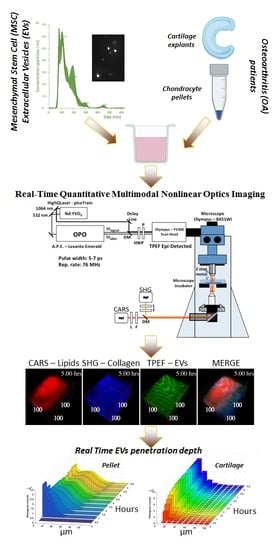
In Vitro Study of Extracellular Vesicles Migration in Cartilage-Derived Osteoarthritis Samples Using Real-Time Quantitative Multimodal Nonlinear Optics Imaging

 , , , and
, , , and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
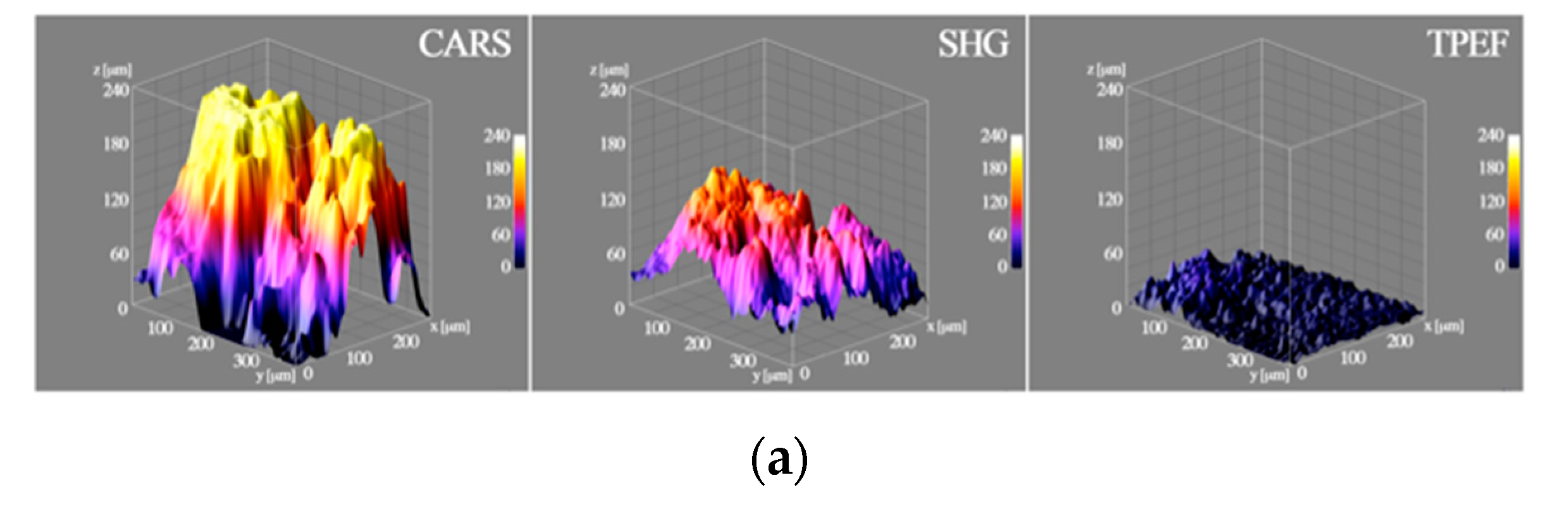{kind=link}
{kind=link}
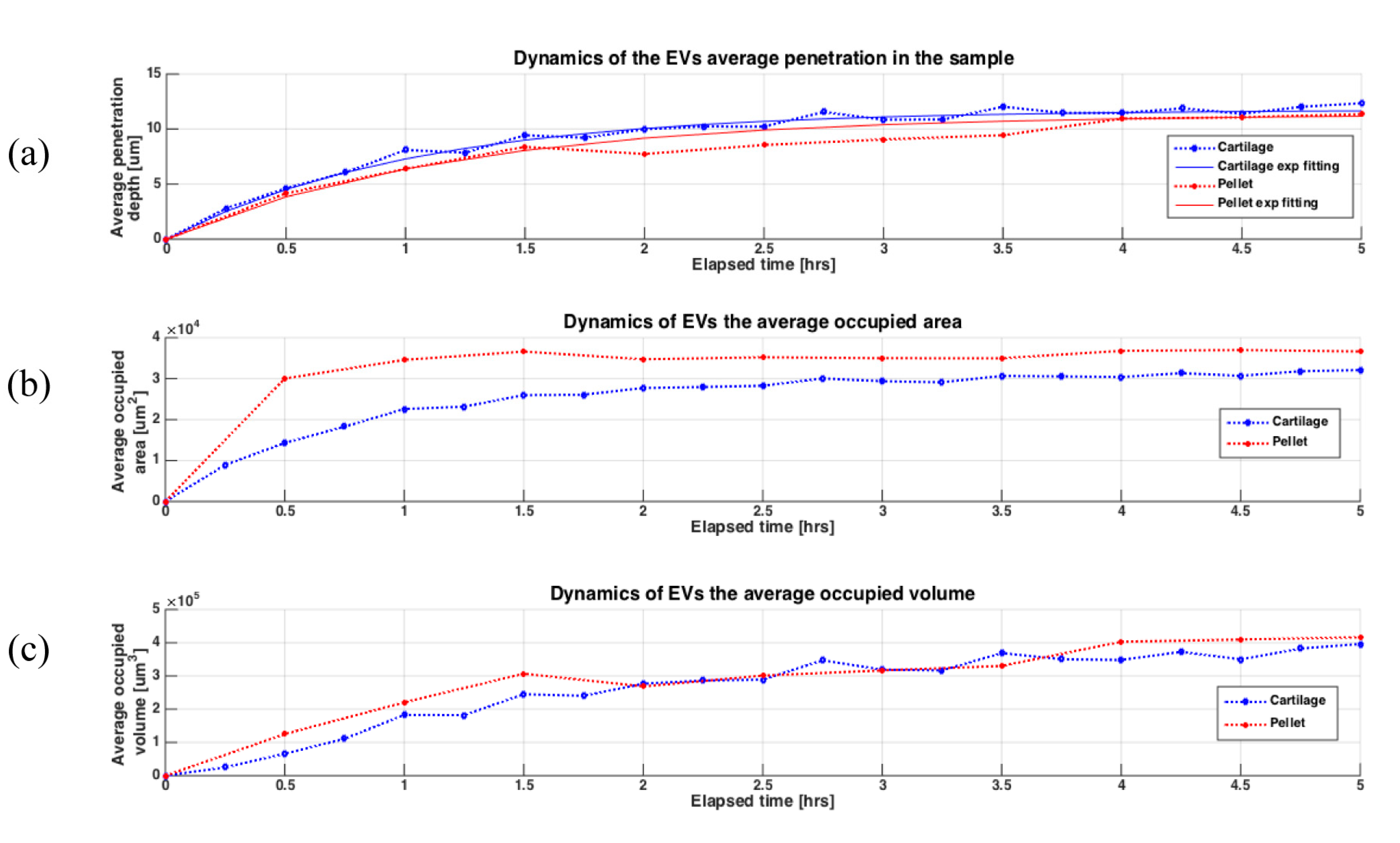{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cartilage and Adipose Tissue Collection, Cell Isolation and Expansion
2.2. ASCs Characterization by Flow Cytometry
2.3. Extracellular Vesicles (EVs) Isolation and Staining
2.4. EVs Quantification and Dimensional Evaluation by Nanoparticle Tracking Analysis (NTA)
2.5. EVs Characterization by Flow Cytometry
2.6. Transmission Electron Microscopy (TEM)
2.7. Chondrocyte Micromass Cultures
2.8. EVs Incorporation Analysis by Standard Immunofluorescence
2.9. EVs Incorporation Detection by Time-Lapse Microscopy
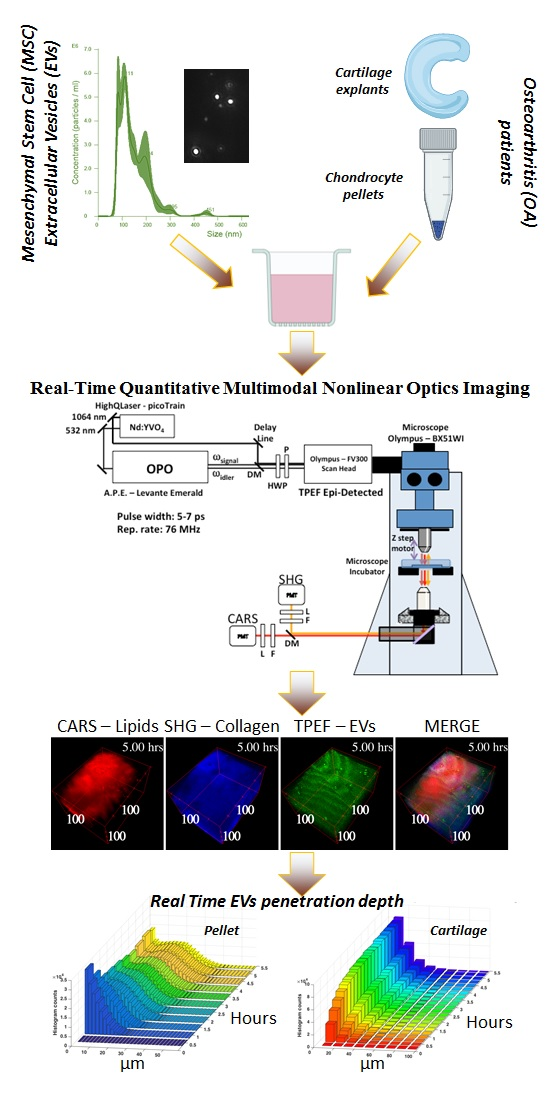
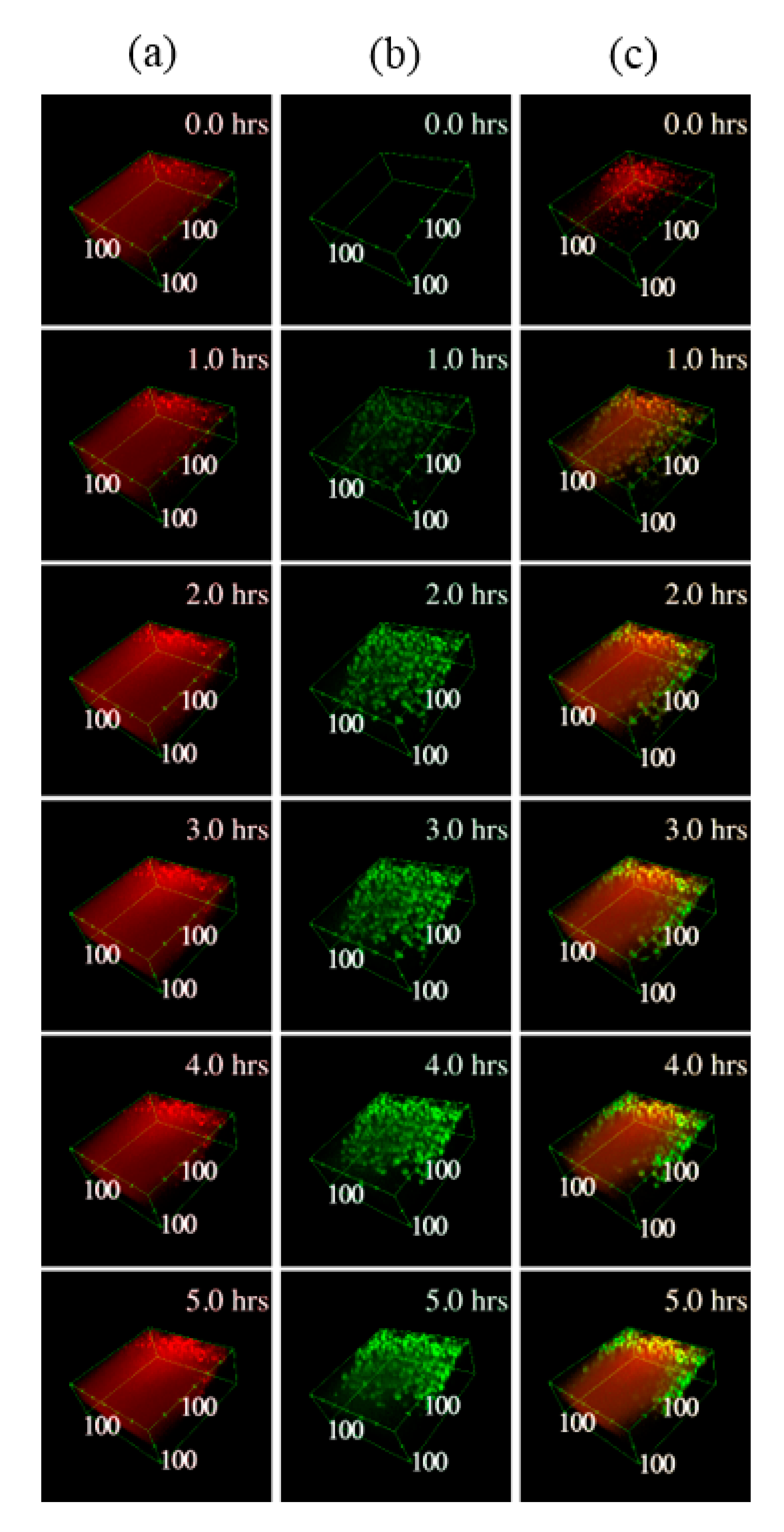
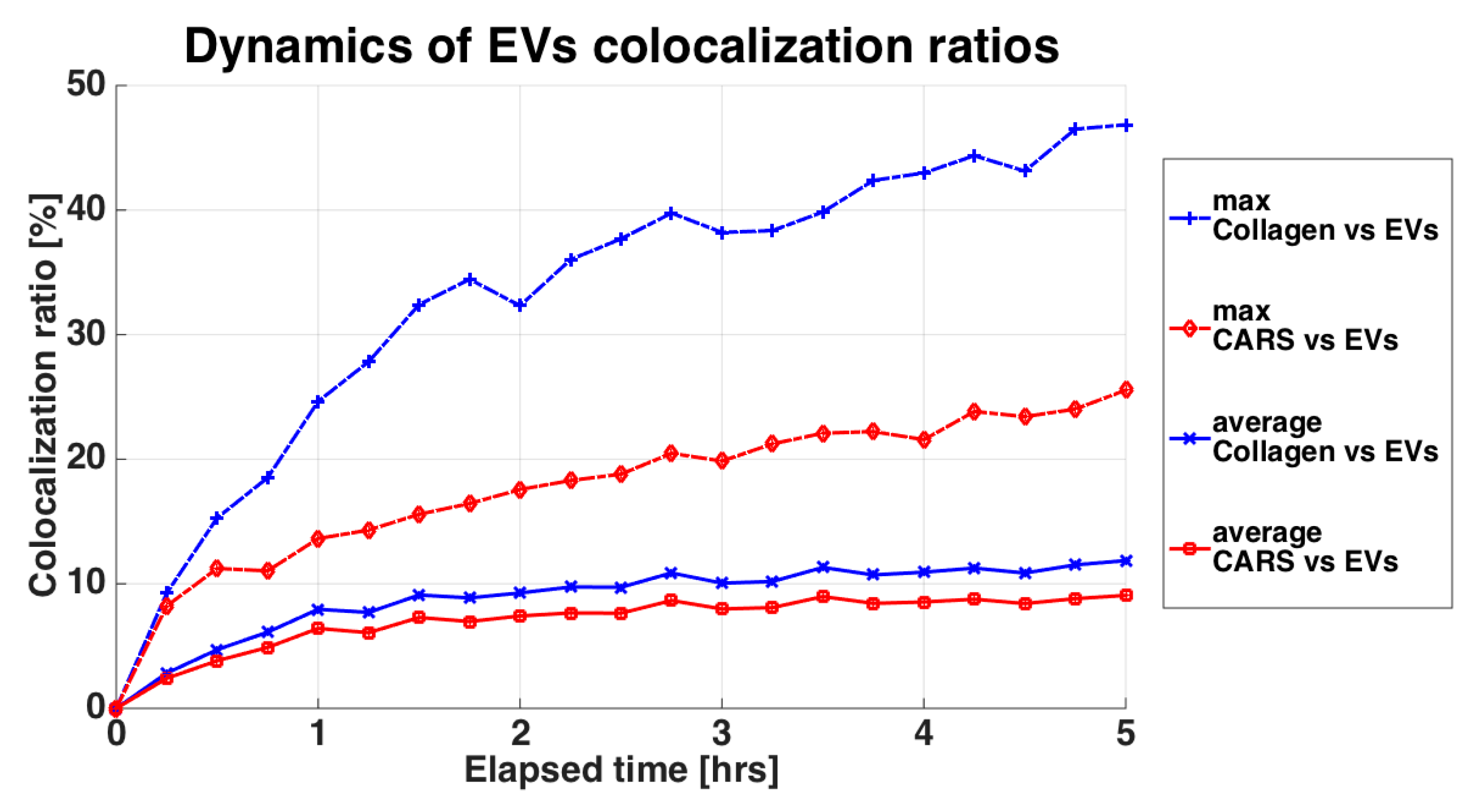
2.10. Multimodal Microscopy Data Analysis
3. Results
3.1. ASC-EVs Showed Characteristic Size, Morphology and MSC-EVs Markers
3.2. Endpoint Incorporation Showed ASC-EVs Full Migration in Chondrocyte Micromass
3.3. Time-Lapse Showed a Fast Penetration and Specific Pattern of ASC-EVs Incorporation in Chondrocyte Micromass and Cartilage Explants
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Baek, G.; Choi, H.; Kim, Y.; Lee, H.C.; Choi, C. Mesenchymal Stem Cell-Derived Extracellular Vesicles as Therapeutics and as a Drug Delivery Platform. Stem Cells Transl. Med. 2019, 8, 880–886. [Google Scholar] [CrossRef] [Green Version]
- Colombini, A.; Perucca Orfei, C.; Kouroupis, D.; Ragni, E.; De Luca, P.; Viganò, M.; Correa, D.; de Girolamo, L. Mesenchymal stem cells in the treatment of articular cartilage degeneration: New biological insights for an old-timer cell. Cytotherapy 2019, 21, 1179–1197. [Google Scholar] [CrossRef] [PubMed]
- Phinney, D.G.; Pittenger, M.F. Concise Review: MSC-Derived Exosomes for Cell-Free Therapy. Stem Cells 2017, 35, 851–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burrello, J.; Monticone, S.; Gai, C.; Gomez, Y.; Kholia, S.; Camussi, G. Stem Cell-Derived Extracellular Vesicles and Immune-Modulation. Front. Cell Dev. Biol. 2016, 4, 83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, L.; Leijten, J.C.H.; Georgi, N.; Post, J.N.; van Blitterswijk, C.A.; Karperien, M. Trophic Effects of Mesenchymal Stem Cells Increase Chondrocyte Proliferation and Matrix Formation. Tissue Eng. Pt A 2011, 17, 1425–1436. [Google Scholar] [CrossRef] [PubMed]
- Cosenza, S.; Ruiz, M.; Toupet, K.; Jorgensen, C.; Noel, D. Mesenchymal stem cells derived exosomes and microparticles protect cartilage and bone from degradation in osteoarthritis. Sci. Rep. 2017, 7, 16214. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Chu, W.C.; Lai, R.C.; Lim, S.K.; Hui, J.H.; Toh, W.S. Exosomes derived from human embryonic mesenchymal stem cells promote osteochondral regeneration. Osteoarthr. Cartil. 2016, 24, 2135–2140. [Google Scholar] [CrossRef] [Green Version]
- Tao, S.C.; Yuan, T.; Zhang, Y.L.; Yin, W.J.; Guo, S.C.; Zhang, C.Q. Exosomes derived from miR-140-5p-overexpressing human synovial mesenchymal stem cells enhance cartilage tissue regeneration and prevent osteoarthritis of the knee in a rat model. Theranostics 2017, 7, 180–195. [Google Scholar] [CrossRef]
- Woo, C.H.; Kim, H.K.; Jung, G.Y.; Jung, Y.J.; Lee, K.S.; Yun, Y.E.; Han, J.; Lee, J.; Kim, W.S.; Choi, J.S.; et al. Small extracellular vesicles from human adipose-derived stem cells attenuate cartilage degeneration. J. Extracell. Vesicles 2020, 9, 1735249. [Google Scholar] [CrossRef] [Green Version]
- Ragni, E.; Lommel, M.; Moro, M.; Crosti, M.; Lavazza, C.; Parazzi, V.; Saredi, S.; Strahl, S.; Lazzari, L. Protein O-mannosylation is crucial for human mesencyhmal stem cells fate. Cell. Mol. Life Sci. CMLS 2016, 73, 445–458. [Google Scholar] [CrossRef]
- Ragni, E.; Perucca Orfei, C.; De Luca, P.; Lugano, G.; Vigano, M.; Colombini, A.; Valli, F.; Zacchetti, D.; Bollati, V.; de Girolamo, L. Interaction with hyaluronan matrix and miRNA cargo as contributors for in vitro potential of mesenchymal stem cell-derived extracellular vesicles in a model of human osteoarthritic synoviocytes. Stem Cell Res. Ther. 2019, 10, 109. [Google Scholar] [CrossRef] [PubMed]
- Vonk, L.A.; van Dooremalen, S.F.J.; Liv, N.; Klumperman, J.; Coffer, P.J.; Saris, D.B.F.; Lorenowicz, M.J. Mesenchymal Stromal/stem Cell-derived Extracellular Vesicles Promote Human Cartilage Regeneration in Vitro. Theranostics 2018, 8, 906–920. [Google Scholar] [CrossRef]
- Hoover, E.E.; Squier, J.A. Advances in multiphoton microscopy technology. Nat. Photonics 2013, 7, 93–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moura, C.C.; Lanham, S.A.; Monfort, T.; Bourdakos, K.N.; Tare, R.S.; Oreffo, R.O.C.; Mahajan, S. Quantitative temporal interrogation in 3D of bioengineered human cartilage using multimodal label-free imaging. Integr. Biol.-UK 2018, 10, 635–645. [Google Scholar] [CrossRef] [Green Version]
- Bergholt, M.S.; St-Pierre, J.P.; Offeddu, G.S.; Parmar, P.A.; Albro, M.B.; Puetzer, J.L.; Oyen, M.L.; Stevens, M.M. Raman Spectroscopy Reveals New Insights into the Zonal Organization of Native and Tissue-Engineered Articular Cartilage. ACS Cent. Sci. 2016, 2, 885–895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stok, K.S.; Noel, D.; Apparailly, F.; Gould, D.; Chernajovsky, Y.; Jorgensen, C.; Muller, R. Quantitative imaging of cartilage and bone for functional assessment of gene therapy approaches in experimental arthritis. J. Tissue Eng. Regen. Med. 2010, 4, 387–394. [Google Scholar] [CrossRef]
- Li, Y.W.; Trivedil, V.; Truong, T.V.; Koos, D.S.; Lansford, R.; Chuong, C.; Warburton, D.; Moats, R.A.; Fraserl, S.E. Dynamic imaging of the growth plate cartilage reveals multiple contributors to skeletal morphogenesis. Nat. Commun. 2015, 6, 6798. [Google Scholar] [CrossRef] [Green Version]
- Periasamy, A.; So, P.T.C.; König, K.; Xie, X.S. Multiphoton Microscopy in the Biomedical Sciences XVII; SPIE-International Society for Optics and Photonics: San Francisco, CA, USA, 2017; pp. 1–410. [Google Scholar]
- Zhang, Z.J.; McCaffery, J.M.; Spencer, R.G.S.; Francomano, C.A. Hyaline cartilage engineered by chondrocytes in pellet culture: Histological, immunohistochemical and ultrastructural analysis in comparison with cartilage explants. J. Anat. 2004, 205, 229–237. [Google Scholar] [CrossRef]
- Johnson, C.I.; Argyle, D.J.; Clements, D.N. In vitro models for the study of osteoarthritis. Vet. J. 2016, 209, 40–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aulthouse, A.L.; Beck, M.; Griffey, E.; Sanford, J.; Arden, K.; Machado, M.A.; Horton, W.A. Expression of the Human Chondrocyte Phenotype Invitro. Vitr. Cell Dev. Biol. 1989, 25, 659–668. [Google Scholar] [CrossRef]
- Hardingham, T.; Tew, S.; Murdoch, A. Tissue engineering: Chondrocytes and cartilage. Arthritis Res. 2002, 4, S63–S68. [Google Scholar] [CrossRef] [PubMed]
- Darling, E.M.; Athanasiou, K.A. Rapid phenotypic changes in passaged articular chondrocyte subpopulations. J. Orthop. Res. 2005, 23, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Lewis, M.C.; MacArthur, B.; Tare, R.S.; Oreffo, R.O.C.; Please, C.P. Extracellular Matrix Deposition in Engineered Micromass Cartilage Pellet Cultures: Measurements and Modelling. PLoS ONE 2016, 11, e0147302. [Google Scholar] [CrossRef]
- Hunziker, E.B.; Quinn, T.M.; Hauselmann, H.J. Quantitative structural organization of normal adult human articular cartilage. Osteoarthr. Cartil. 2002, 10, 564–572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahmoudifar, N.; Doran, P.M. Chondrogenesis and cartilage tissue engineering: The longer road to technology development. Trends Biotechnol. 2012, 30, 166–176. [Google Scholar] [CrossRef]
- Doran, P.M. Cartilage Tissue Engineering: What Have We Learned in Practice? Methods Mol. Biol. 2015, 1340, 3–21. [Google Scholar] [CrossRef]
- Cope, P.J.; Ourradi, K.; Li, Y.; Sharif, M. Models of osteoarthritis: The good, the bad and the promising. Osteoarthr. Cartil. 2019, 27, 230–239. [Google Scholar] [CrossRef] [Green Version]
- Lopa, S.; Colombini, A.; Sansone, V.; Preis, F.W.; Moretti, M. Influence on chondrogenesis of human osteoarthritic chondrocytes in co-culture with donor-matched mesenchymal stem cells from infrapatellar fat pad and subcutaneous adipose tissue. Int. J. Immunopathol. Pharmacol. 2013, 26, 23–31. [Google Scholar] [CrossRef] [Green Version]
- Lopa, S.; Colombini, A.; Stanco, D.; de Girolamo, L.; Sansone, V.; Moretti, M. Donor-matched mesenchymal stem cells from knee infrapatellar and subcutaneous adipose tissue of osteoarthritic donors display differential chondrogenic and osteogenic commitment. Eur. Cells Mater. 2014, 27, 298–311. [Google Scholar] [CrossRef]
- Dominici, M.; Le Blanc, K.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.; Krause, D.; Deans, R.; Keating, A.; Prockop, D.; Horwitz, E. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 2006, 8, 315–317. [Google Scholar] [CrossRef]
- Ragni, E.; Banfi, F.; Barilani, M.; Cherubini, A.; Parazzi, V.; Larghi, P.; Dolo, V.; Bollati, V.; Lazzari, L. Extracellular Vesicle-Shuttled mRNA in Mesenchymal Stem Cell Communication. Stem Cells 2017, 35, 1093–1105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thery, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramos, T.L.; Sanchez-Abarca, L.I.; Muntion, S.; Preciado, S.; Puig, N.; Lopez-Ruano, G.; Hernandez-Hernandez, A.; Redondo, A.; Ortega, R.; Rodriguez, C.; et al. MSC surface markers (CD44, CD73, and CD90) can identify human MSC-derived extracellular vesicles by conventional flow cytometry. Cell Commun. Signal 2016, 14, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Luca, P.; Kouroupis, D.; Viganò, M.; Perucca-Orfei, C.; Kaplan, L.; Zagra, L.; de Girolamo, L.; Correa, D.; Colombini, A. Human Diseased Articular Cartilage Contains a Mesenchymal Stem Cell-Like Population of Chondroprogenitors with Strong Immunomodulatory Responses. J. Clin. Med. 2019, 8, 423. [Google Scholar] [CrossRef] [Green Version]
- Mortati, L.; Divieto, C.; Sassi, M.P. CARS and SHG microscopy to follow collagen production in living human corneal fibroblasts and mesenchymal stem cells in fibrin hydrogel 3D cultures. J. Raman Spectrosc. 2012, 43, 675–680. [Google Scholar] [CrossRef] [Green Version]
- Webber, J.; Clayton, A. How pure are your vesicles? J. Extracell. Vesicles 2013, 2. [Google Scholar] [CrossRef]
- Ragni, E.; Perucca Orfei, C.; De Luca, P.; Colombini, A.; Vigano, M.; de Girolamo, L. Secreted Factors and EV-miRNAs Orchestrate the Healing Capacity of Adipose Mesenchymal Stem Cells for the Treatment of Knee Osteoarthritis. Int. J. Mol. Sci. 2020, 21, 1582. [Google Scholar] [CrossRef] [Green Version]
- Barilani, M.; Peli, V.; Cherubini, A.; Dossena, M.; Dolo, V.; Lazzari, L. NG2 as an Identity and Quality Marker of Mesenchymal Stem Cell Extracellular Vesicles. Cells 2019, 8, 1524. [Google Scholar] [CrossRef] [Green Version]
- Mendt, M.; Rezvani, K.; Shpall, E. Mesenchymal stem cell-derived exosomes for clinical use. Bone Marrow Transplant. 2019, 54, 789–792. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Xu, B.; Xu, H. TGF-beta1 promoted chondrocyte proliferation by regulating Sp1 through MSC-exosomes derived miR-135b. Cell Cycle 2018, 17, 2756–2765. [Google Scholar] [CrossRef] [Green Version]
- Mao, G.; Zhang, Z.; Hu, S.; Zhang, Z.; Chang, Z.; Huang, Z.; Liao, W.; Kang, Y. Exosomes derived from miR-92a-3p-overexpressing human mesenchymal stem cells enhance chondrogenesis and suppress cartilage degradation via targeting WNT5A. Stem Cell Res. Ther. 2018, 9, 247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sohel, M.H. Extracellular/Circulating MicroRNAs: Release Mechanisms, Functions and Challenges. Achiev. Life Sci. 2016, 10, 175–186. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Liu, S.; Huang, J.; Guo, W.; Chen, J.; Zhang, L.; Zhao, B.; Peng, J.; Wang, A.; Wang, Y.; et al. The ECM-cell interaction of cartilage extracellular matrix on chondrocytes. Biomed. Res. Int. 2014, 2014, 648459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ragni, E.; Palombella, S.; Lopa, S.; Talo, G.; Perucca Orfei, C.; De Luca, P.; Moretti, M.; de Girolamo, L. Innovative Visualization and Quantification of Extracellular Vesicles Interaction with and Incorporation in Target Cells in 3D Microenvironments. Cells 2020, 9, 1180. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.; Lyu, X.; Wang, Z.; Jin, H.; Lu, S.; Xing, D.; Hu, X. Cocktail polyprodrug nanoparticles concurrently release cisplatin and peroxynitrite-generating nitric oxide in cisplatin-resistant cancers. Chem. Eng. J. 2020, 402, 126125. [Google Scholar] [CrossRef]
- Guo, X.; Cao, B.; Wang, C.; Lu, S.; Hu, X. In vivo photothermal inhibition of methicillin-resistant Staphylococcus aureus infection by in situ templated formulation of pathogen-targeting phototheranostics. Nanoscale 2020, 12, 7651–7659. [Google Scholar] [CrossRef] [PubMed]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mortati, L.; de Girolamo, L.; Perucca Orfei, C.; Viganò, M.; Brayda-Bruno, M.; Ragni, E.; Colombini, A. In Vitro Study of Extracellular Vesicles Migration in Cartilage-Derived Osteoarthritis Samples Using Real-Time Quantitative Multimodal Nonlinear Optics Imaging. Pharmaceutics 2020, 12, 734. https://doi.org/10.3390/pharmaceutics12080734
Mortati L, de Girolamo L, Perucca Orfei C, Viganò M, Brayda-Bruno M, Ragni E, Colombini A. In Vitro Study of Extracellular Vesicles Migration in Cartilage-Derived Osteoarthritis Samples Using Real-Time Quantitative Multimodal Nonlinear Optics Imaging. Pharmaceutics. 2020; 12(8):734. https://doi.org/10.3390/pharmaceutics12080734
Chicago/Turabian StyleMortati, Leonardo, Laura de Girolamo, Carlotta Perucca Orfei, Marco Viganò, Marco Brayda-Bruno, Enrico Ragni, and Alessandra Colombini. 2020. "In Vitro Study of Extracellular Vesicles Migration in Cartilage-Derived Osteoarthritis Samples Using Real-Time Quantitative Multimodal Nonlinear Optics Imaging" Pharmaceutics 12, no. 8: 734. https://doi.org/10.3390/pharmaceutics12080734