Light-Activated Liposomes Coated with Hyaluronic Acid as a Potential Drug Delivery System

 , , , ,
, , , ,  ,
,
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. DSPE–HA Conjugate
2.3. DSPE–HA Conjugate Analysis
2.4. Light-Activated Liposomes
2.5. Size and Zeta Potential Analysis in Buffer
2.6. Size Analysis by Flow Cytometry in Vitreous and Plasma Samples
2.7. Differential Scanning Calorimetry
2.8. ICG Stability
2.9. Temperature-Induced Release
2.10. Light-Activated Release
2.11. Liposome Stability in Vitreous and Plasma
2.12. Diffusion of Liposomes in Intact Porcine Vitreous
2.13. Protein Corona Structure and Composition
2.14. Statistical Analysis
3. Results
3.1. Analysis of the DSPE–HA Conjugate
3.2. Properties of the HA-Coated Liposomes
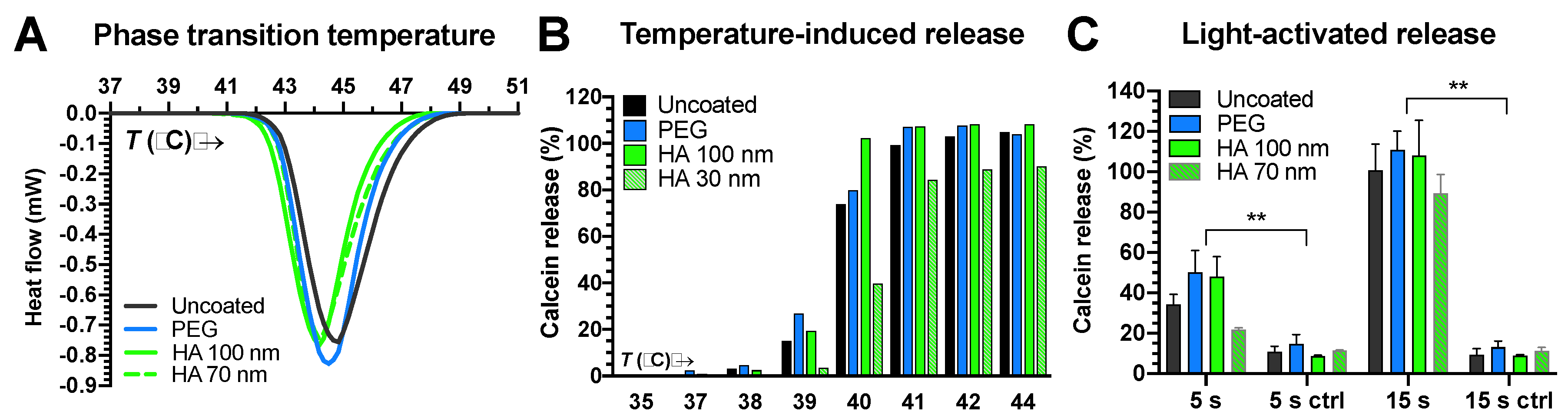
3.3. Stability Studies
3.3.1. ICG Stability and Passive Content Leakage
3.3.2. Light-Activated Content Release in Vitreous and Plasma Samples
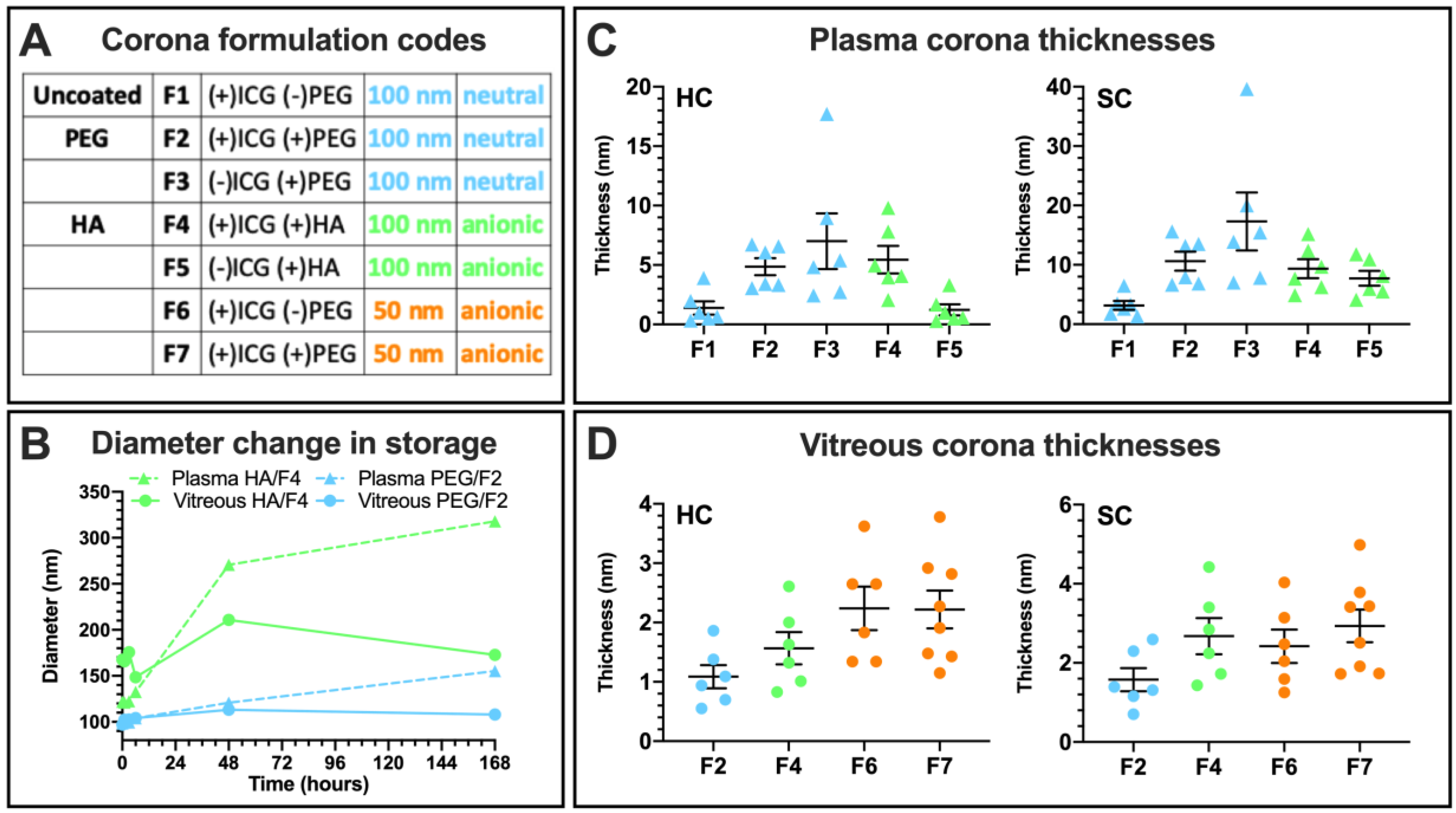
3.3.3. Liposome Size Change
3.4. Protein Corona Formation
3.4.1. Hard and Soft Corona Thickness
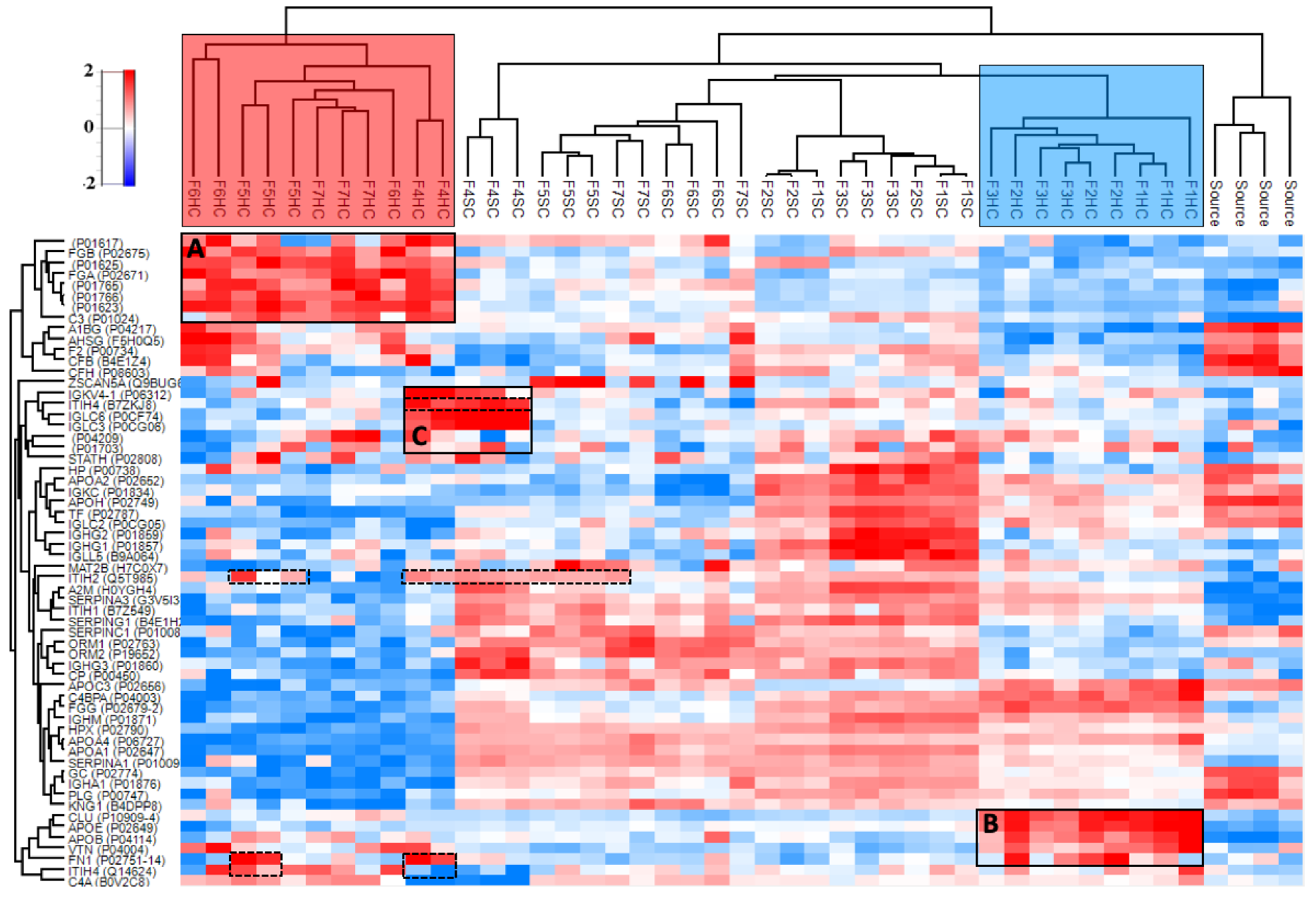
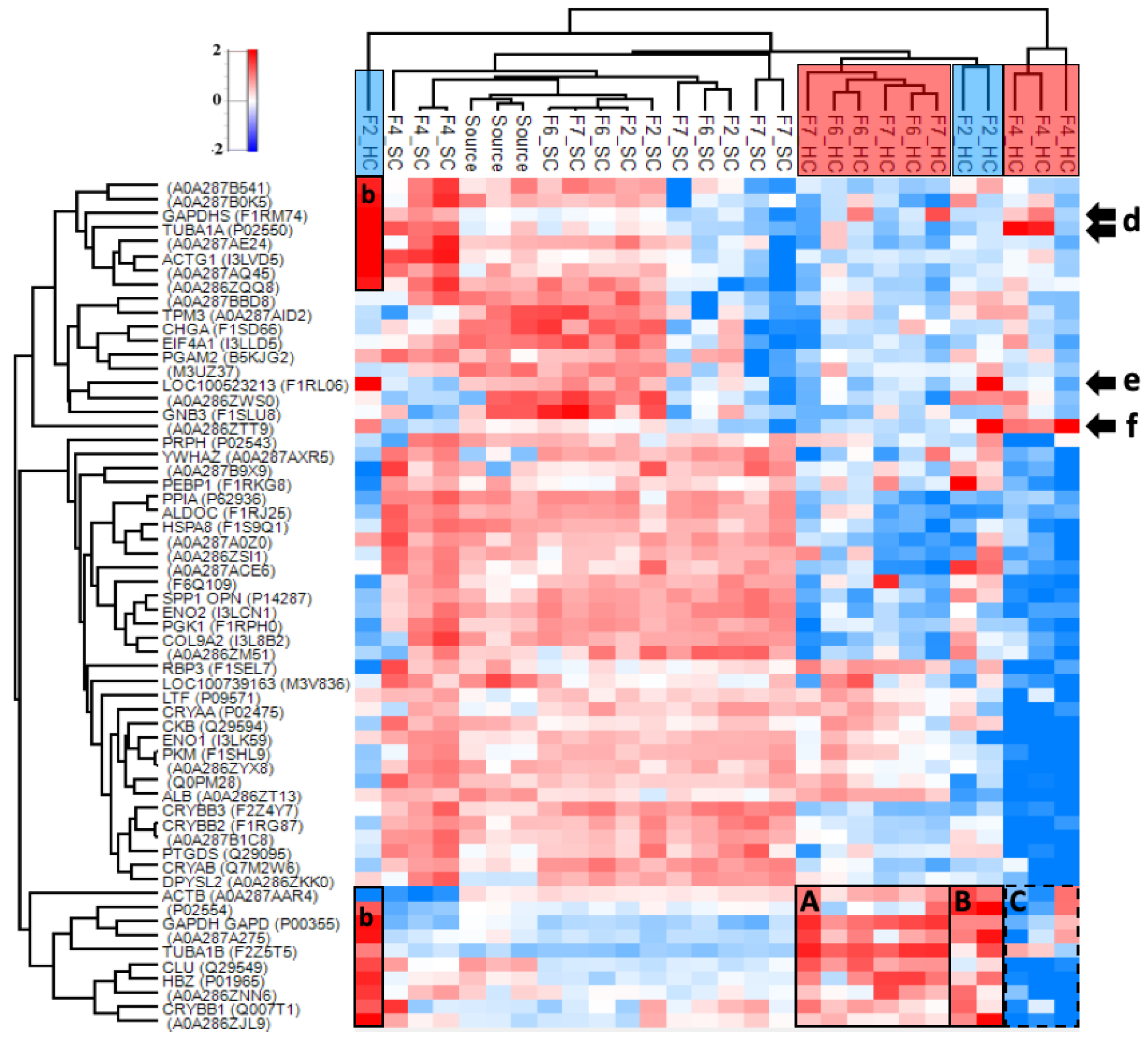
3.4.2. Hard and Soft Corona Composition
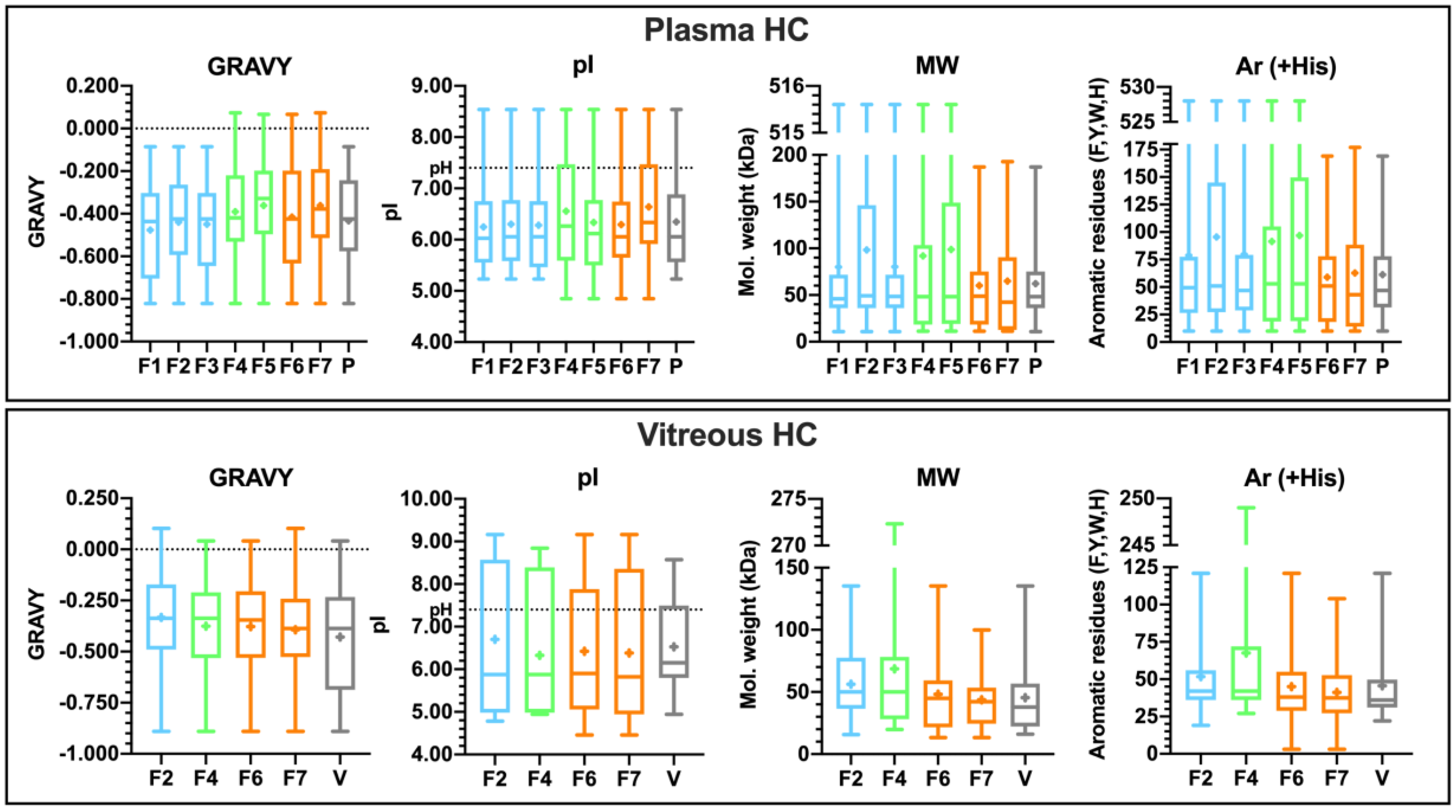
3.4.3. Properties of the Corona-Enriched Proteins
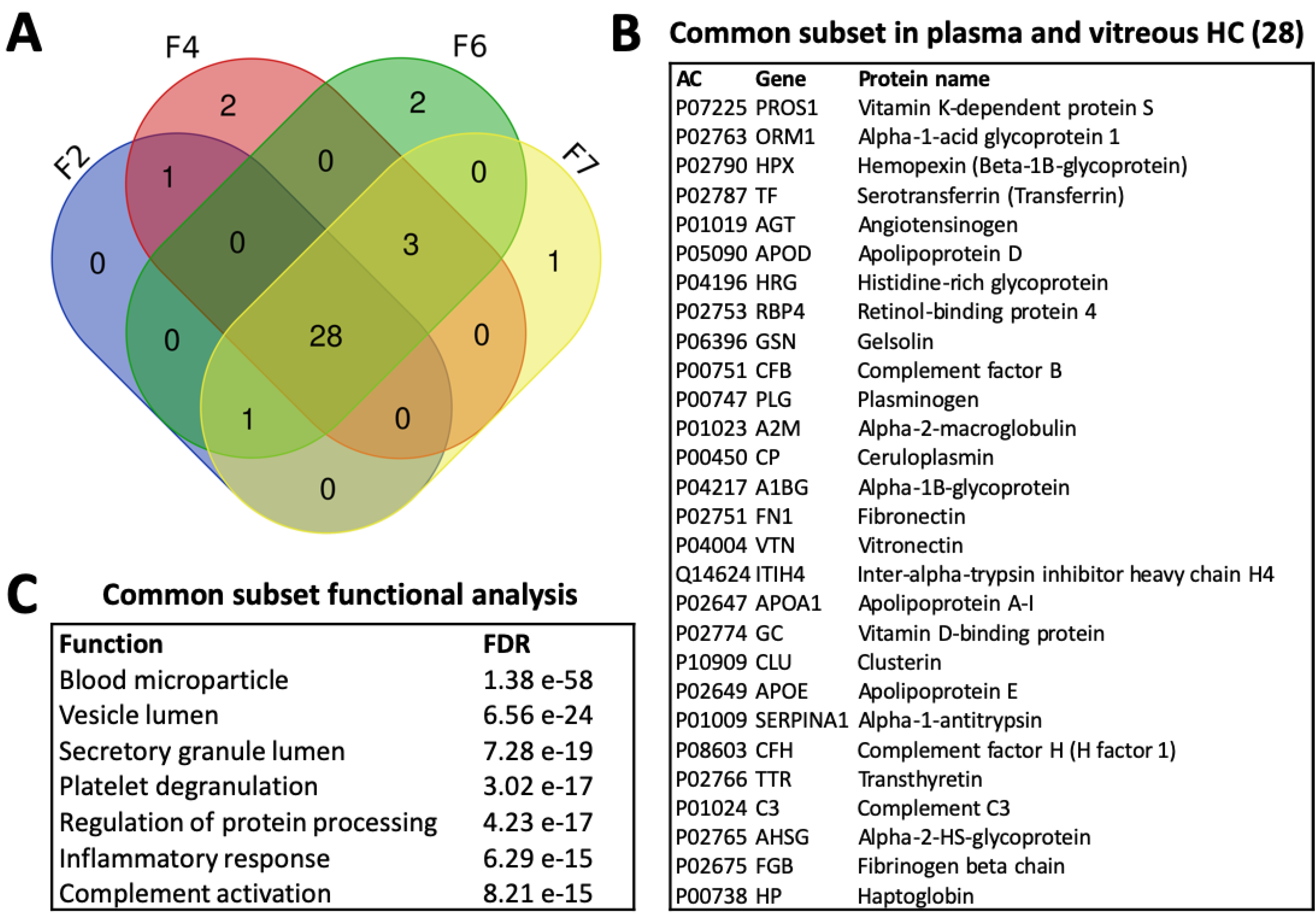
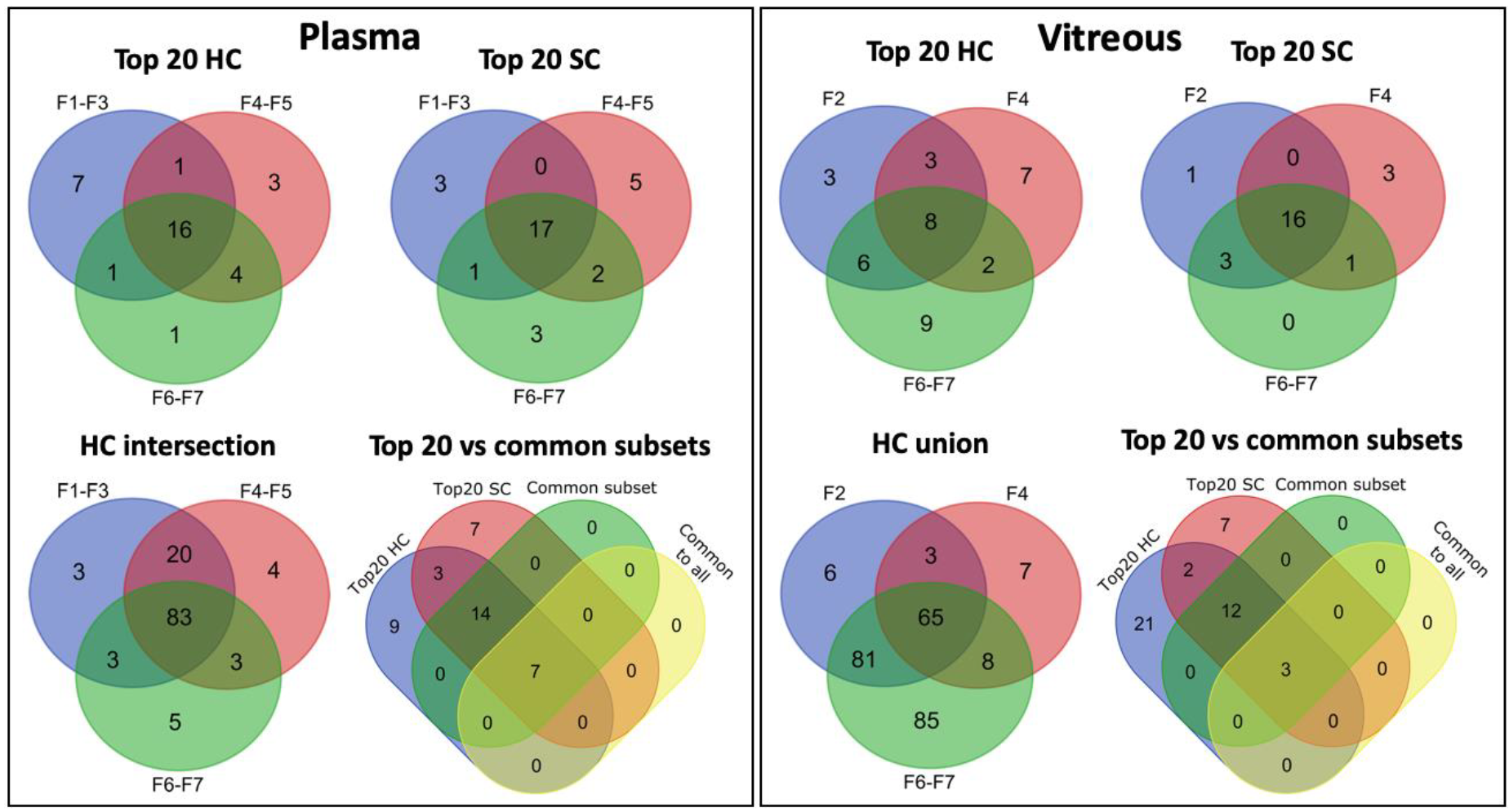
3.4.4. Common Subsets of Hard Corona Proteins in Plasma and Vitreous Samples
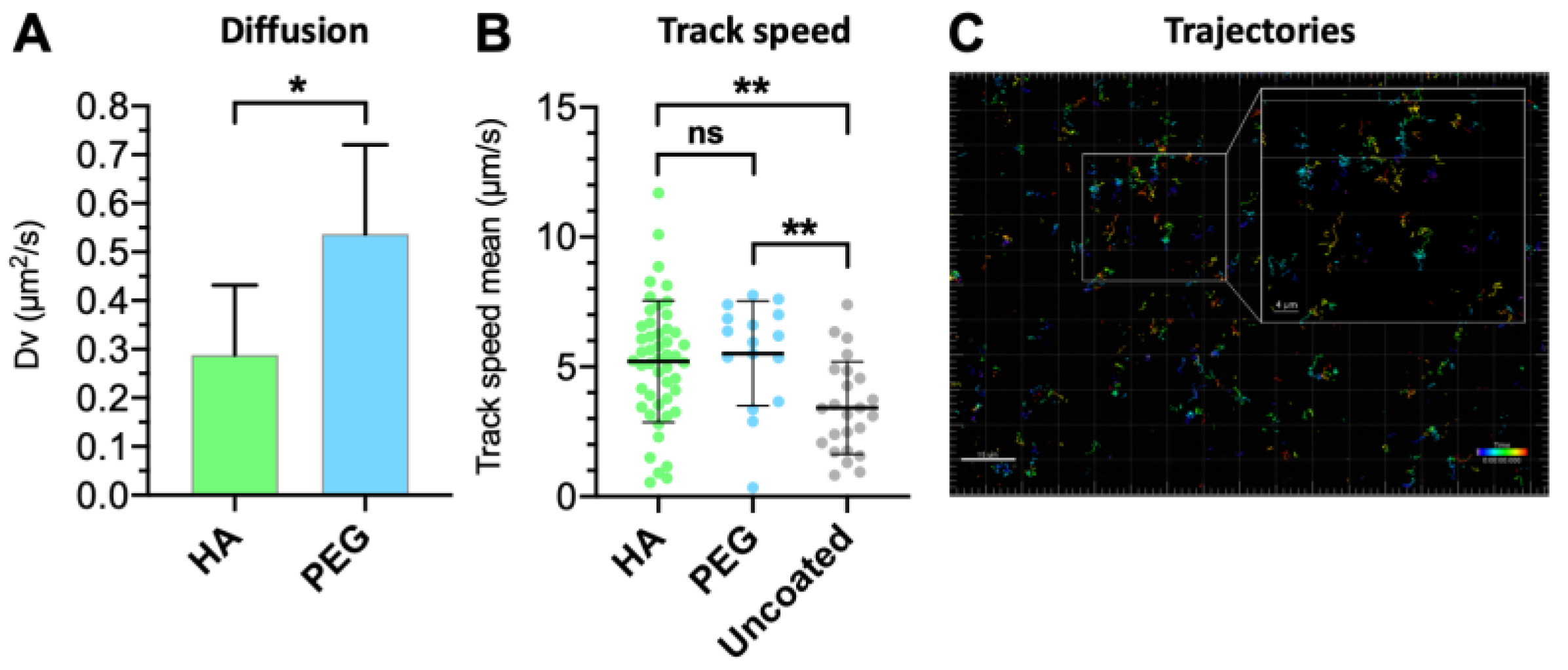
3.5. Vitreal Mobility
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Etheridge, M.L.; Campbell, S.A.; Erdman, A.G.; Haynes, C.L.; Wolf, S.M.; McCullough, J. The big picture on nanomedicine: The state of investigational and approved nanomedicine products. Nanomed. Nanotechnol. Biol. Med. 2013, 9, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Vonarbourg, A.; Passirani, C.; Saulnier, P.; Simard, P.; Leroux, J.-C.; Benoit, J.-P. Evaluation of pegylated lipid nanocapsules versus complement system activation and macrophage uptake. J. Biomed. Mater. Res. Part A 2006, 78A, 620–628. [Google Scholar] [CrossRef]
- Amoozgar, Z.; Yeo, Y. Recent advances in stealth coating of nanoparticle drug delivery systems. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2012, 4, 219–233. [Google Scholar] [CrossRef] [Green Version]
- Romberg, B.; Metselaar, J.M.; Baranyi, L.; Snel, C.J.; Bünger, R.; Hennink, W.E.; Szebeni, J.; Storm, G. Poly(amino acid)s: Promising enzymatically degradable stealth coatings for liposomes. Int. J. Pharm. 2007, 331, 186–189. [Google Scholar] [CrossRef]
- Sroda, K.; Rydlewski, J.; Langner, M.; Kozubek, A.; Grzybek, M.; Sikorski, A.F. Repeated injections of PEG-PE liposomes generate anti-PEG antibodies. Cell. Mol. Biol. Lett. 2005, 10, 37–47. [Google Scholar]
- Yang, Q.; Lai, S.K. Anti-PEG immunity: Emergence, characteristics, and unaddressed questions. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2015, 7, 655–677. [Google Scholar] [CrossRef] [Green Version]
- Knop, K.; Hoogenboom, R.; Fischer, D.; Schubert, U.S. Poly(ethylene glycol) in drug delivery: Pros and cons as well as potential alternatives. Angew. Chemie Int. Ed. 2010, 49, 6288–6308. [Google Scholar] [CrossRef] [PubMed]
- Kaneda, Y.; Tsutsumi, Y.; Yoshioka, Y.; Kamada, H.; Yamamoto, Y.; Kodaira, H.; Tsunoda, S.I.; Okamoto, T.; Mukai, Y.; Shibata, H.; et al. The use of PVP as a polymeric carrier to improve the plasma half-life of drugs. Biomaterials 2004, 25, 3259–3266. [Google Scholar] [CrossRef] [PubMed]
- Engler, A.C.; Ke, X.; Gao, S.; Chan, J.M.W.; Coady, D.J.; Ono, R.J.; Lubbers, R.; Nelson, A.; Yang, Y.Y.; Hedrick, J.L. Hydrophilic polycarbonates: Promising degradable alternatives to poly(ethylene glycol)-based stealth materials. Macromolecules 2015, 48, 1673–1678. [Google Scholar] [CrossRef]
- Kang, E.; Aklyoshi, K.; Sunamoto, J. Surface coating of liposomes with hydrophobized polysaccharides. J. Bioact. Compat. Polym. 1997, 12, 14–26. [Google Scholar] [CrossRef]
- Magarkar, A.; Róg, T.; Bunker, A. A computational study suggests that replacing PEG with PMOZ may increase exposure of hydrophobic targeting moiety. Eur. J. Pharm. Sci. 2017, 103, 128–135. [Google Scholar] [CrossRef]
- Wickens, J.M.; Alsaab, H.O.; Kesharwani, P.; Bhise, K.; Amin, M.C.I.M.; Tekade, R.K.; Gupta, U.; Iyer, A.K. Recent advances in hyaluronic acid-decorated nanocarriers for targeted cancer therapy. Drug Discov. Today 2017, 22, 665–680. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Deng, C.; Fu, Y.; Sun, X.; Gong, T.; Zhang, Z. Repeated Administration of Hyaluronic Acid Coated Liposomes with Improved Pharmacokinetics and Reduced Immune Response. Mol. Pharm. 2016, 13, 1800–1808. [Google Scholar] [CrossRef] [PubMed]
- Eliaz, R.E.; Szoka, J. Liposome-encapsulated doxorubicin targeted to CD44: A strategy to kill CD44-overexpressing tumor cells. Cancer Res. 2001, 61, 2592–2601. [Google Scholar]
- Peer, D.; Florentin, A.; Margalit, R. Hyaluronan is a key component in cryoprotection and formulation of targeted unilamellar liposomes. Biochim. Biophys. Acta Biomembr. 2003, 1612, 76–82. [Google Scholar] [CrossRef] [Green Version]
- Torchilin, V.P. Recent advances with liposomes as pharmaceutical carriers. Nat. Rev. Drug Discov. 2005, 4, 145–160. [Google Scholar] [CrossRef]
- Gómez-Hens, A.; Fernández-Romero, J.M. Analytical methods for the control of liposomal delivery systems. TrAC Trends Anal. Chem. 2006, 25, 167–178. [Google Scholar] [CrossRef]
- Lajunen, T.; Nurmi, R.; Kontturi, L.-S.; Viitala, L.; Yliperttula, M.; Murtomäki, L.; Urtti, A. Light activated liposomes: Functionality and prospects in ocular drug delivery. J. Control. Release 2016, 244, 157–166. [Google Scholar] [CrossRef]
- Alvarez-Lorenzo, C.; Bromberg, L.; Concheiro, A. Light-sensitive intelligent drug delivery systems. Photochem. Photobiol. 2009, 85, 848–860. [Google Scholar] [CrossRef] [PubMed]
- Lajunen, T.; Kontturi, L.-S.; Viitala, L.; Manna, M.; Cramariuc, O.; Róg, T.; Bunker, A.; Laaksonen, T.; Viitala, T.; Murtomäki, L.; et al. Indocyanine green-loaded liposomes for light-triggered drug release. Mol. Pharm. 2016, 13, 2095–2107. [Google Scholar] [CrossRef] [PubMed]
- Lajunen, T.; Nurmi, R.; Wilbie, D.; Ruoslahti, T.; Johansson, N.G.; Korhonen, O.; Rog, T.; Bunker, A.; Ruponen, M.; Urtti, A. The effect of light sensitizer localization on the stability of indocyanine green liposomes. J. Control. Release 2018, 284, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Viitala, L.; Pajari, S.; Lajunen, T.; Kontturi, L.S.; Laaksonen, T.; Kuosmanen, P.; Viitala, T.; Urtti, A.; Murtoma-ki, L. Photothermally triggered lipid bilayer phase transition and drug release from gold nanorod and indocyanine green encapsulated liposomes. Langmuir 2016, 32, 4554–4563. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency. Guideline on the Evaluation of the Pharmacokinetics of Medicinal Products in Patients with Impaired Hepatic Function; European Medicines Agency: London, UK, 2005.
- U.S. Food and Drug Administration. Product insert NDA 11-525-S-017: IC-GreenTM, Indocyanine Green for Injection; Silver Springs: Rockville, MD, USA, 2006.
- Kraft, J.C.; Ho, R.J.Y. Interactions of indocyanine green and lipid in enhancing near-infrared fluorescence properties: The basis for near-infrared imaging in vivo. Biochemistry 2014, 53, 1275–1283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monopoli, M.P.; Åberg, C.; Salvati, A.; Dawson, K.A.; Åberg, C.; Salvati, A.; Dawson, K.A.; Aberg, C.; Salvati, A.; Dawson, K.A. Biomolecular coronas provide the biological identity of nanosized materials. Nat. Nanotechnol. 2012, 7, 779–786. [Google Scholar] [CrossRef] [PubMed]
- Ke, P.C.; Lin, S.; Parak, W.J.; Davis, T.P.; Caruso, F. A Decade of the Protein Corona. ACS Nano 2017, 11, 11773–11776. [Google Scholar] [CrossRef]
- Kari, O.K.; Ndika, J.; Parkkila, P.; Louna, A.; Lajunen, T.; Puustinen, A.; Viitala, T.; Alenius, H.; Urtti, A. In situ analysis of liposome hard and soft protein corona structure and composition in a single label-free workflow. Nanoscale 2020, 12, 1728–1741. [Google Scholar] [CrossRef] [Green Version]
- Tavakoli, S.; Kari, O.K.; Turunen, T.; Lajunen, T.; Schmitt, M.; Lehtinen, J.; Tasaka, F.; Parkkila, P.; Ndika, J.; Viitala, T.; et al. Diffusion and protein corona formation of lipid-based nanoparticles in vitreous humor: Profiling and pharmacokinetic considerations. Mol. Pharm. 2020. [Google Scholar] [CrossRef]
- Jo, D.H.; Kim, J.H.; Son, J.G.; Dan, K.S.; Song, S.H.; Lee, T.G.; Kim, J.H. Nanoparticle-protein complexes mimicking corona formation in ocular environment. Biomaterials 2016, 109, 23–31. [Google Scholar] [CrossRef]
- Rimpelä, A.-K.; Schmitt, M.; Latonen, S.; Hagström, M.; Antopolsky, M.; Manzanares, J.A.; Kidron, H.; Urtti, A. Drug Distribution to Retinal Pigment Epithelium: Studies on Melanin Binding, Cellular Kinetics, and Single Photon Emission Computed Tomography/Computed Tomography Imaging. Mol. Pharm. 2016. [Google Scholar] [CrossRef]
- Yao, H.J.; Sun, L.; Liu, Y.; Jiang, S.; Pu, Y.; Li, J.; Zhang, Y. Monodistearoylphosphatidylethanolamine-hyaluronic acid functionalization of single-walled carbon nanotubes for targeting intracellular drug delivery to overcome multidrug resistance of cancer cells. Carbon N. Y. 2016, 96, 362–376. [Google Scholar] [CrossRef]
- Saxena, V.; Sadoqi, M.; Shao, J. Degradation Kinetics of Indocyanine Green in Aqueous Solution. J. Pharm. Sci. 2003, 92, 2090–2097. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.; Hein, M.Y.; Luber, C.A.; Paron, I.; Nagaraj, N.; Mann, M. Accurate Proteome-wide Label-free Quantification by Delayed Normalization and Maximal Peptide Ratio Extraction, Termed MaxLFQ. Mol. Cell. Proteomics 2014, 13, 2513–2526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Saadat, E.; Amini, M.; Dinarvand, R.; Dorkoosh, F.A. Polymeric micelles based on hyaluronic acid and phospholipids: Design, characterization, and cytotoxicity. J. Appl. Polym. Sci. 2014, 131, 1–8. [Google Scholar] [CrossRef]
- Al-Ahmady, Z.S.; Hadjidemetriou, M.; Gubbins, J.; Kostarelos, K. Formation of protein corona in vivo affects drug release from temperature-sensitive liposomes. J. Control. Release 2018, 276, 157–167. [Google Scholar] [CrossRef]
- Choi, K.Y.; Min, K.H.; Yoon, H.Y.; Kim, K.; Park, J.H.; Kwon, I.C.; Choi, K.; Jeong, S.Y. PEGylation of hyaluronic acid nanoparticles improves tumor targetability in vivo. Biomaterials 2011, 32, 1880–1889. [Google Scholar] [CrossRef]
- Rege, K.; Medintz, I.L. Methods in Bioengineering: Nanoscale Bioengineering and Nanomedicine; Artech House: Norwood, MA, USA, 2009; ISBN 9781596934115. [Google Scholar]
- Soo Choi, H.; Liu, W.; Misra, P.; Tanaka, E.; Zimmer, J.P.; Itty Ipe, B.; Bawendi, M.G.; Frangioni, J.V. Renal clearance of quantum dots. Nat. Biotechnol. 2007, 25, 1165–1170. [Google Scholar] [CrossRef] [Green Version]
- Hadjidemetriou, M.; Al-Ahmady, Z.; Mazza, M.; Collins, R.F.; Dawson, K.; Kostarelos, K. In Vivo Biomolecule Corona around Blood-Circulating, Clinically Used and Antibody-Targeted Lipid Bilayer Nanoscale Vesicles. ACS Nano 2015, 9, 8142–8156. [Google Scholar] [CrossRef]
- Tenzer, S.; Docter, D.; Kuharev, J.; Musyanovych, A.; Fetz, V.; Hecht, R.; Schlenk, F.; Fischer, D.; Kiouptsi, K.; Reinhardt, C.; et al. Rapid formation of plasma protein corona critically affects nanoparticle pathophysiology. Nat. Nanotechnol. 2013, 8, 772–781. [Google Scholar] [CrossRef]
- Dong, Y.; Arif, A.; Olsson, M.; Cali, V.; Hardman, B.; Dosanjh, M.; Lauer, M.; Midura, R.J.; Hascall, V.C.; Brown, K.L.; et al. Endotoxin free hyaluronan and hyaluronan fragments do not stimulate TNF-α, interleukin-12 or upregulate co-stimulatory molecules in dendritic cells or macrophages. Sci. Rep. 2016, 6, 1–15. [Google Scholar] [CrossRef]
- Mahmoudi, M. Debugging Nano–Bio Interfaces: Systematic Strategies to Accelerate Clinical Translation of Nanotechnologies. Trends Biotechnol. 2018, 36, 755–769. [Google Scholar] [CrossRef] [PubMed]
- Bishop, P. Structural Macromolecules and Supramolecular Organisation of the Vitreous Gel. Prog. Retin. Eye Res. 2000, 19, 323–344. [Google Scholar] [CrossRef]
- Xu, Q.; Boylan, N.J.; Suk, J.S.; Wang, Y.-Y.; Nance, E.A.; Yang, J.-C.; McDonnell, P.J.; Cone, R.A.; Duh, E.J.; Hanes, J. Nanoparticle diffusion in, and microrheology of, the bovine vitreous ex vivo. J. Control. Release 2013, 167, 76–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Käsdorf, B.T.; Arends, F.; Lieleg, O. Diffusion Regulation in the Vitreous Humor. Biophys. J. 2015, 109, 2171–2181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Havugimana, P.C.; Hart, G.T.; Nepusz, T.; Yang, H.; Turinsky, A.L.; Li, Z.; Wang, P.I.; Boutz, D.R.; Fong, V.; Phanse, S.; et al. A census of human soluble protein complexes. Cell 2012, 150, 1068–1081. [Google Scholar] [CrossRef] [Green Version]
- Skeie, J.M.; Roybal, C.N.; Mahajan, V.B. Proteomic insight into the molecular function of the vitreous. PLoS ONE 2015, 10, 1–19. [Google Scholar] [CrossRef]
- Bertrand, N.; Grenier, P.; Mahmoudi, M.; Lima, E.M.; Appel, E.A.; Dormont, F.; Lim, J.-M.; Karnik, R.; Langer, R.; Farokhzad, O.C. Mechanistic understanding of in vivo protein corona formation on polymeric nanoparticles and impact on pharmacokinetics. Nat. Commun. 2017, 8, 777. [Google Scholar] [CrossRef]
- Shannahan, J.H.; Lai, X.; Ke, P.C.; Podila, R.; Brown, J.M.; Witzmann, F.A. Silver Nanoparticle Protein Corona Composition in Cell Culture Media. PLoS ONE 2013, 8. [Google Scholar] [CrossRef]
- Tsuda, A.; Konduru, N.V. The role of natural processes and surface energy of inhaled engineered nanoparticles on aggregation and corona formation. NanoImpact 2016, 2, 38–44. [Google Scholar] [CrossRef] [Green Version]
- Kuznetsova, I.M.; Turoverov, K.K.; Uversky, V.N. What macromolecular crowding can do to a protein. Int. J. Mol. Sci. 2014, 15, 23090–23140. [Google Scholar] [CrossRef] [Green Version]
- Digiacomo, L.; Giulimondi, F.; Mahmoudi, M.; Caracciolo, G. Effect of molecular crowding on the biological identity of liposomes: An overlooked factor at the bio-nano interface. Nanoscale Adv. 2019, 1, 2518–2522. [Google Scholar] [CrossRef] [Green Version]
- Dewavrin, J.; Hamzavi, N.; Shim, V.P.W.; Raghunath, M. Tuning the architecture of three-dimensional collagen hydrogels by physiological macromolecular crowding. Acta Biomater. 2014, 10, 4351–4359. [Google Scholar] [CrossRef] [PubMed]
- Nel, A.E.; Mädler, L.; Velegol, D.; Xia, T.; Hoek, E.M.V.; Somasundaran, P.; Klaessig, F.; Castranova, V.; Thompson, M. Understanding biophysicochemical interactions at the nano-bio interface. Nat. Mater. 2009, 8, 543–557. [Google Scholar] [CrossRef] [PubMed]
- Walkey, C.D.; Chan, W.C.W. Understanding and controlling the interaction of nanomaterials with proteins in a physiological environment. Chem. Soc. Rev. 2012, 41, 2780–2799. [Google Scholar] [CrossRef]
- Swindle, K.E.; Hamilton, P.D.; Ravi, N. In situ formation of hydrogels as vitreous substitutes: Viscoelastic comparison to porcine vitreous. J. Biomed. Mater. Res. Part A 2008, 87, 656–665. [Google Scholar] [CrossRef]
- Kelly, P.M.; Åberg, C.; Polo, E.; O’Connell, A.; Cookman, J.; Fallon, J.; Krpetić, Ž.; Dawson, K.A. Mapping protein binding sites on the biomolecular corona of nanoparticles. Nat. Nanotechnol. 2015, 10, 472–479. [Google Scholar] [CrossRef]
- Raesch, S.S.; Tenzer, S.; Storck, W.; Rurainski, A.; Selzer, D.; Ruge, C.A.; Perez-Gil, J.; Schaefer, U.F.; Lehr, C.M. Proteomic and Lipidomic Analysis of Nanoparticle Corona upon Contact with Lung Surfactant Reveals Differences in Protein, but Not Lipid Composition. ACS Nano 2015, 9, 11872–11885. [Google Scholar] [CrossRef]
- Müller, J.; Prozeller, D.; Ghazaryan, A.; Kokkinopoulou, M.; Mailänder, V.; Morsbach, S.; Landfester, K. Beyond the protein corona – lipids matter for biological response of nanocarriers. Acta Biomater. 2018, 71, 420–431. [Google Scholar] [CrossRef]
- Simon, J.; Müller, L.K.; Kokkinopoulou, M.; Lieberwirth, I.; Morsbach, S.; Landfester, K.; Mailänder, V. Exploiting the biomolecular corona: Pre-coating of nanoparticles enables controlled cellular interactions. Nanoscale 2018, 10, 10731–10739. [Google Scholar] [CrossRef] [Green Version]
- del Amo, E.M.; Rimpelä, A.-K.; Heikkinen, E.; Kari, O.K.; Ramsay, E.; Lajunen, T.; Schmitt, M.; Pelkonen, L.; Bhattacharya, M.; Richardson, D.; et al. Pharmacokinetic aspects of retinal drug delivery. Prog. Retin. Eye Res. 2016, 1–52. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Size (nm) | Zeta Potential (mV) | ICG Absorbance (a.u.) | |
|---|---|---|---|
| Uncoated | 102 ± 24 | −0.44 ± 1.32 | 0.72 ± 0.08 |
| PEG | 119 ± 30 | −2.94 ± 0.69 | 0.90 ± 0.13 |
| HA | 104 ± 25 | −11.27 ± 0.21 | 0.88 ± 0.12 |
| HA (30 nm extrusion) | 68 ± 16 | n.a. | 0.86 ± 0.03 |
| Formulation | Mean Diameter (nm) | PdI | Dv (μm2/s) | Dw (μm2/s) | Dw/Dv |
|---|---|---|---|---|---|
| HA | 116.1 | 0.008 | 0.28 ± 0.14 | 5.68 | 20.2 |
| PEG * | 107.6 | 0.086 | 0.47 ± 0.25 | 6.12 | 13.0 |
| Uncoated * | 125.8 | 0.035 | 0.33 ± 0.17 | 5.27 | 16.0 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kari, O.K.; Tavakoli, S.; Parkkila, P.; Baan, S.; Savolainen, R.; Ruoslahti, T.; Johansson, N.G.; Ndika, J.; Alenius, H.; Viitala, T.; et al. Light-Activated Liposomes Coated with Hyaluronic Acid as a Potential Drug Delivery System. Pharmaceutics 2020, 12, 763. https://doi.org/10.3390/pharmaceutics12080763
Kari OK, Tavakoli S, Parkkila P, Baan S, Savolainen R, Ruoslahti T, Johansson NG, Ndika J, Alenius H, Viitala T, et al. Light-Activated Liposomes Coated with Hyaluronic Acid as a Potential Drug Delivery System. Pharmaceutics. 2020; 12(8):763. https://doi.org/10.3390/pharmaceutics12080763
Chicago/Turabian StyleKari, Otto K., Shirin Tavakoli, Petteri Parkkila, Simone Baan, Roosa Savolainen, Teemu Ruoslahti, Niklas G. Johansson, Joseph Ndika, Harri Alenius, Tapani Viitala, and et al. 2020. "Light-Activated Liposomes Coated with Hyaluronic Acid as a Potential Drug Delivery System" Pharmaceutics 12, no. 8: 763. https://doi.org/10.3390/pharmaceutics12080763