Spontaneous In Situ Formation of Liposomes from Inert Porous Microparticles for Oral Drug Delivery

 , , , and
, , , and
Abstract
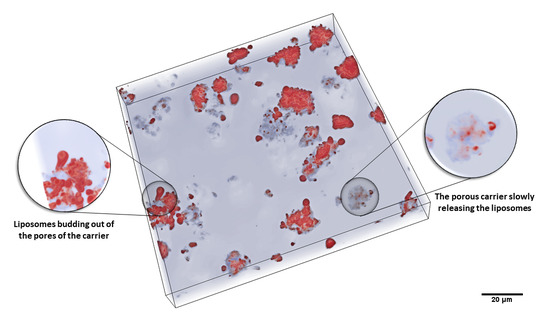
:
1. Introduction
2. Materials and Methods
2.1. Loading of Nifedipine and DMPC in FCC
2.2. Quantification of the Drug Content in Formulations
2.3. Screening for an Absence of External Crystallization
2.4. Liposomal Size Measurement
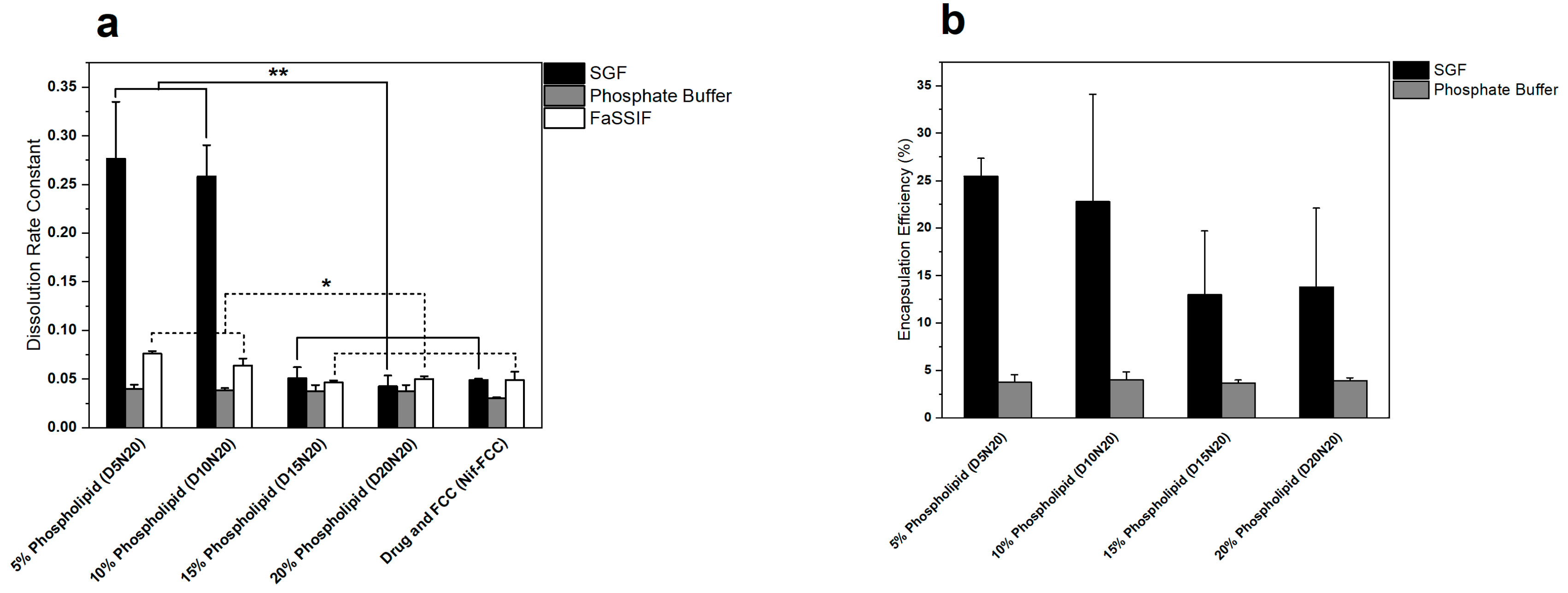
2.5. Quantification of Nifedipine eEncapsulation in Liposomes
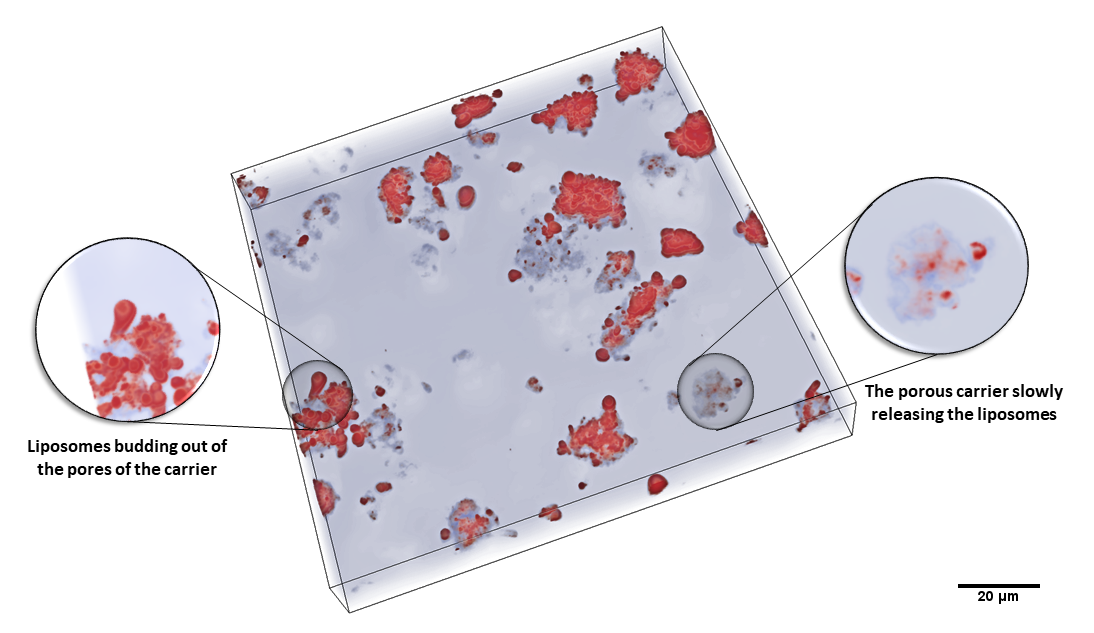
2.6. Liposome Imaging by Cryo-Electron Microscopy (Cryo-EM) and Fluorescent Microscopy
2.7. In Vitro Dissolution
2.8. Differential Scanning Calorimetry
2.9. X-ray Powder Diffraction
2.10. Surface Area Measurements
2.11. Water Sorption Measurements
3. Results
3.1. Characteristics of the Liposomal System after Hydration of the Powder Formulations
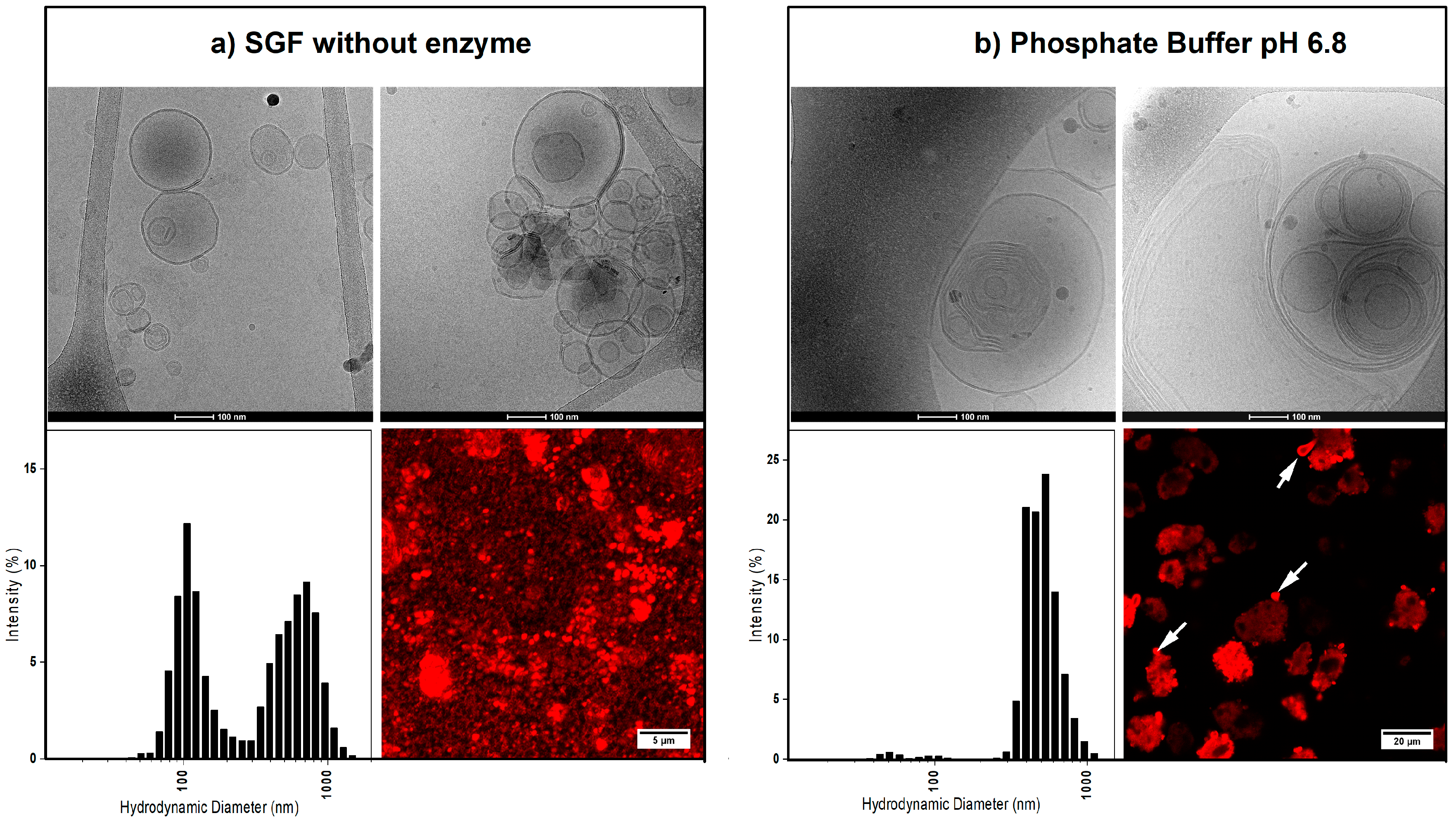
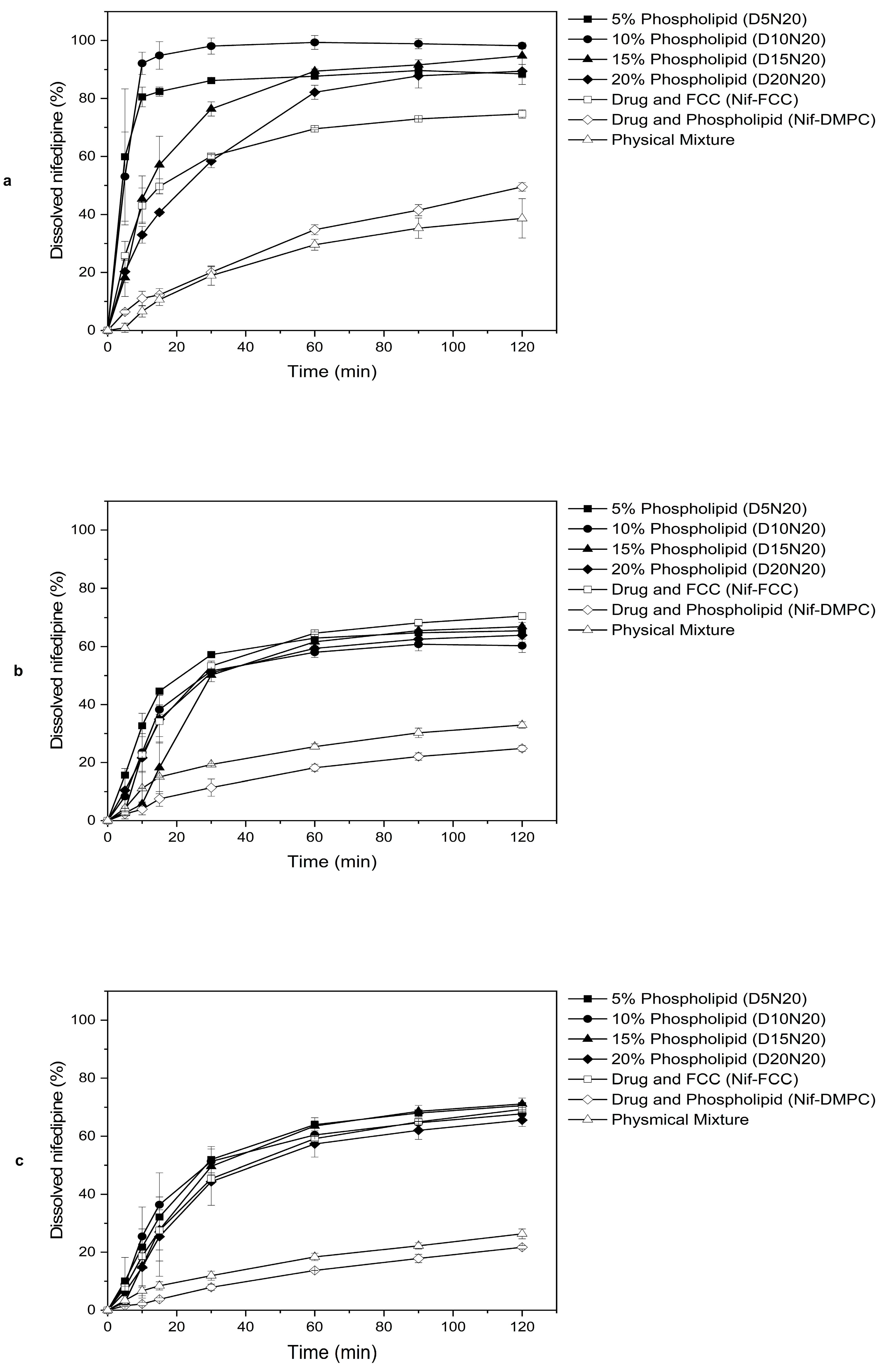
3.2. In Vitro Dissolution
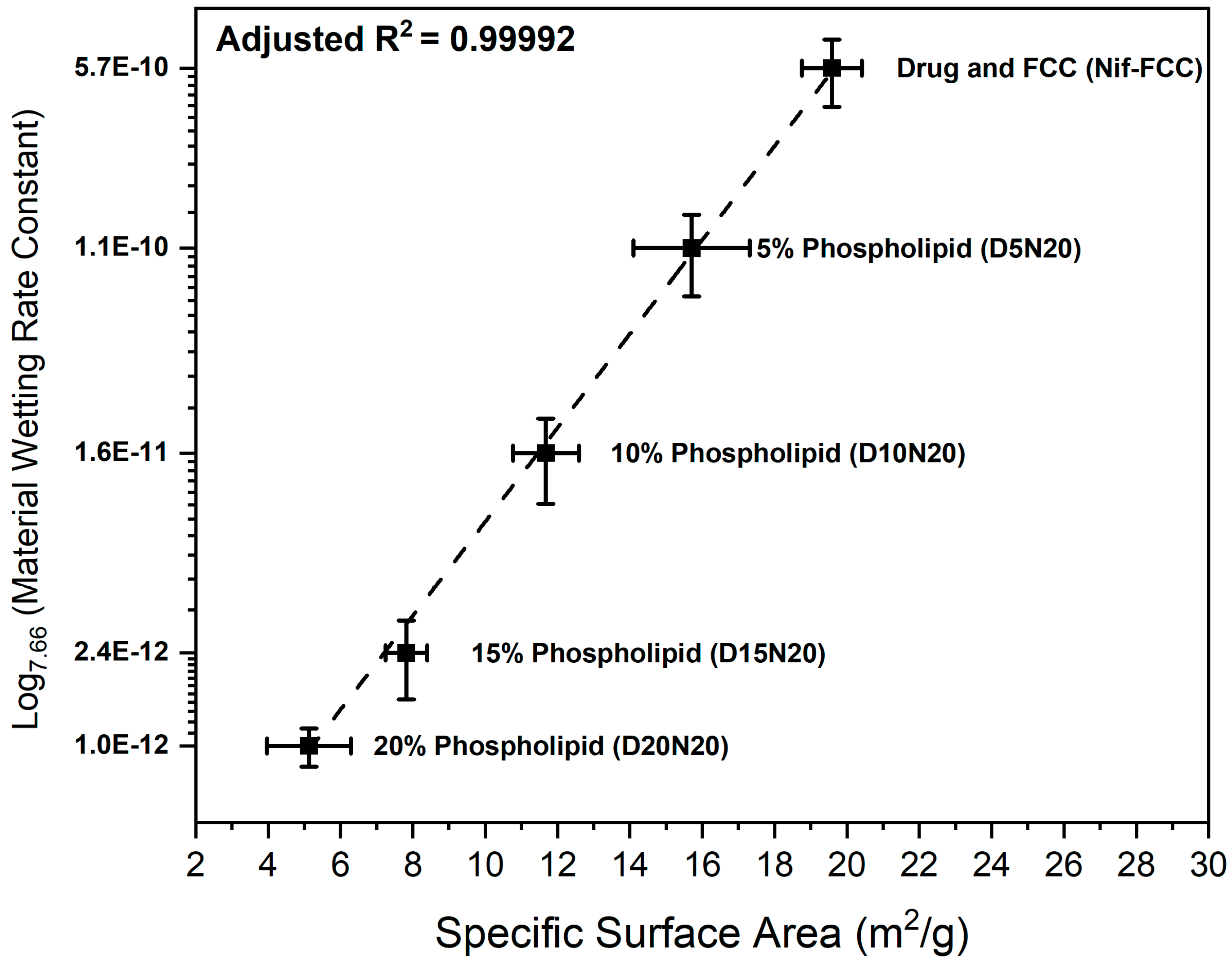
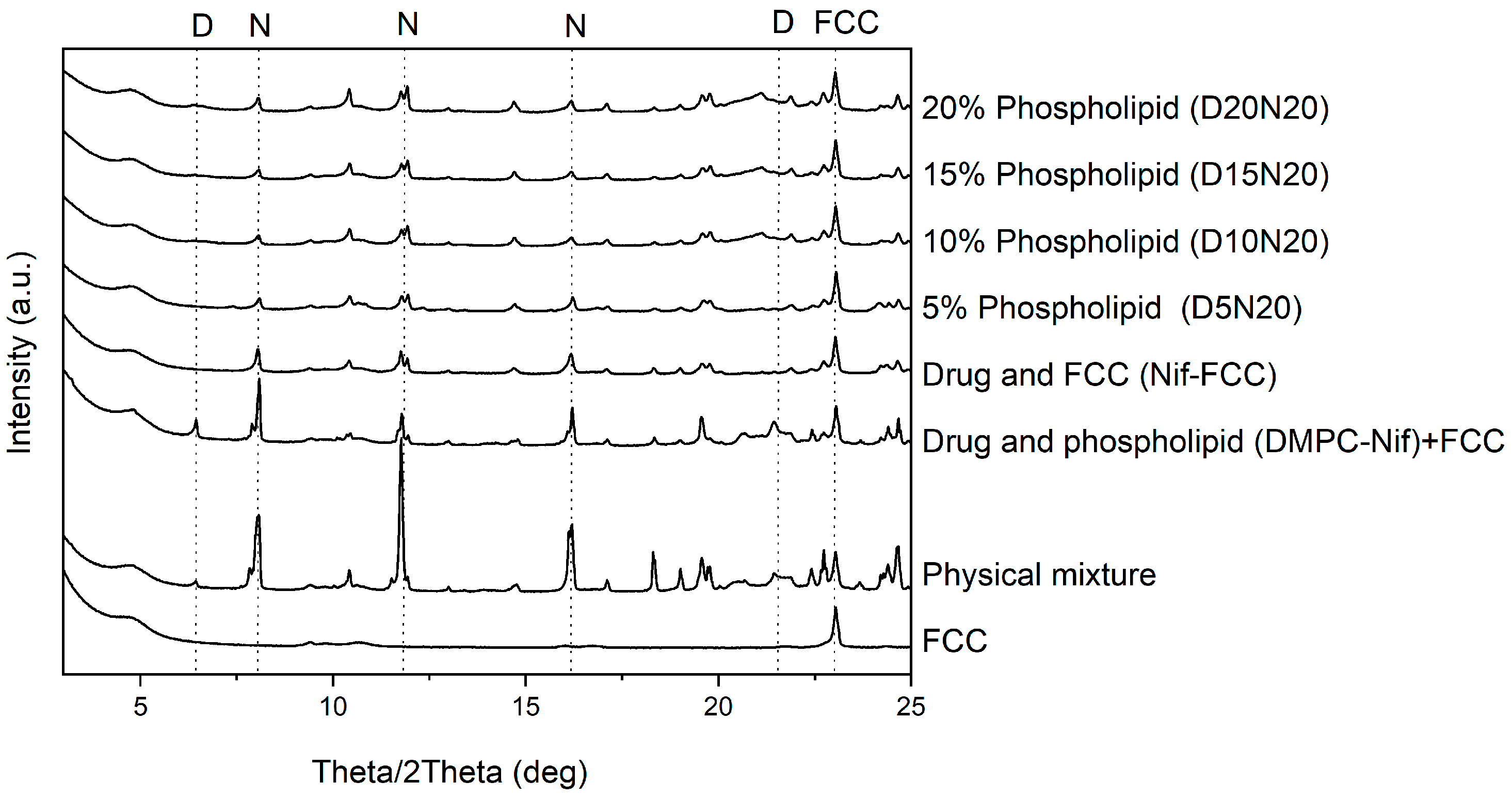
3.3. Characterization of the Dry Powder Formulations
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dapergolas, G.; Gregoriadis, G. Hypoglycqmic effect of liposome-entrapped insulin administered intragastrically into rats. Lancet 1976, 308, 824–827. [Google Scholar] [CrossRef]
- Patel, H.; Ryman, B.E. Oral administration of insulin by encapsulation within liposomes. FEBS Lett. 1976, 62, 60–63. [Google Scholar] [CrossRef] [Green Version]
- Tang, W.-L.; Chen, W.C.; Diako, C.; Ross, C.; Li, S.-D.; Tang, W.-H. Development of a rapidly dissolvable oral pediatric formulation for mefloquine using liposomes. Mol. Pharm. 2017, 14, 1969–1979. [Google Scholar] [CrossRef]
- Uhl, P.; Pantze, S.; Storck, P.; Parmentier, J.; Witzigmann, D.; Hofhaus, G.; Huwyler, J.; Mier, W.; Fricker, G. Oral delivery of vancomycin by tetraether lipid liposomes. Eur. J. Pharm. Sci. 2017, 108, 111–118. [Google Scholar] [CrossRef]
- Song, K.-H.; Chung, S.-J.; Shim, C.-K. Enhanced intestinal absorption of salmon calcitonin (sCT) from proliposomes containing bile salts. J. Control. Release 2005, 106, 298–308. [Google Scholar] [CrossRef]
- Lee, M.-K. Liposomes for enhanced bioavailability of water-insoluble drugs: In Vivo evidence and recent approaches. Pharmaceutics 2020, 12, 264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zidan, A.S.; Spinks, C.B.; Habib, M.J.; Khan, M.A. Formulation and transport properties of tenofovir loaded liposomes through Caco-2 cell model. J. Liposome Res. 2013, 23, 318–326. [Google Scholar] [CrossRef] [PubMed]
- Uhl, P.; Helm, F.; Hofhaus, G.; Brings, S.; Kaufman, C.; Leotta, K.; Urban, S.; Haberkorn, U.; Mier, W.; Fricker, G. A liposomal formulation for the oral application of the investigational hepatitis B drug Myrcludex B. Eur. J. Pharm. Biopharm. 2016, 103, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, A.; Kawada, J. Effects of oral administration of positively charged insulin liposomes on alloxan diabetic rats: Preliminary study. Endocrinol. Jpn. 1979, 26, 337–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H. Polyethylene glycol-coated liposomes for oral delivery of recombinant human epidermal growth factor. Int. J. Pharm. 2003, 258, 11–19. [Google Scholar] [CrossRef]
- Liu, W.; Ye, A.; Liu, W.; Liu, C.; Han, J.; Singh, H. Behaviour of liposomes loaded with bovine serum albumin during in vitro digestion. Food Chem. 2015, 175, 16–24. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Lu, Y.; Qi, J.; Zhu, Q.; Chen, Z.; Wu, W. Adapting liposomes for oral drug delivery. Acta Pharm. Sin. B 2018, 9, 36–48. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Lu, Y.; Qi, J. Oral delivery of liposomes. Ther. Deliv. 2015, 6, 1239–1241. [Google Scholar] [CrossRef] [PubMed]
- Van Hoogevest, P. Review—An update on the use of oral phospholipid excipients. Eur. J. Pharm. Sci. 2017, 108, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Franze, S.; Selmin, F.; Samaritani, E.; Minghetti, P.; Cilurzo, F. Lyophilization of liposomal formulations: Still necessary, still challenging. Pharmaceutics 2018, 10, 139. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Han, D.; Cai, C.; Tang, X. An overview of liposome lyophilization and its future potential. J. Control. Release 2010, 142, 299–311. [Google Scholar] [CrossRef]
- Brinkmann-Trettenes, U.; Bauer-Brandl, A. Solid phospholipid nano-particles: Investigations into formulation and dissolution properties of griseofulvin. Int. J. Pharm. 2014, 467, 42–47. [Google Scholar] [CrossRef]
- Sweeney, L.G.; Wang, Z.; Loebenberg, R.; Wong, J.P.; Lange, C.F.; Finlay, W.H. Spray-freeze-dried liposomal ciprofloxacin powder for inhaled aerosol drug delivery. Int. J. Pharm. 2005, 305, 180–185. [Google Scholar] [CrossRef]
- Otake, K.; Imura, T.; Sakai, H.; Abe, M. Development of a new preparation method of liposomes using supercritical carbon dioxide. Langmuir 2001, 17, 3898–3901. [Google Scholar] [CrossRef]
- Imura, T.; Otake, K.; Hashimoto, S.; Gotoh, T.; Yuasa, M.; Yokoyama, S.; Sakai, H.; Rathman, J.F.; Abe, M. Preparation and physicochemical properties of various soybean lecithin liposomes using supercritical reverse phase evaporation method. Colloids Surf. B Biointerfaces 2003, 27, 133–140. [Google Scholar] [CrossRef]
- Misra, A.; Jinturkar, K.; Patel, D.; Lalani, J.; Chougule, M. Recent advances in liposomal dry powder formulations: Preparation and evaluation. Expert Opin. Drug Deliv. 2009, 6, 71–89. [Google Scholar] [CrossRef] [PubMed]
- Ingvarsson, P.T.; Yang, M.; Nielsen, H.M.; Rantanen, J.; Foged, C. Stabilization of liposomes during drying. Expert Opin. Drug Deliv. 2011, 8, 375–388. [Google Scholar] [CrossRef] [PubMed]
- Jannin, V.; Musakhanian, J.; Marchaud, D. Approaches for the development of solid and semi-solid lipid-based formulations. Adv. Drug Deliv. Rev. 2008, 60, 734–746. [Google Scholar] [CrossRef] [PubMed]
- Payne, N.I.; Timmins, P.; Ambrose, C.V.; Ward, M.D.; Ridgway, F. Proliposomes: A Novel solution to an old problem. J. Pharm. Sci. 1986, 75, 325–329. [Google Scholar] [CrossRef]
- Jia, J.; Zhang, K.; Zhou, X.; Ma, J.; Liu, X.; Xiang, A.; Ge, F. Berberine-loaded solid proliposomes prepared using solution enhanced dispersion by supercritical CO2: Sustained release and bioavailability enhancement. J. Drug Deliv. Sci. Technol. 2019, 51, 356–363. [Google Scholar] [CrossRef]
- Khan, I.; Yousaf, S.; Subramanian, S.; Alhnan, M.A.; Ahmed, W.; Elhissi, A. Proliposome tablets manufactured using a slurry-driven lipid-enriched powders: Development, characterization and stability evaluation. Int. J. Pharm. 2018, 538, 250–262. [Google Scholar] [CrossRef]
- Weng, W.; Wang, Q.; Wei, C.; Man, N.; Zhang, K.; Wei, Q.; Adu-Frimpong, M.; Toreniyazov, E.; Ji, H.; Yu, J.-N.; et al. Preparation, characterization, pharmacokinetics and anti-hyperuricemia activity studies of myricitrin-loaded proliposomes. Int. J. Pharm. 2019, 572, 118735. [Google Scholar] [CrossRef]
- Fong, S.Y.K.; Brandl, M.; Bauer-Brandl, A. Phospholipid-based solid drug formulations for oral bioavailability enhancement: A meta-analysis. Eur. J. Pharm. Sci. 2015, 80, 89–110. [Google Scholar] [CrossRef]
- Vanić, Ž.; Planinšek, O.; Škalko-Basnet, N.; Tho, I. Tablets of pre-liposomes govern In Situ formation of liposomes: Concept and potential of the novel drug delivery system. Eur. J. Pharm. Biopharm. 2014, 88, 443–454. [Google Scholar] [CrossRef]
- Chu, C.; Tong, S.-S.; Xu, Y.; Wang, L.; Fu, M.; Ge, Y.-R.; Yu, J.-N.; Xu, X.-M. Proliposomes for oral delivery of dehydrosilymarin: Preparation and evaluation In Vitro and In Vivo. Acta Pharmacol. Sin. 2011, 32, 973–980. [Google Scholar] [CrossRef] [Green Version]
- Bobbala, S.K.R.; Veerareddy, P.R. Formulation, evaluation, and pharmacokinetics of isradipine proliposomes for oral delivery. J. Liposome Res. 2012, 22, 285–294. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; He, L.; Nie, S.; Guan, J.; Zhang, X.; Yang, X.; Pan, W. Optimized preparation of vinpocetine proliposomes by a novel method and In Vivo evaluation of its pharmacokinetics in New Zealand rabbits. J. Control. Release 2009, 140, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Janga, K.Y.; Jukanti, R.; Velpula, A.; Sunkavalli, S.; Bandari, S.; Kandadi, P.; Veerareddy, P.R. Bioavailability enhancement of zaleplon via proliposomes: Role of surface charge. Eur. J. Pharm. Biopharm. 2012, 80, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Velpula, A.; Jukanti, R.; Janga, K.Y.; Sunkavalli, S.; Bandari, S.; Kandadi, P.; Veerareddy, P.R. Proliposome powders for enhanced intestinal absorption and bioavailability of raloxifene hydrochloride: Effect of surface charge. Drug Dev. Ind. Pharm. 2012, 39, 1895–1906. [Google Scholar] [CrossRef] [PubMed]
- Roth, R.; Schoelkopf, J.; Huwyler, J.; Puchkov, M. Functionalized calcium carbonate microparticles for the delivery of proteins. Eur. J. Pharm. Biopharm. 2018, 122, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Ridgway, C.; Gane, P.A.; Schoelkopf, J. Modified calcium carbonate coatings with rapid absorption and extensive liquid uptake capacity. Colloids Surf. A Physicochem. Eng. Asp. 2004, 236, 91–102. [Google Scholar] [CrossRef]
- Farzan, M.; Roth, R.; Québatte, G.; Schoelkopf, J.; Huwyler, J.; Puchkov, M. Loading of porous functionalized calcium carbonate microparticles: Distribution analysis with focused ion beam electron microscopy and mercury porosimetry. Pharmaceutics 2019, 11, 32. [Google Scholar] [CrossRef] [Green Version]
- Preisig, D.; Haid, D.; Varum, F.J.; Bravo, R.; Alles, R.; Huwyler, J.; Puchkov, M. Drug loading into porous calcium carbonate microparticles by solvent evaporation. Eur. J. Pharm. Biopharm. 2014, 87, 548–558. [Google Scholar] [CrossRef]
- Preisig, D.; Roth, R.; Tognola, S.; Varum, F.J.; Bravo, R.; Cetinkaya, Y.; Huwyler, J.; Puchkov, M. Mucoadhesive microparticles for local treatment of gastrointestinal diseases. Eur. J. Pharm. Biopharm. 2016, 105, 156–165. [Google Scholar] [CrossRef]
- Stirnimann, T.; Atria, S.; Schoelkopf, J.; Gane, P.; Alles, R.; Huwyler, J.; Puchkov, M. Compaction of functionalized calcium carbonate, a porous and crystalline microparticulate material with a lamellar surface. Int. J. Pharm. 2014, 466, 266–275. [Google Scholar] [CrossRef]
- Stirnimann, T.; Di Maiuta, N.; Gerard, D.E.; Alles, R.; Huwyler, J.; Puchkov, M. Functionalized calcium carbonate as a novel pharmaceutical excipient for the preparation of orally dispersible tablets. Pharm. Res. 2013, 30, 1915–1925. [Google Scholar] [CrossRef] [PubMed]
- Wagner-Hattler, L.; Wyss, K.; Schoelkopf, J.; Huwyler, J.; Puchkov, M. In Vitro characterization and mouthfeel study of functionalized calcium carbonate in orally disintegrating tablets. Int. J. Pharm. 2017, 534, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Eberle, V.A.; Schoelkopf, J.; Gane, P.A.; Alles, R.; Huwyler, J.; Puchkov, M. Floating gastroretentive drug delivery systems: Comparison of experimental and simulated dissolution profiles and floatation behavior. Eur. J. Pharm. Sci. 2014, 58, 34–43. [Google Scholar] [CrossRef] [PubMed]
- The United States Pharmacopeial Convention. The United States Pharmacopeia 2018; United States Pharmacopeial Convention: Rockville, MD, USA, 2017. [Google Scholar]
- FaSSIF/FeSSIF/FaSSGF Biorelevant Dissolution Media. Biorelevant. Available online: https://biorelevant.com/fassif-fessif-fassgf/buy/ (accessed on 22 June 2020).
- Wolfram Language Documentation, Numerical Operations on Data. Available online: https://reference.wolfram.com/language/tutorial/NumericalOperationsOnData.html#19037 (accessed on 22 April 2020).
- Khan, K.A.; Rhodes, C.T. Effect of compaction pressure on the dissolution efficiency of some direct compression systems. Pharm. Acta Helv. 1972, 47, 594–607. [Google Scholar] [PubMed]
- Ting, J.; Navale, T.S.; Bates, F.S.; Reineke, T.M. Design of tunable multicomponent polymers as modular vehicles to solubilize highly lipophilic drugs. Macromolecules 2014, 47, 6554–6565. [Google Scholar] [CrossRef]
- Vitez, I.M. Utilization of DSC for pharmaceutical crystal form quantitation. J. Therm. Anal. Calorim. 2004, 78, 33–45. [Google Scholar] [CrossRef]
- Shah, B.; Kakumanu, V.K.; Bansal, A.K. Analytical techniques for quantification of amorphous/crystalline phases in pharmaceutical solids. J. Pharm. Sci. 2006, 95, 1641–1665. [Google Scholar] [CrossRef]
- Washburn, E.W. The dynamics of capillary flow. Phys. Rev. 1921, 17, 273–283. [Google Scholar] [CrossRef]
- Lucas, R. Ueber das zeitgesetz des kapillaren aufstiegs von flüssigkeiten. Colloid Polym. Sci. 1918, 23, 15–22. [Google Scholar] [CrossRef]
- Krüss Tensiometer K100 Instruction Manual; Krüss GmbH: Hamburg, Germany, 2005.
- Stuart, M.C.A.; Boekema, E.J. Two distinct mechanisms of vesicle-to-micelle and micelle-to-vesicle transition are mediated by the packing parameter of phospholipid–detergent systems. Biochim. Biophys. Acta (BBA) Biomembr. 2007, 1768, 2681–2689. [Google Scholar] [CrossRef] [Green Version]
- Marsh, D. Handbook of Lipid Bilayers, 2nd ed.; CRC Press, Taylor & Francis Group: Boca Raton, FL, USA, 2013. [Google Scholar]
- Israelachvili, J.N. Soft and biological structures. In Intermolecular and Surface Forces, 3rd ed.; Israelachvili, J.N., Ed.; Academic Press: San Diego, CA, USA, 2011; pp. 535–576. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulation Code | DMPC (w/w%) | Nifedipine (w/w%) | FCC (w/w%) |
|---|---|---|---|
| D5N10 | 5 | 10 | 85 |
| D5N15 | 5 | 15 | 80 |
| D5N20 | 5 | 20 | 75 |
| D10N10 | 10 | 10 | 80 |
| D10N15 | 10 | 15 | 75 |
| D10N20 | 10 | 20 | 70 |
| D15N10 | 15 | 10 | 75 |
| D15N15 | 15 | 15 | 70 |
| D15N20 | 15 | 20 | 65 |
| D20N10 | 20 | 10 | 70 |
| D20N15 | 20 | 15 | 65 |
| D20N20 | 20 | 20 | 60 |
| Nif-FCC (control) | 0 | 20 | 80 |
| DMPC-Nif (control) | 50 | 50 | 0 |
| DMPC-FCC (control) | 20 | 0 | 80 |
| Physical mixture | 20 | 20 | 60 |
| Formulation Code | SGF | Phosphate Buffer | FaSSIF |
|---|---|---|---|
| D5N20 | 91.53 ± 1.33 | 79.66 ± 4.33 | 86.40 ± 1.33 |
| D10N20 | 89.74 ± 2.96 | 79.35 ± 1.31 | 84.88 ± 1.02 |
| D15N20 | 84.23 ± 1.06 | 76.26 ± 2.19 | 78.09 ± 1.10 |
| D20N20 | 77.45 ± 1.37 | 74.95 ± 6.76 | 80.34 ± 0.005 |
| Nif-FCC | 84.19 ± 1.13 | 73.66 ± 1.95 | 80.19 ± 2.01 |
| Nif-DMPC | 60.12 ± 9.79 | 57.23 ± 1.42 | 64.80 ± 3.25 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Farzan, M.; Québatte, G.; Strittmatter, K.; Hilty, F.M.; Schoelkopf, J.; Huwyler, J.; Puchkov, M. Spontaneous In Situ Formation of Liposomes from Inert Porous Microparticles for Oral Drug Delivery. Pharmaceutics 2020, 12, 777. https://doi.org/10.3390/pharmaceutics12080777
Farzan M, Québatte G, Strittmatter K, Hilty FM, Schoelkopf J, Huwyler J, Puchkov M. Spontaneous In Situ Formation of Liposomes from Inert Porous Microparticles for Oral Drug Delivery. Pharmaceutics. 2020; 12(8):777. https://doi.org/10.3390/pharmaceutics12080777
Chicago/Turabian StyleFarzan, Maryam, Gabriela Québatte, Katrin Strittmatter, Florentine Marianne Hilty, Joachim Schoelkopf, Jörg Huwyler, and Maxim Puchkov. 2020. "Spontaneous In Situ Formation of Liposomes from Inert Porous Microparticles for Oral Drug Delivery" Pharmaceutics 12, no. 8: 777. https://doi.org/10.3390/pharmaceutics12080777