] |
| D2A21 (phase III) | Membrane disruption | Gram (+), (−); fungi; Gram (+), (−); S. aureus; E. coli; P. aeruginosa (MIC: 4 µg/mL) | No side effects reported | Burn wound infections; topical | [60] |
| DPK-060 (phase II) | Membrane disruption and immunomodulation | Gram (+), (−); fungi; S. aureus (MIC: 4.6 µg/mL) | No side effects reported | Acute external otitis; ear drops | [23] |
| EA-230 (phase IIb) | Immunomodulation | Gram (−) | Safety reported | Sepsis and renal failure protection; IV | [61] |
| Histatin (phase I) | Membrane disruption | Gram (−); C. albicans (MIC: 4–16 μg/mL) | No side effects reported | Treatment of P. aeruginosa infections and oral candidiasis | [62,63] |
| hLF1-11 (phase I/II) | Membrane disruption | Gram(+), (−); fungi; Staphylococcus spp. (including MRSA), Streptococcus mitis (MIC: 1.6–6.3 μg/mL); A. baumannii, Pseudomonas spp., Klebsiella spp., E. coli (MIC: 6.3–12.5 μg/mL); Candida spp. (MIC >12.5 μg/mL) | Little discomfort at the injection site | LPS-related fungal infections; IV | [64,65] |
| IDR-1 (phase I) | Immunomodulation | MRSA; VRSA | No side effects reported | Infection prevention in immunocompromised patients | [62] |
| IMX942/SGX942 (IDR-1 derivative; phase III) | Immunomodulation | Gram (+), (−) | Safety reported | Treatment of nosocomial infections, neutropenia | [62] |
| LL-37 (phase IIb) | Barrel-stave mechanism of membrane disruption; inhibit 5 LPS binding | Bacteria, fungi and viral pathogens; P. aeruginosa (MIC: 64 μg/mL) | Cytotoxic | Diabetic foot ulcers; chronic middle ear infection | [25,66] |
| LTX-109 (Lytixar, phase II) | Membrane disruption and cell lysis | Gram (+); MRSA; VISA; VRSA (MIC: 2–4 µg/mL) | Itching, pain and burning effects | Treatment of nasal MRSA infections; nasal and topical | [67,68] |
| Mel4 (phase II/III) | Membrane disruption | Gram (+), (−); P. aeruginosa (MIC: 62.5–250 μg/mL) | No cytotoxicity and no resistance reported; no staining of human cornea | Contact lenses | [69,70] |
| Melimine (phase I/II) | Membrane disruption | Gram (+), (−); P. aeruginosa (MIC: 250–500 μg/mL) | No cytotoxicity and no resistance reported; staining of human cornea | Contact lenses | [69,70] |
| MX-226 (Omiganan®; phase III) | Cell disruption | Gram (+), (−); MRSA (MIC: 2–8 μg/mL); E. coli (MIC: 8–16 μg/mL); C. albicans (MIC: 64 μg/mL) | Not reported | Prevention of device-related infections; topical | [71] |
| Novexatin (NP213; phase IIb) | Membrane disruption | Fungi | Not reported | Treatment of dermatophyte fungal infections | [57] |
| Opebacan (rBPI21, neuprex; phase II) | Membrane disruption | Gram (+), (−) | Fever reported | Meningococcal, wound, and burn infections; IV | [15] |
| OP-145 (LL-37 derived; phase II) | Membrane disruption | Gram (+) | Lytic to human cells at high concentrations | Treatment of chronic bacterial middle ear infection; ear drops | [72] |
| PAC113 (P113; histatin 5 analog; phase IIb) | Membrane disruption and immunomodulation | Gram (+), (−); Candida spp.; A. baumannii (MIC: 38 μM); P. aeruginosa (MIC: 47 μM); E. cloacae (MIC: 90 μM) | No cytotoxicity reported | Oral candidiasis in HIV patients and prevention of bacterial periodontal disease; topical | [23,73] |
| p2TA (AB103; phase III) | Immunomodulation | Gram (−) | No adverse effects reported | Necrotizing soft tissue infections; IV | [74] |
| Ramoplanin (NTI-851; phase II) | Membrane disruption and cell wall synthesis | Gram (+); C. difficile; 4 MSSA (MIC: 2–4 μg/mL) | Low local tolerability when injected IV | Treatment of C. difficile-associated infections; oral | [75,76] |
| FDA Approved | |||||
| Anidulafungin (Eraxis™) in 2006 | Inhibition of (1,3)-β-d-glucan synthase | Fungi; C. albicans and K. crusei (MIC: 0.06 μg/mL) | Hypersensitivity; hepatic effects | Treatment of Candida infections, especially esophageal candidiasis; IV infusion | [77,78] |
| Caspofungin (Cancidas) in 2001 | Inhibition of β (1,3)-d-glucan production | Fungi; C. albicans; (MIC: 0.06 μg/mL); K. crusei (MIC: 0.25 μg/mL) | Hypersensitivity; hepatic effects | Treatment of esophageal candidiasis; IV | [79] |
| Dalbavancin (Dalvance™) in 2014 | Inhibition of bacterial cell wall synthesis | Gram (+); S. aureus (MIC: ≤0.008–0.5 μg/mL) | May cause nausea, headache, and diarrhea | Treatment of complicated skin and skin structure infections (cSSSI); IV injection | [80,81] |
| Daptomycin (Cubicin®) in 2003 | Membrane lytic | Gram (+); MRSA (MIC: 0.25–1 µg/mL); VISA (MIC: 1–8 µg/mL); VRSA (MIC: 0.12–1 µg/mL) | Not approved for pediatric patients | Treatment of cSSSI; IV injection | [68,82] |
| Gramicidin (Neosporin®) in 1955 | Pore-forming; aggregation; membrane disruption | Gram (+), (−); E. faecalis (MIC: 8–16 μg/mL) | Hemolytic activity | Treatment of bacterial conjunctivitis; ointment | [44,83] |
| Polymyxins (Polymyxin E = colistin) in 1964 | Membrane disruption | Gram (−); P. aeruginosa (MIC: 8 μg/mL); E. coli (0.5 μg/mL) | Used only as “last-resort” due to neuro- and nephrotoxic effects and neuromuscular blockage | Skin burns, for washing of wounds after operations, in superficial eye infections, and to protect minor wounds against infections; cream, ear and eye drops | [84,85] |
| Oritavancin (Orbactiv®) in 2014 | Inhibition of bacterial cell wall synthesis and disruption of bacterial membrane 1 | Gram (+); MRSA and MSSA (MIC: ≤0.008–0.25 μg/mL) | Long-term treatment is ambiguous | Treatment of (cSSSI); IV | [86,87] |
| Telavancin (Vibativ™ and Vibativ®) in 2009 | Inhibition of bacterial cell wall synthesis and disruption of bacterial membrane 1 | Gram (+); VRSA (MIC: 2–4 μg/mL); VISA (MIC: 0.25–1 μg/mL) | May induce acute kidney injury | Treatment of cSSSI; IV | [86,88] |
| Vancomycin (Vancocin® HCl) in 2016 | Inhibition of bacterial cell wall synthesis | Gram (+); Candida spp. (MIC: >256 µg/mL); MRSA (MIC: 0.5–2 µg/mL); VISA (MIC: 4–8 µg/mL); VRSA (MIC: 32–>64 µg/mL) | May cause nephrotoxicity | Treatment of severe MRSA infections; IV and oral | [68,82] |
| Advantages | Disadvantages |
|---|---|
| Several AMPs show broad and simultaneous activities against bacteria, fungi, viruses, and in case of infection with multiple microorganisms, one AMP could be efficient to overcome this issue. | AMPs are susceptible to proteolytic degradation, which leads to the loss of biological activity. |
| Natural AMPs are already found in high doses at the site of infection. | Some AMPs can be toxic to mammal cells at high concentrations. |
| Some AMPs have wound-healing and angiogenesis promotion properties, which are essential in case of hard-to-heal and infected wounds, such as diabetes and foot ulcer. | AMPs can induce hypersensitivity reactions after application. |
| AMPs show as well anti-inflammatory properties by modulating immune cytokines, which are responsible for the inflammatory response. | AMPs can be influenced by pH variation and at low concentration of salt can be destabilized, leading to loss of activity. |
| AMPs exhibit therapeutic antimicrobial activity at extremely low concentrations in the microscale and sometimes nanoscale range. | AMP’s production and purification costs are high. |
| Resistance to AMPs is very low and only in a limited number of AMPs. | |
| AMPs showed to be time-efficient; some AMPs can act within a few seconds to a few minutes. | |
| AMPs inhibit biofilm formation, which is especially important to prevent bacterial growth on medical devices. | |
| AMPs show synergism when chemically coupled to polymers, encapsulated into different delivery systems or simultaneously applied with antimicrobial agents. | |
| AMPs can act synergistically with antibiotics. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Patrulea, V.; Borchard, G.; Jordan, O. An Update on Antimicrobial Peptides (AMPs) and Their Delivery Strategies for Wound Infections. Pharmaceutics 2020, 12, 840. https://doi.org/10.3390/pharmaceutics12090840
Patrulea V, Borchard G, Jordan O. An Update on Antimicrobial Peptides (AMPs) and Their Delivery Strategies for Wound Infections. Pharmaceutics. 2020; 12(9):840. https://doi.org/10.3390/pharmaceutics12090840
Chicago/Turabian StylePatrulea, Viorica, Gerrit Borchard, and Olivier Jordan. 2020. "An Update on Antimicrobial Peptides (AMPs) and Their Delivery Strategies for Wound Infections" Pharmaceutics 12, no. 9: 840. https://doi.org/10.3390/pharmaceutics12090840