Preclinical Development of Autologous Hematopoietic Stem Cell-Based Gene Therapy for Immune Deficiencies: A Journey from Mouse Cage to Bed Side

 , , ,
, , , 
Abstract
:1. Introduction
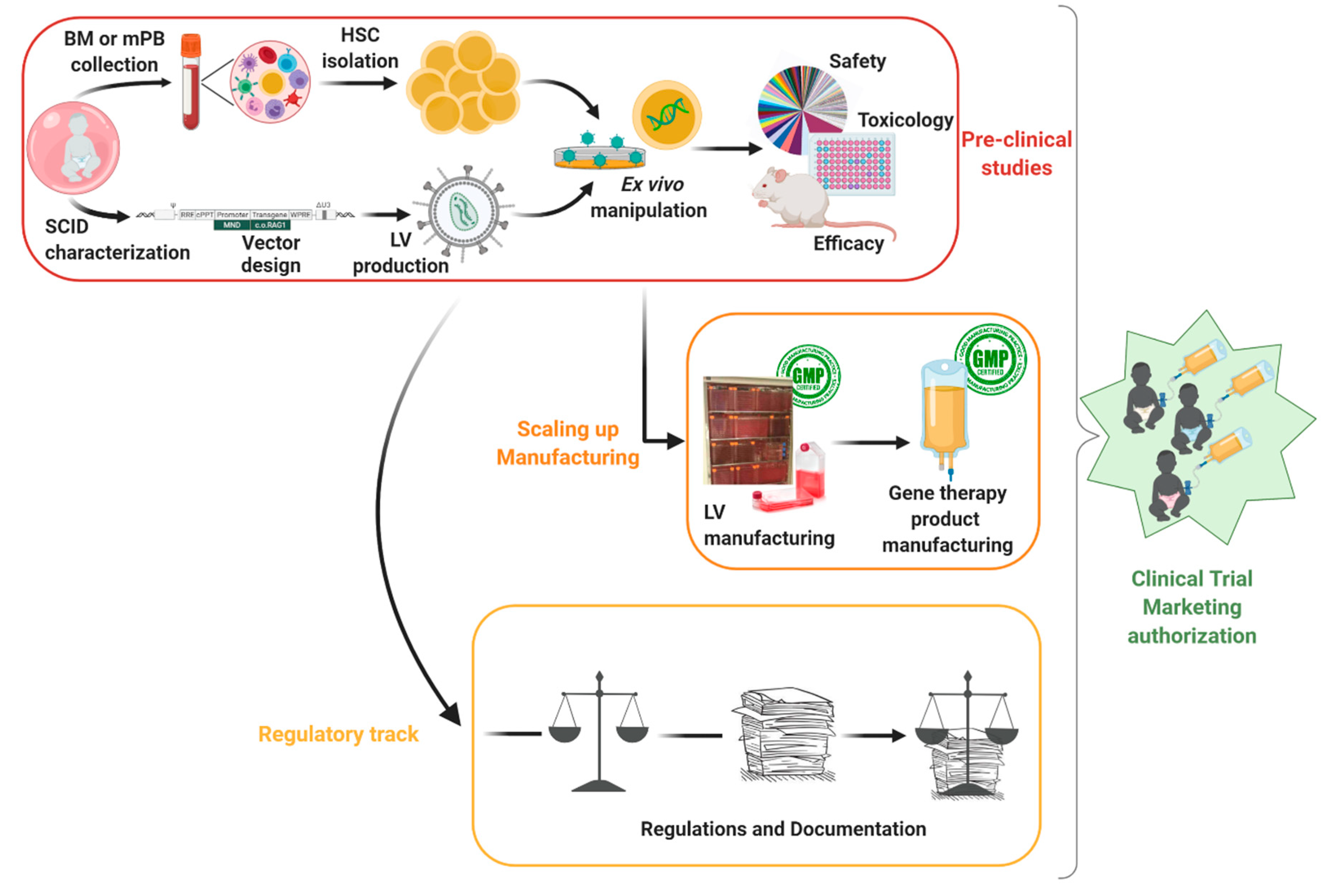
2. Product Development
2.1. Target Cells of Interest
2.2. Vector Design: Balancing Insertion Site and Therapeutic Expression
3. Proof-Of-Concept
3.1. Ex Vivo Manipulation: Transduction Efficiency
3.2. Call for Suitable Models to Test the Efficacy of Gene Therapy
3.2.1. Animal Models
3.2.2. In Vitro Models
3.3. Safety and Toxicology Assessment for Gene Therapy
4. Pharmaceutical and Clinical Development Phases: From Mouse to Human Treatment
4.1. Scaling Up: GMP Protocols and Manufacturing
4.1.1. GMP Compliant Virus Manufacturing
4.1.2. GMP Gene Therapy Product Manufacturing
4.2. Regulatory Hurdles
5. Conclusions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ADA | Adenosine Deaminase Deficiency |
| ATMP | Advanced Therapy Medicinal Product |
| BM | Bone Marrow |
| CAR | Chimeric Antigen Receptor |
| CAT | Committee for Advanced Therapies |
| CCMO | Centrale Commissie Mensgebonden Onderzoek |
| CGD | Chronic Granulomatous Disease |
| CHMP | Committee for Human Medicinal Products |
| CTA | Clinical Trial Application |
| ddPCR | Digital Droplet PCR |
| DL1 | Delta-Ligand 1 |
| EC | European Commission |
| EMA | European Medicines Agency |
| EU | European Union |
| FDA | U.S. Food and Drug Administration |
| FTOC | Fetal Thymus Organ Culture |
| GMO | Genetically Modified Organism |
| GMP | Good Manufacturing Practice |
| GvHD | Graft versus Host Disease |
| HLA | Human Leukocyte Antigen |
| HSC | Hematopoietic Stem Cell |
| HSPC | Hematopoietic Stem Progenitor cell |
| IB | Investigator Brochure |
| IMPD | Investigational Medicinal Product Dossier |
| iPSCS | Induced Pluripotent Stem Cell |
| IVIM | In Vitro Immortalization Assay |
| LAM-PCR | Linear Amplification Mediated PCR |
| LSK | Lin−Sca+cKit+ cells |
| LTR | Long Terminal Repeat |
| LUMC | Leiden University Medical Center |
| LV | Lentiviral vector |
| MA | Marketing Authorization |
| mPB | Mobilized Peripheral Blood |
| NSG | NOD Scid Gamma |
| nrLAM-PCR | Non-restrictive LAM-PCR |
| PCR | Polymerase Chain Reaction |
| PRIME | Priority Medicines |
| RAG1/2 | Recombinase-Activating Gene 1/2 |
| RV | Retroviral Vector |
| SAGA | Surrogate Assay for Genotoxicity Assessment |
| SCID | Severe Combined Immunodeficiency |
| SIN | Self-Inactivating |
| TE | Transduction enhancer |
| USA | United States of America |
| VCN | Vector Copy Number |
| VHP | Voluntary Harmonized Procedure |
| WAS | Wiskott–Aldrich Syndrome |
| WPRE | Woodchuck hepatitis virus Post-transcriptional Regulatory Element |
References
- Gene Therapy Clinical Trials Worldwide. Available online: http://www.abedia.com/wiley/index.html (accessed on 31 May 2020).
- Shahryari, A.; Saghaeian Jazi, M.; Mohammadi, S.; Razavi Nikoo, H.; Nazari, Z.; Hosseini, E.S.; Burtscher, I.; Mowla, S.J.; Lickert, H. Development and Clinical Translation of Approved Gene Therapy Products for Genetic Disorders. Front. Genet. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Aiuti, A.; Vai, S.; Mortellaro, A.; Casorati, G.; Ficara, F.; Andolfi, G.; Ferrari, G.; Tabucchi, A.; Carlucci, F.; Ochs, H.D.; et al. Immune reconstitution in ADA-SCID after PBL gene therapy and discontinuation of enzyme replacement. Nat. Med. 2002, 8, 423–425. [Google Scholar] [CrossRef] [PubMed]
- Aiuti, A.; Cassani, B.; Andolfi, G.; Mirolo, M.; Biasco, L.; Recchia, A.; Urbinati, F.; Valacca, C.; Scaramuzza, S.; Aker, M.; et al. Multilineage hematopoietic reconstitution without clonal selection in ADA-SCID patients treated with stem cell gene therapy. J. Clin. Investig. 2007, 117, 2233–2240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrua, F.; Aiuti, A. Twenty-Five Years of Gene Therapy for ADA-SCID: From Bubble Babies to an Approved Drug. Hum. Gene Ther. 2017, 28, 972–981. [Google Scholar] [CrossRef] [PubMed]
- Cavazzana-Calvo, M. Gene Therapy of Human Severe Combined Immunodeficiency (SCID)-X1 Disease. Science 2000, 288, 669–672. [Google Scholar] [CrossRef]
- Gaspar, H.B.; Parsley, K.L.; Howe, S.; King, D.; Gilmour, K.C.; Sinclair, J.; Brouns, G.; Schmidt, M.; Von Kalle, C.; Barington, T.; et al. Gene therapy of X-linked severe combined immunodeficiency by use of a pseudotyped gammaretroviral vector. Lancet 2004, 364, 2181–2187. [Google Scholar] [CrossRef]
- De Ravin, S.S.; Wu, X.; Moir, S.; Kardava, L.; Anaya-O’Brien, S.; Kwatemaa, N.; Littel, P.; Theobald, N.; Choi, U.; Su, L.; et al. Lentiviral hematopoietic stem cell gene therapy for X-linked severe combined immunodeficiency. Sci. Transl. Med. 2016, 8, 335ra357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poletti, V.; Charrier, S.; Corre, G.; Gjata, B.; Vignaud, A.; Zhang, F.; Rothe, M.; Schambach, A.; Gaspar, H.B.; Thrasher, A.J.; et al. Preclinical Development of a Lentiviral Vector for Gene Therapy of X-Linked Severe Combined Immunodeficiency. Mol. Ther. Methods Clin. Dev. 2018, 9, 257–269. [Google Scholar] [CrossRef] [Green Version]
- Benjelloun, F.; Garrigue, A.; Demerens-De Chappedelaine, C.; Soulas-Sprauel, P.; Malassis-Séris, M.; Stockholm, D.; Hauer, J.; Blondeau, J.; Rivière, J.; Lim, A.; et al. Stable and Functional Lymphoid Reconstitution in Artemis-deficient Mice Following Lentiviral Artemis Gene Transfer into Hematopoietic Stem Cells. Mol. Ther. 2008, 16, 1490–1499. [Google Scholar] [CrossRef]
- Punwani, D.; Kawahara, M.; Yu, J.; Sanford, U.; Roy, S.; Patel, K.; Carbonaro, D.A.; Karlen, A.D.; Khan, S.; Cornetta, K.; et al. Lentivirus Mediated Correction of Artemis-Deficient Severe Combined Immunodeficiency. Hum. Gene Ther. 2017, 28, 112–124. [Google Scholar] [CrossRef]
- Charrier, S.; Lagresle-Peyrou, C.; Poletti, V.; Rothe, M.; Cédrone, G.; Gjata, B.; Mavilio, F.; Fischer, A.; Schambach, A.; De Villartay, J.-P.; et al. Biosafety Studies of a Clinically Applicable Lentiviral Vector for the Gene Therapy of Artemis-SCID. Mol. Ther. Methods Clin. Dev. 2019, 15, 232–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aiuti, A.; Biasco, L.; Scaramuzza, S.; Ferrua, F.; Cicalese, M.P.; Baricordi, C.; Dionisio, F.; Calabria, A.; Giannelli, S.; Castiello, M.C.; et al. Lentiviral Hematopoietic Stem Cell Gene Therapy in Patients with Wiskott-Aldrich Syndrome. Science 2013, 341, 1233151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braun, C.J.; Boztug, K.; Paruzynski, A.; Witzel, M.; Schwarzer, A.; Rothe, M.; Modlich, U.; Beier, R.; Gohring, G.; Steinemann, D.; et al. Gene Therapy for Wiskott-Aldrich Syndrome—Long-Term Efficacy and Genotoxicity. Sci. Transl. Med. 2014, 6, 227ra233. [Google Scholar] [CrossRef] [PubMed]
- Ferrua, F.; Cicalese, M.P.; Galimberti, S.; Giannelli, S.; Dionisio, F.; Barzaghi, F.; Migliavacca, M.; Bernardo, M.E.; Calbi, V.; Assanelli, A.A.; et al. Lentiviral haemopoietic stem/progenitor cell gene therapy for treatment of Wiskott-Aldrich syndrome: Interim results of a non-randomised, open-label, phase 1/2 clinical study. Lancet Haematol. 2019, 6, e239–e253. [Google Scholar] [CrossRef] [Green Version]
- Brendel, C.; Rothe, M.; Santilli, G.; Charrier, S.; Stein, S.; Kunkel, H.; Abriss, D.; Müller-Kuller, U.; Gaspar, B.; Modlich, U.; et al. Non-Clinical Efficacy and Safety Studies on G1XCGD, a Lentiviral Vector for Ex Vivo Gene Therapy of X-Linked Chronic Granulomatous Disease. Hum. Gene Ther. Clin. Dev. 2018, 29, 69–79. [Google Scholar] [CrossRef]
- Kohn, D.B.; Booth, C.; Kang, E.M.; Pai, S.-Y.; Shaw, K.L.; Santilli, G.; Armant, M.; Buckland, K.F.; Choi, U.; De Ravin, S.S.; et al. Lentiviral gene therapy for X-linked chronic granulomatous disease. Nat. Med. 2020, 26, 200–206. [Google Scholar] [CrossRef]
- Friedmann, T.; Roblin, R. Gene Therapy for Human Genetic Disease? Science 1972, 175, 949–955. [Google Scholar] [CrossRef]
- Lim, B.; Apperley, J.F.; Orkin, S.H.; Williams, D.A. Long-term expression of human adenosine deaminase in mice transplanted with retrovirus-infected hematopoietic stem cells. Proc. Natl. Acad. Sci. USA 1989, 86, 8892–8896. [Google Scholar] [CrossRef] [Green Version]
- Bortin, M.M. Severe Combined Immunodeficiency Disease. JAMA 1977, 238, 591. [Google Scholar] [CrossRef]
- Grunebaum, E. Bone Marrow Transplantation for Severe Combined Immune Deficiency. JAMA 2006, 295, 508. [Google Scholar] [CrossRef] [Green Version]
- Gennery, A.R.; Slatter, M.A.; Grandin, L.; Taupin, P.; Cant, A.J.; Veys, P.; Amrolia, P.J.; Gaspar, H.B.; Davies, E.G.; Friedrich, W.; et al. Transplantation of hematopoietic stem cells and long-term survival for primary immunodeficiencies in Europe: Entering a new century, do we do better? J. Allergy Clin. Immunol. 2010, 126, 602–610.e611. [Google Scholar] [CrossRef] [PubMed]
- Haddad, E.; Logan, B.R.; Griffith, L.M.; Buckley, R.H.; Parrott, R.E.; Prockop, S.E.; Small, T.N.; Chaisson, J.; Dvorak, C.C.; Murnane, M.; et al. SCID genotype and 6-month post-transplant CD4 count predict survival and immune recovery: A PIDTC retrospective study. Blood 2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shamriz, O.; Chandrakasan, S. Update on Advances in Hematopoietic Cell Transplantation for Primary Immunodeficiency Disorders. Immunol. Allergy Clin. N. Am. 2019, 39, 113–128. [Google Scholar] [CrossRef] [PubMed]
- Castagnoli, R.; Delmonte, O.M.; Calzoni, E.; Notarangelo, L.D. Hematopoietic Stem Cell Transplantation in Primary Immunodeficiency Diseases: Current Status and Future Perspectives. Front. Pediatr. 2019, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gennery, A.R.; Lankester, A. Long Term Outcome and Immune Function after Hematopoietic Stem Cell Transplantation for Primary Immunodeficiency. Front. Pediatr. 2019, 7. [Google Scholar] [CrossRef] [PubMed]
- Touzot, F.; Moshous, D.; Creidy, R.; Neven, B.; Frange, P.; Cros, G.; Caccavelli, L.; Blondeau, J.; Magnani, A.; Luby, J.-M.; et al. Faster T-cell development following gene therapy compared with haploidentical HSCT in the treatment of SCID-X1. Blood 2015, 125, 3563–3569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aiuti, A. Correction of ADA-SCID by Stem Cell Gene Therapy Combined with Nonmyeloablative Conditioning. Science 2002, 296, 2410–2413. [Google Scholar] [CrossRef] [Green Version]
- Aiuti, A.; Cattaneo, F.; Galimberti, S.; Benninghoff, U.; Cassani, B.; Callegaro, L.; Scaramuzza, S.; Andolfi, G.; Mirolo, M.; Brigida, I.; et al. Gene Therapy for Immunodeficiency Due to Adenosine Deaminase Deficiency. N. Engl. J. Med. 2009, 360, 447–458. [Google Scholar] [CrossRef] [Green Version]
- Cassani, B.; Montini, E.; Maruggi, G.; Ambrosi, A.; Mirolo, M.; Selleri, S.; Biral, E.; Frugnoli, I.; Hernandez-Trujillo, V.; Di Serio, C.; et al. Integration of retroviral vectors induces minor changes in the transcriptional activity of T cells from ADA-SCID patients treated with gene therapy. Blood 2009, 114, 3546–3556. [Google Scholar] [CrossRef] [Green Version]
- Gaspar, H.B.; Cooray, S.; Gilmour, K.C.; Parsley, K.L.; Zhang, F.; Adams, S.; Bjorkegren, E.; Bayford, J.; Brown, L.; Davies, E.G.; et al. Hematopoietic Stem Cell Gene Therapy for Adenosine Deaminase-Deficient Severe Combined Immunodeficiency Leads to Long-Term Immunological Recovery and Metabolic Correction. Sci. Transl. Med. 2011, 3, 97ra80. [Google Scholar] [CrossRef]
- Candotti, F.; Shaw, K.L.; Muul, L.; Carbonaro, D.; Sokolic, R.; Choi, C.; Schurman, S.H.; Garabedian, E.; Kesserwan, C.; Jagadeesh, G.J.; et al. Gene therapy for adenosine deaminase-deficient severe combined immune deficiency: Clinical comparison of retroviral vectors and treatment plans. Blood 2012, 120, 3635–3646. [Google Scholar] [CrossRef] [PubMed]
- Cicalese, M.P.; Ferrua, F.; Castagnaro, L.; Pajno, R.; Barzaghi, F.; Giannelli, S.; Dionisio, F.; Brigida, I.; Bonopane, M.; Casiraghi, M.; et al. Update on the safety and efficacy of retroviral gene therapy for immunodeficiency due to adenosine deaminase deficiency. Blood 2016, 128, 45–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aiuti, A.; Roncarolo, M.G.; Naldini, L. Gene therapy for ADA-SCID, the first marketing approval of an ex vivo gene therapy in Europe: Paving the road for the next generation of advanced therapy medicinal products. EMBO Mol. Med. 2017, 9, 737–740. [Google Scholar] [CrossRef]
- Shaw, K.L.; Garabedian, E.; Mishra, S.; Barman, P.; Davila, A.; Carbonaro, D.; Shupien, S.; Silvin, C.; Geiger, S.; Nowicki, B.; et al. Clinical efficacy of gene-modified stem cells in adenosine deaminase–deficient immunodeficiency. J. Clin. Investig. 2017, 127, 1689–1699. [Google Scholar] [CrossRef] [PubMed]
- Kohn, D.B.; Hershfield, M.S.; Puck, J.M.; Aiuti, A.; Blincoe, A.; Gaspar, H.B.; Notarangelo, L.D.; Grunebaum, E. Consensus approach for the management of severe combined immune deficiency caused by adenosine deaminase deficiency. J. Allergy Clin. Immunol. 2019, 143, 852–863. [Google Scholar] [CrossRef] [PubMed]
- Carriglio, N.; Klapwijk, J.; Hernandez, R.J.; Vezzoli, M.; Chanut, F.; Lowe, R.; Draghici, E.; Nord, M.; Albertini, P.; Cristofori, P.; et al. Good Laboratory Practice Preclinical Safety Studies for GSK2696273 (MLV Vector-Based Ex Vivo Gene Therapy for Adenosine Deaminase Deficiency Severe Combined Immunodeficiency) in NSG Mice. Hum. Gene Ther. Clin. Dev. 2017, 28, 17–27. [Google Scholar] [CrossRef]
- Kohn, D.B.; Gaspar, H.B. How We Manage Adenosine Deaminase-Deficient Severe Combined Immune Deficiency (ADA SCID). J. Clin. Immunol. 2017, 37, 351–356. [Google Scholar] [CrossRef]
- Kohn, D.; Shaw, K.; Garabedian, E.; Carbonaro-Sarracino, D.; Moore, T.; De Oliveira, S.; Crooks, G.; Tse, J.; Shupien, S.; Terrazas, D.; et al. Lentiviral Gene Therapy with Autologous Hematopoietic Stem and Progenitor Cells (HSPCs) for the Treatment of Severe Combined Immune Deficiency Due to Adenosine Deaminase Deficiency (ADA-SCID): Results in an Expanded Cohort. Blood 2019, 134, 3345. [Google Scholar] [CrossRef]
- Cowan, M.J.; Yu, J.; Facchino, J.; Chag, S.; Fraser-Browne, C.; Long-Boyle, J.; Kawahara, M.; Sanford, U.; Oh, J.; Teoh, S.; et al. Early Outcome of a Phase I/II Clinical Trial (NCT03538899) of Gene-Corrected Autologous CD34+ Hematopoietic Cells and Low-Exposure Busulfan in Newly Diagnosed Patients with Artemis-Deficient Severe Combined Immunodeficiency (ART-SCID). Biol. Blood Marrow Transplant. 2020, 26, S88–S89. [Google Scholar] [CrossRef]
- Ott, M.G.; Schmidt, M.; Schwarzwaelder, K.; Stein, S.; Siler, U.; Koehl, U.; Glimm, H.; Kühlcke, K.; Schilz, A.; Kunkel, H.; et al. Correction of X-linked chronic granulomatous disease by gene therapy, augmented by insertional activation of MDS1-EVI1, PRDM16 or SETBP1. Nat. Med. 2006, 12, 401–409. [Google Scholar] [CrossRef]
- Kang, H.J.; Bartholomae, C.C.; Paruzynski, A.; Arens, A.; Kim, S.; Yu, S.S.; Hong, Y.; Joo, C.-W.; Yoon, N.-K.; Rhim, J.-W.; et al. Retroviral Gene Therapy for X-linked Chronic Granulomatous Disease: Results from Phase I/II Trial. Mol. Ther. 2011, 19, 2092–2101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hacein-Bey Abina, S.; Gaspar, H.B.; Blondeau, J.; Caccavelli, L.; Charrier, S.; Buckland, K.; Picard, C.; Six, E.; Himoudi, N.; Gilmour, K.; et al. Outcomes Following Gene Therapy in Patients with Severe Wiskott-Aldrich Syndrome. JAMA 2015, 313, 1550. [Google Scholar] [CrossRef] [PubMed]
- Morris, E.C.; Fox, T.; Chakraverty, R.; Tendeiro, R.; Snell, K.; Rivat, C.; Grace, S.; Gilmour, K.; Workman, S.; Buckland, K.; et al. Gene therapy for Wiskott-Aldrich syndrome in a severely affected adult. Blood 2017, 130, 1327–1335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hacein-Bey-Abina, S.; Le Deist, F.; Carlier, F.; Bouneaud, C.; Hue, C.; De Villartay, J.-P.; Thrasher, A.J.; Wulffraat, N.; Sorensen, R.; Dupuis-Girod, S.; et al. Sustained Correction of X-Linked Severe Combined Immunodeficiency by ex Vivo Gene Therapy. N. Engl. J. Med. 2002, 346, 1185–1193. [Google Scholar] [CrossRef]
- Hacein-Bey-Abina, S.; Hauer, J.; Lim, A.; Picard, C.; Wang, G.P.; Berry, C.C.; Martinache, C.; Rieux-Laucat, F.; Latour, S.; Belohradsky, B.H.; et al. Efficacy of Gene Therapy for X-Linked Severe Combined Immunodeficiency. N. Engl. J. Med. 2010, 363, 355–364. [Google Scholar] [CrossRef] [Green Version]
- Hacein-Bey-Abina, S.; Pai, S.-Y.; Gaspar, H.B.; Armant, M.; Berry, C.C.; Blanche, S.; Bleesing, J.; Blondeau, J.; De Boer, H.; Buckland, K.F.; et al. A Modified γ-Retrovirus Vector for X-Linked Severe Combined Immunodeficiency. N. Engl. J. Med. 2014, 371, 1407–1417. [Google Scholar] [CrossRef] [Green Version]
- Clarke, E.L.; Connell, A.J.; Six, E.; Kadry, N.A.; Abbas, A.A.; Hwang, Y.; Everett, J.K.; Hofstaedter, C.E.; Marsh, R.; Armant, M.; et al. T cell dynamics and response of the microbiota after gene therapy to treat X-linked severe combined immunodeficiency. Genome Med. 2018, 10. [Google Scholar] [CrossRef] [Green Version]
- Mamcarz, E.; Zhou, S.; Lockey, T.; Abdelsamed, H.; Cross, S.J.; Kang, G.; Ma, Z.; Condori, J.; Dowdy, J.; Triplett, B.; et al. Lentiviral Gene Therapy Combined with Low-Dose Busulfan in Infants with SCID-X1. N. Engl. J. Med. 2019, 380, 1525–1534. [Google Scholar] [CrossRef]
- US Department of Health and Human Services. Human Gene Therapy for Rare Diseases: Guidance for Industry; Office of Medical Products and Tobacco, Center for Biologics Evaluation and Research: Silver Spring, MD, USA, 2020.
- EMA. Guideline on the Non-Clinical Studies Required before First Clinical Use of Gene Therapy Medicinal Products; European Medicines Agency (EMA): London, UK, 2008.
- Pike-Overzet, K.; Rodijk, M.; Ng, Y.Y.; Baert, M.R.; Lagresle-Peyrou, C.; Schambach, A.; Zhang, F.; Hoeben, R.C.; Hacein-Bey-Abina, S.; Lankester, A.C.; et al. Correction of murine Rag1 deficiency by self-inactivating lentiviral vector-mediated gene transfer. Leukemia 2011, 25, 1471–1483. [Google Scholar] [CrossRef]
- Pike-Overzet, K.; Baum, C.; Bredius, R.G.M.; Cavazzana, M.; Driessen, G.-J.; Fibbe, W.E.; Gaspar, H.B.; Hoeben, R.C.; Lagresle-Peyrou, C.; Lankester, A.; et al. Successful RAG1-SCID gene therapy depends on the level of RAG1 expression. J. Allergy Clin. Immunol. 2014, 134, 242–243. [Google Scholar] [CrossRef]
- Garcia-Perez, L.; van Eggermond, M.; van Roon, L.; Vloemans, S.A.; Cordes, M.; Schambach, A.; Rothe, M.; Berghuis, D.; Lagresle-Peyrou, C.; Cavazzana, M.; et al. Successful Preclinical Development of Gene Therapy for Recombinase-Activating Gene-1-Deficient SCID. Mol. Ther. Methods Clin. Dev. 2020, 17, 666–682. [Google Scholar] [CrossRef] [PubMed]
- Becker, A.J.; McCulloch, E.A.; Till, J.E. Cytological Demonstration of the Clonal Nature of Spleen Colonies Derived from Transplanted Mouse Marrow Cells. Nature 1963, 197, 452–454. [Google Scholar] [CrossRef] [PubMed]
- Jordan, C.T.; Lemischka, I.R. Clonal and systemic analysis of long-term hematopoiesis in the mouse. Genome Res. 1990, 4, 220–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrison, S.J.; Weissman, I.L. The long-term repopulating subset of hematopoietic stem cells is deterministic and isolatable by phenotype. Immunity 1994, 1, 661–673. [Google Scholar] [CrossRef]
- Morrison, S.J.; Wandycz, A.M.; Hemmati, H.D.; Wright, D.E.; Weissman, I.L. Identification of a lineage of multipotent hematopoietic progenitors. Development 1997, 124, 1929–1939. [Google Scholar]
- Uchida, N. Searching for hematopoietic stem cells: Evidence that Thy-1.1lo Lin- Sca-1+ cells are the only stem cells in C57BL/Ka-Thy-1.1 bone marrow. J. Exp. Med. 1992, 175, 175–184. [Google Scholar] [CrossRef] [Green Version]
- Uchida, N.; Fleming, W.H.; Alpern, E.J.; Weissman, I.L. Heterogeneity of hematopoietic stem cells. Curr. Opin. Immunol. 1993, 5, 177–184. [Google Scholar] [CrossRef]
- Baum, C.M.; Weissman, I.L.; Tsukamoto, A.S.; Buckle, A.M.; Peault, B. Isolation of a candidate human hematopoietic stem-cell population. Proc. Natl. Acad. Sci. USA 1992, 89, 2804–2808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhatia, M.; Bonnet, D.; Kapp, U.; Wang, J.C.Y.; Murdoch, B.; Dick, J.E. Quantitative Analysis Reveals Expansion of Human Hematopoietic Repopulating Cells after Short-term Ex Vivo Culture. J. Exp. Med. 1997, 186, 619–624. [Google Scholar] [CrossRef]
- Majeti, R.; Park, C.Y.; Weissman, I.L. Identification of a Hierarchy of Multipotent Hematopoietic Progenitors in Human Cord Blood. Cell Stem Cell 2007, 1, 635–645. [Google Scholar] [CrossRef] [Green Version]
- Notta, F.; Doulatov, S.; Laurenti, E.; Poeppl, A.; Jurisica, I.; Dick, J.E. Isolation of Single Human Hematopoietic Stem Cells Capable of Long-Term Multilineage Engraftment. Science 2011, 333, 218–221. [Google Scholar] [CrossRef] [PubMed]
- Fares, I.; Chagraoui, J.; Lehnertz, B.; Macrae, T.; Mayotte, N.; Tomellini, E.; Aubert, L.; Roux, P.P.; Sauvageau, G. EPCR expression marks UM171-expanded CD34+ cord blood stem cells. Blood 2017, 129, 3344–3351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgan, R.A.; Gray, D.; Lomova, A.; Kohn, D.B. Hematopoietic Stem Cell Gene Therapy: Progress and Lessons Learned. Cell Stem Cell 2017, 21, 574–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soni, S.; Kohn, D.B. Chemistry, manufacturing and controls for gene modified hematopoietic stem cells. Cytotherapy 2019, 21, 358–366. [Google Scholar] [CrossRef] [PubMed]
- Brave, M.; Farrell, A.; Ching Lin, S.; Ocheltree, T.; Pope Miksinski, S.; Lee, S.-L.; Saber, H.; Fourie, J.; Tornoe, C.; Booth, B.; et al. FDA Review Summary: Mozobil in Combination with Granulocyte Colony-Stimulating Factor to Mobilize Hematopoietic Stem Cells to the Peripheral Blood for Collection and Subsequent Autologous Transplantation. Oncology 2010, 78, 282–288. [Google Scholar] [CrossRef] [PubMed]
- Tajer, P.; Pike-Overzet, K.; Arias, S.; Havenga, M.; Staal, F. Ex Vivo Expansion of Hematopoietic Stem Cells for Therapeutic Purposes: Lessons from Development and the Niche. Cells 2019, 8, 169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sauvageau, G.; Iscove, N.N.; Humphries, R.K. In vitro and in vivo expansion of hematopoietic stem cells. Oncogene 2004, 23, 7223–7232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofmeister, C.C.; Zhang, J.; Knight, K.L.; Le, P.; Stiff, P.J. Ex vivo expansion of umbilical cord blood stem cells for transplantation: Growing knowledge from the hematopoietic niche. Bone Marrow Transplant. 2007, 39, 11–23. [Google Scholar] [CrossRef] [Green Version]
- Zonari, E.; Desantis, G.; Petrillo, C.; Boccalatte, F.E.; Lidonnici, M.R.; Kajaste-Rudnitski, A.; Aiuti, A.; Ferrari, G.; Naldini, L.; Gentner, B. Efficient Ex Vivo Engineering and Expansion of Highly Purified Human Hematopoietic Stem and Progenitor Cell Populations for Gene Therapy. Stem Cell Rep. 2017. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Geiger, H. HSC Niche Biology and HSC Expansion Ex Vivo. Trends Mol. Med. 2017, 23, 799–819. [Google Scholar] [CrossRef]
- Masiuk, K.E.; Brown, D.; Laborada, J.; Hollis, R.P.; Urbinati, F.; Kohn, D.B. Improving Gene Therapy Efficiency through the Enrichment of Human Hematopoietic Stem Cells. Mol. Ther. 2017, 25, 2163–2175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baldwin, K.; Urbinati, F.; Romero, Z.; Campo-Fernandez, B.; Kaufman, M.L.; Cooper, A.R.; Masiuk, K.; Hollis, R.P.; Kohn, D.B. Enrichment of Human Hematopoietic Stem/Progenitor Cells Facilitates Transduction for Stem Cell Gene Therapy. Stem Cells 2015, 33, 1532–1542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scala, S.; Basso-Ricci, L.; Dionisio, F.; Pellin, D.; Giannelli, S.; Salerio, F.A.; Leonardelli, L.; Cicalese, M.P.; Ferrua, F.; Aiuti, A.; et al. Dynamics of genetically engineered hematopoietic stem and progenitor cells after autologous transplantation in humans. Nat. Med. 2018, 24, 1683–1690. [Google Scholar] [CrossRef] [PubMed]
- Athanasopoulos, T.; Munye, M.M.; Yáñez-Muñoz, R.J. Nonintegrating Gene Therapy Vectors. Hematol. Oncol. Clin. N. Am. 2017, 31, 753–770. [Google Scholar] [CrossRef] [PubMed]
- Biasco, L.; Rothe, M.; Schott, J.W.; Schambach, A. Integrating Vectors for Gene Therapy and Clonal Tracking of Engineered Hematopoiesis. Hematol. Oncol. Clin. N. Am. 2017, 31, 737–752. [Google Scholar] [CrossRef]
- Hacein-Bey-Abina, S. LMO2-Associated Clonal T Cell Proliferation in Two Patients after Gene Therapy for SCID-X1. Science 2003, 302, 415–419. [Google Scholar] [CrossRef]
- Hacein-Bey-Abina, S.; Garrigue, A.; Wang, G.P.; Soulier, J.; Lim, A.; Morillon, E.; Clappier, E.; Caccavelli, L.; Delabesse, E.; Beldjord, K.; et al. Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J. Clin. Investig. 2008, 118, 3132–3142. [Google Scholar] [CrossRef]
- Howe, S.J.; Mansour, M.R.; Schwarzwaelder, K.; Bartholomae, C.; Hubank, M.; Kempski, H.; Brugman, M.H.; Pike-Overzet, K.; Chatters, S.J.; De Ridder, D.; et al. Insertional mutagenesis combined with acquired somatic mutations causes leukemogenesis following gene therapy of SCID-X1 patients. J. Clin. Investig. 2008, 118, 3143–3150. [Google Scholar] [CrossRef]
- Kotterman, M.A.; Chalberg, T.W.; Schaffer, D.V. Viral Vectors for Gene Therapy: Translational and Clinical Outlook. Annu. Rev. Biomed. Eng. 2015, 17, 63–89. [Google Scholar] [CrossRef] [Green Version]
- Naldini, L.; Blomer, U.; Gallay, P.; Ory, D.; Mulligan, R.; Gage, F.H.; Verma, I.M.; Trono, D. In Vivo Gene Delivery and Stable Transduction of Nondividing Cells by a Lentiviral Vector. Science 1996, 272, 263–267. [Google Scholar] [CrossRef] [Green Version]
- Dull, T.; Zufferey, R.; Kelly, M.; Mandel, R.J.; Nguyen, M.; Trono, D.; Naldini, L. A Third-Generation Lentivirus Vector with a Conditional Packaging System. J. Virol. 1998, 72, 8463–8471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montini, E.; Cesana, D.; Schmidt, M.; Sanvito, F.; Bartholomae, C.C.; Ranzani, M.; Benedicenti, F.; Sergi, L.S.; Ambrosi, A.; Ponzoni, M.; et al. The genotoxic potential of retroviral vectors is strongly modulated by vector design and integration site selection in a mouse model of HSC gene therapy. J. Clin. Investig. 2009, 119, 964–975. [Google Scholar] [CrossRef] [PubMed]
- Schambach, A.; Galla, M.; Maetzig, T.; Loew, R.; Baum, C. Improving Transcriptional Termination of Self-inactivating Gamma-retroviral and Lentiviral Vectors. Mol. Ther. 2007, 15, 1167–1173. [Google Scholar] [CrossRef] [PubMed]
- Schambach, A.; Bohne, J.; Baum, C.; Hermann, F.G.; Egerer, L.; Von Laer, D.; Giroglou, T. Woodchuck hepatitis virus post-transcriptional regulatory element deleted from X protein and promoter sequences enhances retroviral vector titer and expression. Gene Ther. 2006, 13, 641–645. [Google Scholar] [CrossRef] [Green Version]
- Montini, E.; Cesana, D.; Schmidt, M.; Sanvito, F.; Ponzoni, M.; Bartholomae, C.; Sergi, L.S.; Benedicenti, F.; Ambrosi, A.; Di Serio, C.; et al. Hematopoietic stem cell gene transfer in a tumor-prone mouse model uncovers low genotoxicity of lentiviral vector integration. Nat. Biotechnol. 2006, 24, 687–696. [Google Scholar] [CrossRef]
- Modlich, U.; Navarro, S.; Zychlinski, D.; Maetzig, T.; Knoess, S.; Brugman, M.H.; Schambach, A.; Charrier, S.; Galy, A.; Thrasher, A.J.; et al. Insertional Transformation of Hematopoietic Cells by Self-inactivating Lentiviral and Gammaretroviral Vectors. Mol. Ther. 2009, 17, 1919–1928. [Google Scholar] [CrossRef]
- Ginn, S.L.; Amaya, A.K.; Alexander, I.E.; Edelstein, M.; Abedi, M.R. Gene therapy clinical trials worldwide to 2017: An update. J. Gene Med. 2018, 20, e3015. [Google Scholar] [CrossRef]
- Sastry, L.; Johnson, T.; Hobson, M.J.; Smucker, B.; Cornetta, K. Titering lentiviral vectors: Comparison of DNA, RNA and marker expression methods. Gene Ther. 2002, 9, 1155–1162. [Google Scholar] [CrossRef] [Green Version]
- Sena-Esteves, M.; Gao, G. Titration of Lentivirus Vectors. Cold Spring Harb. Protoc. 2018, 2018, 095695. [Google Scholar] [CrossRef]
- Heffner, G.C.; Bonner, M.; Christiansen, L.; Pierciey, F.J.; Campbell, D.; Smurnyy, Y.; Zhang, W.; Hamel, A.; Shaw, S.; Lewis, G.; et al. Prostaglandin E2 Increases Lentiviral Vector Transduction Efficiency of Adult Human Hematopoietic Stem and Progenitor Cells. Mol. Ther. 2018, 26, 320–328. [Google Scholar] [CrossRef] [Green Version]
- Lewis, G.; Christiansen, L.; McKenzie, J.; Luo, M.; Pasackow, E.; Smurnyy, Y.; Harrington, S.; Gregory, P.; Veres, G.; Negre, O.; et al. Staurosporine Increases Lentiviral Vector Transduction Efficiency of Human Hematopoietic Stem and Progenitor Cells. Mol. Ther. Methods Clin. Dev. 2018, 9, 313–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrillo, C.; Cesana, D.; Piras, F.; Bartolaccini, S.; Naldini, L.; Montini, E.; Kajaste-Rudnitski, A. Cyclosporin A and Rapamycin Relieve Distinct Lentiviral Restriction Blocks in Hematopoietic Stem and Progenitor Cells. Mol. Ther. 2015, 23, 352–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schott, J.W.; León-Rico, D.; Ferreira, C.B.; Buckland, K.F.; Santilli, G.; Armant, M.A.; Schambach, A.; Cavazza, A.; Thrasher, A.J. Enhancing Lentiviral and Alpharetroviral Transduction of Human Hematopoietic Stem Cells for Clinical Application. Mol. Ther. Methods Clin. Dev. 2019, 14, 134–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, H.-T.; Okumura, T.; Yatsuda, Y.; Ito, S.; Nakauchi, H.; Otsu, M. Application of Droplet Digital PCR for Estimating Vector Copy Number States in Stem Cell Gene Therapy. Hum. Gene Ther. Methods 2016, 27, 197–208. [Google Scholar] [CrossRef] [Green Version]
- Manoj, P. Droplet digital PCR technology promises new applications and research areas. Mitochondrial DNA 2016, 27, 742–746. [Google Scholar] [CrossRef]
- Taylor, S.C.; Laperriere, G.; Germain, H. Droplet Digital PCR versus qPCR for gene expression analysis with low abundant targets: From variable nonsense to publication quality data. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [Green Version]
- Scherr, M.; Battmer, K.; Blömer, U.; Ganser, A.; Grez, M. Quantitative Determination of Lentiviral Vector Particle Numbers by Real-Time PCR. Biotechniques 2001, 31, 520–526. [Google Scholar] [CrossRef] [Green Version]
- Lana, M.G.; Strauss, B.E. Production of Lentivirus for the Establishment of CAR-T Cells; Springer: New York, NY, USA, 2020; pp. 61–67. [Google Scholar]
- Delville, M.; Soheili, T.; Bellier, F.; Durand, A.; Denis, A.; Lagresle-Peyrou, C.; Cavazzana, M.; Andre-Schmutz, I.; Six, E. A Nontoxic Transduction Enhancer Enables Highly Efficient Lentiviral Transduction of Primary Murine T Cells and Hematopoietic Stem Cells. Mol. Ther. Methods Clin. Dev. 2018, 10, 341–347. [Google Scholar] [CrossRef] [Green Version]
- Perlman, R.L. Mouse Models of Human Disease: An Evolutionary Perspective. Evol. Med. Public Health 2016, eow014. [Google Scholar] [CrossRef] [Green Version]
- Wolfe, J.H. Gene Therapy in Large Animal Models of Human Genetic Diseases. ILAR J. 2009, 50, 107–111. [Google Scholar] [CrossRef] [Green Version]
- Vite, C.H.; McGowan, J.C.; Niogi, S.N.; Passini, M.A.; Drobatz, K.J.; Haskins, M.E.; Wolfe, J.H. Effective gene therapy for an inherited CNS disease in a large animal model. Ann. Neurol. 2005, 57, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.-H.; Cheng, P.-H.; Banta, H.; Piotrowska-Nitsche, K.; Yang, J.-J.; Cheng, E.C.H.; Snyder, B.; Larkin, K.; Liu, J.; Orkin, J.; et al. Towards a transgenic model of Huntington’s disease in a non-human primate. Nature 2008, 453, 921–924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Chamberlain, J.S.; Tapscott, S.J.; Storb, R. Gene Therapy in Large Animal Models of Muscular Dystrophy. ILAR J. 2009, 50, 187–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, A.W.S.; Yang, S.-H. Generation of transgenic monkeys with human inherited genetic disease. Methods 2009, 49, 78–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagree, M.S.; Scalia, S.; McKillop, W.M.; Medin, J.A. An update on gene therapy for lysosomal storage disorders. Expert Opin. Biol. Ther. 2019, 19, 655–670. [Google Scholar] [CrossRef]
- Gagliardi, C.; Bunnell, B.A. Large Animal Models of Neurological Disorders for Gene Therapy. ILAR J. 2009, 50, 128–143. [Google Scholar] [CrossRef] [Green Version]
- Acland, G.M.; Aguirre, G.D.; Ray, J.; Zhang, Q.; Aleman, T.S.; Cideciyan, A.V.; Pearce-Kelling, S.E.; Anand, V.; Zeng, Y.; Maguire, A.M.; et al. Gene therapy restores vision in a canine model of childhood blindness. Nat. Genet. 2001, 28, 92–95. [Google Scholar] [CrossRef]
- Bennett, J. Gene therapy for Leber congenital amaurosis. Novartis. Found. Symp. 2004, 255, 195–202. [Google Scholar]
- Rogers, C.S.; Abraham, W.M.; Brogden, K.A.; Engelhardt, J.F.; Fisher, J.T.; McCray, P.B., Jr.; McLennan, G.; Meyerholz, D.K.; Namati, E.; Ostedgaard, L.S.; et al. The porcine lung as a potential model for cystic fibrosis. Am. J. Physiol. Lung Cell Mol. Physiol. 2008, 295, L240–L263. [Google Scholar] [CrossRef] [Green Version]
- Ito, T.; Sendai, Y.; Yamazaki, S.; Seki-Soma, M.; Hirose, K.; Watanabe, M.; Fukawa, K.; Nakauchi, H. Generation of Recombination Activating Gene-1-Deficient Neonatal Piglets: A Model of T and B Cell Deficient Severe Combined Immune Deficiency. PLoS ONE 2014, 9, e113833. [Google Scholar] [CrossRef]
- Suzuki, S.; Iwamoto, M.; Hashimoto, M.; Suzuki, M.; Nakai, M.; Fuchimoto, D.; Sembon, S.; Eguchi-Ogawa, T.; Uenishi, H.; Onishi, A. Generation and characterization of RAG2 knockout pigs as animal model for severe combined immunodeficiency. Vet. Immunol. Immunopathol. 2016, 178, 37–49. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, M.A.; Hong, K.; Kim, J.H.; Choi, Y. Severe combined immunodeficiency pig as an emerging animal model for human diseases and regenerative medicines. BMB Rep. 2019, 52, 625–634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blackburn, M.R.; Datta, S.K.; Kellems, R.E. Adenosine Deaminase-deficient Mice Generated Using a Two-stage Genetic Engineering Strategy Exhibit a Combined Immunodeficiency. J. Biol. Chem. 1998, 273, 5093–5100. [Google Scholar] [CrossRef] [Green Version]
- Carbonaro, D.; Jin, X.; Petersen, D.; Wang, X.; Dorey, F.; Kil, K.; Aldrich, M.; Blackburn, M.; Kellems, R.; Kohn, D. In Vivo Transduction by Intravenous Injection of a Lentiviral Vector Expressing Human ADA into Neonatal ADA Gene Knockout Mice: A Novel Form of Enzyme Replacement Therapy for ADA Deficiency. Mol. Ther. 2006, 13, 1110–1120. [Google Scholar] [CrossRef] [PubMed]
- Thornhill, S.I.; Schambach, A.; Howe, S.J.; Ulaganathan, M.; Grassman, E.; Williams, D.; Schiedlmeier, B.; Sebire, N.J.; Gaspar, H.B.; Kinnon, C.; et al. Self-inactivating Gammaretroviral Vectors for Gene Therapy of X-linked Severe Combined Immunodeficiency. Mol. Ther. 2008, 16, 590–598. [Google Scholar] [CrossRef]
- Mombaerts, P.; Iacomini, J.; Johnson, R.S.; Herrup, K.; Tonegawa, S.; Papaioannou, V.E. RAG-1-deficient mice have no mature B and T lymphocytes. Cell 1992, 68, 869–877. [Google Scholar] [CrossRef]
- Shinkai, Y. RAG-2-deficient mice lack mature lymphocytes owing to inability to initiate V(D)J rearrangement. Cell 1992, 68, 855–867. [Google Scholar] [CrossRef]
- Marrella, V.; Poliani, P.L.; Casati, A.; Rucci, F.; Frascoli, L.; Gougeon, M.-L.; Lemercier, B.; Bosticardo, M.; Ravanini, M.; Battaglia, M.; et al. A hypomorphic R229Q Rag2 mouse mutant recapitulates human Omenn syndrome. J. Clin. Investig. 2007, 117, 1260–1269. [Google Scholar] [CrossRef] [Green Version]
- Khiong, K.; Murakami, M.; Kitabayashi, C.; Ueda, N.; Sawa, S.-I.; Sakamoto, A.; Kotzin, B.L.; Rozzo, S.J.; Ishihara, K.; Verella-Garcia, M.; et al. Homeostatically proliferating CD4+ T cells are involved in the pathogenesis of an Omenn syndrome murine model. J. Clin. Investig. 2007, 117, 1270–1281. [Google Scholar] [CrossRef] [Green Version]
- Giblin, W.; Chatterji, M.; Westfield, G.; Masud, T.; Theisen, B.; Cheng, H.L.; Devido, J.; Alt, F.W.; Ferguson, D.O.; Schatz, D.G.; et al. Leaky severe combined immunodeficiency and aberrant DNA rearrangements due to a hypomorphic RAG1 mutation. Blood 2009, 113, 2965–2975. [Google Scholar] [CrossRef] [Green Version]
- Ott De Bruin, L.M.; Bosticardo, M.; Barbieri, A.; Lin, S.G.; Rowe, J.H.; Poliani, P.L.; Ching, K.; Eriksson, D.; Landegren, N.; Kämpe, O.; et al. HypomorphicRag1mutations alter the preimmune repertoire at early stages of lymphoid development. Blood 2018, 132, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Puel, A.; Leonard, W.J. Mutations in the gene for the IL-7 receptor result in T – B + NK + severe combined immunodeficiency disease. Curr. Opin. Immunol. 2000, 12, 468–473. [Google Scholar] [CrossRef]
- Shultz, L.D.; Lyons, B.L.; Burzenski, L.M.; Gott, B.; Chen, X.; Chaleff, S.; Kotb, M.; Gillies, S.D.; King, M.; Mangada, J.; et al. Human Lymphoid and Myeloid Cell Development in NOD/LtSz-scid IL2Rγnull Mice Engrafted with Mobilized Human Hemopoietic Stem Cells. J. Immunol. 2005, 174, 6477–6489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coughlan, A.M.; Harmon, C.; Whelan, S.; O′Brien, E.; O′Reilly, V.; Crotty, P.; Kelly, P.; Ryan, M.; Hickey, F.; O′Farrelly, C.; et al. Myeloid engraftment in humanized mice: Impact of GCSF treatment and transgenic mouse strain. Stem Cells Dev. 2016. [Google Scholar] [CrossRef] [PubMed]
- Wiekmeijer, A.-S.; Pike-Overzet, K.; Brugman, M.H.; Salvatori, D.C.F.; Egeler, R.M.; Bredius, R.G.M.; Fibbe, W.E.; Staal, F.J.T. Sustained Engraftment of Cryopreserved Human Bone Marrow CD34+ Cells in Young Adult NSG Mice. Biores. Open Access 2014, 3, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Walsh, N.C.; Kenney, L.L.; Jangalwe, S.; Aryee, K.-E.; Greiner, D.L.; Brehm, M.A.; Shultz, L.D. Humanized Mouse Models of Clinical Disease. Annu. Rev. Pathol. Mech. Dis. 2017, 12, 187–215. [Google Scholar] [CrossRef] [Green Version]
- Yong, K.S.M.; Her, Z.; Chen, Q. Humanized Mice as Unique Tools for Human-Specific Studies. Arch. Immunol. Ther. Exp. 2018, 66, 245–266. [Google Scholar] [CrossRef] [Green Version]
- Wiekmeijer, A.-S.; Pike-Overzet, K.; Ijspeert, H.; Brugman, M.H.; Wolvers-Tettero, I.L.M.; Lankester, A.C.; Bredius, R.G.M.; Van Dongen, J.J.M.; Fibbe, W.E.; Langerak, A.W.; et al. Identification of checkpoints in human T-cell development using severe combined immunodeficiency stem cells. J. Allergy Clin. Immunol. 2016, 137, 517–526. [Google Scholar] [CrossRef]
- Fenwick, N.; Griffin, G.; Gauthier, C. The welfare of animals used in science: How the “Three Rs“ ethic guides improvements. Can. Vet. J. 2009, 50, 523–530. [Google Scholar]
- Hare, K.J.; Jenkinson, E.J.; Anderson, G. In vitromodels of T cell development. Semin. Immunol. 1999, 11, 3–12. [Google Scholar] [CrossRef]
- Jaleco, A.C.; Neves, H.; Hooijberg, E.; Gameiro, P.; Clode, N.; Haury, M.; Henrique, D.; Parreira, L. Differential Effects of Notch Ligands Delta-1 and Jagged-1 in Human Lymphoid Differentiation. J. Exp. Med. 2001, 194, 991–1002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- La Motte-Mohs, R.N. Induction of T-cell development from human cord blood hematopoietic stem cells by Delta-like 1 in vitro. Blood 2005, 105, 1431–1439. [Google Scholar] [CrossRef] [PubMed]
- Six, E.M.; Benjelloun, F.; Garrigue, A.; Bonhomme, D.; Morillon, E.; Rouiller, J.; Cacavelli, L.; Blondeau, J.; Beldjord, K.; Hacein-Bey-Abina, S.; et al. Cytokines and culture medium have a major impact on human in vitro T-cell differentiation. Blood Cells Mol. Dis. 2011, 47, 72–78. [Google Scholar] [CrossRef] [PubMed]
- De Pooter, R.; Zúñiga-Pflücker, J.C. T-cell potential and development in vitro: The OP9-DL1 approach. Curr. Opin. Immunol. 2007, 19, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Seet, C.S.; He, C.; Bethune, M.T.; Li, S.; Chick, B.; Gschweng, E.H.; Zhu, Y.; Kim, K.; Kohn, D.B.; Baltimore, D.; et al. Generation of mature T cells from human hematopoietic stem and progenitor cells in artificial thymic organoids. Nat. Methods 2017, 14, 521–530. [Google Scholar] [CrossRef]
- Miller, P.G.; Shuler, M.L. Design and demonstration of a pumpless 14 compartment microphysiological system. Biotechnol. Bioeng. 2016, 113, 2213–2227. [Google Scholar] [CrossRef]
- De Mello, C.P.P.; Rumsey, J.; Slaughter, V.; Hickman, J.J. A human-on-a-chip approach to tackling rare diseases. Drug Discov. Today 2019, 24, 2139–2151. [Google Scholar] [CrossRef]
- Chhatta, A.R.; Cordes, M.; Hanegraaf, M.A.J.; Vloemans, S.; Cupedo, T.; Cornelissen, J.J.; Carlotti, F.; Salvatori, D.; Pike-Overzet, K.; Fibbe, W.E.; et al. De novo generation of a functional human thymus from induced pluripotent stem cells. J. Allergy Clin. Immunol. 2019, 144, 1416–1419. [Google Scholar] [CrossRef] [Green Version]
- Baum, C.; Modlich, U.; Göhring, G.; Schlegelberger, B. Concise Review: Managing Genotoxicity in the Therapeutic Modification of Stem Cells. Stem Cells 2011, 29, 1479–1484. [Google Scholar] [CrossRef]
- Biasco, L.; Rothe, M.; Büning, H.; Schambach, A. Analyzing the Genotoxicity of Retroviral Vectors in Hematopoietic Cell Gene Therapy. Mol. Ther. Methods Clin. Dev. 2018, 8, 21–30. [Google Scholar] [CrossRef] [Green Version]
- WHO. WHO Guidelines on Nonclinical Evaluation of Vaccines; WHO, Ed.; Technical Report Series; World Health Organization: Geneva, Switzerland, 2005; pp. 31–63. [Google Scholar]
- Schmidt, M.; Hoffmann, G.; Wissler, M.; Lemke, N.; Müßig, A.; Glimm, H.; Williams, D.A.; Ragg, S.; Hesemann, C.-U.; Von Kalle, C. Detection and Direct Genomic Sequencing of Multiple Rare Unknown Flanking DNA in Highly Complex Samples. Hum. Gene Ther. 2001, 12, 743–749. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.; Schwarzwaelder, K.; Bartholomae, C.; Zaoui, K.; Ball, C.; Pilz, I.; Braun, S.; Glimm, H.; Von Kalle, C. High-resolution insertion-site analysis by linear amplification–mediated PCR (LAM-PCR). Nat. Methods 2007, 4, 1051–1057. [Google Scholar] [CrossRef] [PubMed]
- Gabriel, R.; Eckenberg, R.; Paruzynski, A.; Bartholomae, C.C.; Nowrouzi, A.; Arens, A.; Howe, S.J.; Recchia, A.; Cattoglio, C.; Wang, W.; et al. Comprehensive genomic access to vector integration in clinical gene therapy. Nat. Med. 2009, 15, 1431–1436. [Google Scholar] [CrossRef]
- Cesana, D.; Ranzani, M.; Volpin, M.; Bartholomae, C.; Duros, C.; Artus, A.; Merella, S.; Benedicenti, F.; Sergi Sergi, L.; Sanvito, F.; et al. Uncovering and Dissecting the Genotoxicity of Self-inactivating Lentiviral Vectors In Vivo. Mol. Ther. 2014, 22, 774–785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Modlich, U.; Bohne, J.; Schmidt, M.; Von Kalle, C.; Knoss, S.; Schambach, A.; Baum, C. Cell-culture assays reveal the importance of retroviral vector design for insertional genotoxicity. Blood 2006, 108, 2545–2553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, Y.; Jenkins, N.A.; Copeland, N.G. Insertional mutagenesis identifies genes that promote the immortalization of primary bone marrow progenitor cells. Blood 2005, 106, 3932–3939. [Google Scholar] [CrossRef] [Green Version]
- Schwarzer, A.; Talbot, S.R.; Dittrich-Breiholz, O.; Thrasher, A.J.; Gaspar, B.; Modlich, U.; Baum, C.; Schambach, A.; Rothe, M. New Molecular Surrogate Assay for Genotoxicity Assessment of Gene Therapy Vectors (SAGA). Blood 2016, 128, 4710. [Google Scholar] [CrossRef]
- Modlich, U.; Surth, J.; Zychlinski, D.; Meyer, J.; Brendel, C.; Grez, M.; Arumugam, P.; Malik, P.; Grassman, E.; Schambach, A.; et al. Use of the In Vitro Immortalization Assay to Quantify the Impact of Integration Spectrum and Vector Design on Insertional Mutagenesis. Blood 2011, 118, 3123. [Google Scholar] [CrossRef]
- Porteus, M.H. A New Class of Medicines through DNA Editing. N. Engl. J. Med. 2019, 380, 947–959. [Google Scholar] [CrossRef]
- Schiroli, G.; Ferrari, S.; Conway, A.; Jacob, A.; Capo, V.; Albano, L.; Plati, T.; Castiello, M.C.; Sanvito, F.; Gennery, A.R.; et al. Preclinical modeling highlights the therapeutic potential of hematopoietic stem cell gene editing for correction of SCID-X1. Sci. Transl. Med. 2017, 9, eaan0820. [Google Scholar] [CrossRef]
- Pavel-Dinu, M.; Wiebking, V.; Dejene, B.T.; Srifa, W.; Mantri, S.; Nicolas, C.E.; Lee, C.; Bao, G.; Kildebeck, E.J.; Punjya, N.; et al. Gene correction for SCID-X1 in long-term hematopoietic stem cells. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gándara, C.; Affleck, V.; Stoll, E.A. Manufacture of Third-Generation Lentivirus for Preclinical Use, with Process Development Considerations for Translation to Good Manufacturing Practice. Hum. Gene Ther. Methods 2018, 29, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, H.J.; Leroux-Carlucci, M.A.; Sion, C.J.M.; Mitrophanous, K.A.; Radcliffe, P.A. Development of inducible EIAV-based lentiviral vector packaging and producer cell lines. Gene Ther. 2009, 16, 805–814. [Google Scholar] [CrossRef] [PubMed]
- Sanber, K.S.; Knight, S.B.; Stephen, S.L.; Bailey, R.; Escors, D.; Minshull, J.; Santilli, G.; Thrasher, A.J.; Collins, M.K.; Takeuchi, Y. Construction of stable packaging cell lines for clinical lentiviral vector production. Sci. Rep. 2015, 5, 9021. [Google Scholar] [CrossRef]
- Manceur, A.P.; Kim, H.; Misic, V.; Andreev, N.; Dorion-Thibaudeau, J.; Lanthier, S.; Bernier, A.; Tremblay, S.; Gélinas, A.-M.; Broussau, S.; et al. Scalable Lentiviral Vector Production Using Stable HEK293SF Producer Cell Lines. Hum. Gene Ther. Methods 2017, 28, 330–339. [Google Scholar] [CrossRef]
- Tomás, H.A.; Rodrigues, A.F.; Carrondo, M.J.T.; Coroadinha, A.S. LentiPro26: Novel stable cell lines for constitutive lentiviral vector production. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef] [Green Version]
- McCarron, A.; Donnelley, M.; McIntyre, C.; Parsons, D. Challenges of up-scaling lentivirus production and processing. J. Biotechnol. 2016, 240, 23–30. [Google Scholar] [CrossRef]
- Tapia, F.; Vogel, T.; Genzel, Y.; Behrendt, I.; Hirschel, M.; Gangemi, J.D.; Reichl, U. Production of high-titer human influenza A virus with adherent and suspension MDCK cells cultured in a single-use hollow fiber bioreactor. Vaccine 2014, 32, 1003–1011. [Google Scholar] [CrossRef]
- Wang, X.; Olszewska, M.; Qu, J.; Wasielewska, T.; Bartido, S.; Hermetet, G.; Sadelain, M.; Rivière, I. Large-scale clinical-grade retroviral vector production in a fixed-bed bioreactor. J. Immunother. 2015, 38, 127–135. [Google Scholar] [CrossRef]
- Scherr, M.; Battmer, K.; Eder, M.; Schüle, S.; Hohenberg, H.; Ganser, A.; Grez, M.; Blömer, U. Efficient gene transfer into the CNS by lentiviral vectors purified by anion exchange chromatography. Gene Ther. 2002, 9, 1708–1714. [Google Scholar] [CrossRef] [Green Version]
- Segura, M.M.; Garnier, A.; Durocher, Y.; Coelho, H.; Kamen, A. Production of lentiviral vectors by large-scale transient transfection of suspension cultures and affinity chromatography purification. Biotechnol. Bioeng. 2007, 98, 789–799. [Google Scholar] [CrossRef] [PubMed]
- Transfiguracion, J.; Jaalouk, D.E.; Ghani, K.; Galipeau, J.; Kamen, A.A. Size-Exclusion Chromatography Purification of High-Titer Vesicular Stomatitis Virus G Glycoprotein-Pseudotyped Retrovectors for Cell and Gene Therapy Applications. Hum. Gene Ther. 2003, 14, 1139–1153. [Google Scholar] [CrossRef] [PubMed]
- Geraerts, M.; Michiels, M.; Baekelandt, V.; Debyser, Z.; Gijsbers, R. Upscaling of lentiviral vector production by tangential flow filtration. J. Gene Med. 2005, 7, 1299–1310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blake, J.M.; Nicoud, I.B.; Weber, D.; Voorhies, H.; Guthrie, K.A.; Heimfeld, S.; Delaney, C. Improved immunomagnetic enrichment of CD34+ cells from umbilical cord blood using the CliniMACS cell separation system. Cytotherapy 2012, 14, 818–822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adair, J.E.; Waters, T.; Haworth, K.G.; Kubek, S.P.; Trobridge, G.D.; Hocum, J.D.; Heimfeld, S.; Kiem, H.-P. Semi-automated closed system manufacturing of lentivirus gene-modified haematopoietic stem cells for gene therapy. Nat. Commun. 2016, 7, 13173. [Google Scholar] [CrossRef]
- Hurley, P.; Jurmeister, S.; Parsley, K.L. Advanced Therapy Medicinal Products—An Evolving Regulatory Landscape; Regulatory Affairs Professionals Society: Rockville, MD, USA, 2017. [Google Scholar]
- De Wilde, S.; Coppens, D.G.M.; Hoekman, J.; De Bruin, M.L.; Leufkens, H.G.M.; Guchelaar, H.-J.; Meij, P. EU decision-making for marketing authorization of advanced therapy medicinal products: A case study. Drug Discov. Today 2018, 23, 1328–1333. [Google Scholar] [CrossRef] [PubMed]
- White, M.; Whittaker, R.; Gándara, C.; Stoll, E.A. A Guide to Approaching Regulatory Considerations for Lentiviral-Mediated Gene Therapies. Hum. Gene Ther. Methods 2017, 28, 163–176. [Google Scholar] [CrossRef] [Green Version]
- Coppens, D.G.M.; De Wilde, S.; Guchelaar, H.J.; De Bruin, M.L.; Leufkens, H.G.M.; Meij, P.; Hoekman, J. A decade of marketing approval of gene and cell-based therapies in the United States, European Union and Japan: An evaluation of regulatory decision-making. Cytotherapy 2018, 20, 769–778. [Google Scholar] [CrossRef]
- Detela, G.; Lodge, A. EU Regulatory Pathways for ATMPs: Standard, Accelerated and Adaptive Pathways to Marketing Authorisation. Mol. Ther. Methods Clin. Dev. 2019, 13, 205–232. [Google Scholar] [CrossRef] [Green Version]
- European Medecines Agency (EMA). Available online: https://www.ema.europa.eu/en/human-regulatory/overview/advanced-therapy-medicinal-products-overview (accessed on 20 April 2020).
- Kenter, M.J.H. Is it time to reform oversight of clinical gene therapy in the EU? Br. J. Clin. Pharm. 2019, 85, 8–10. [Google Scholar] [CrossRef]
- “REGULATION (EU) No 536/2014 OF THE EUROPEAN PARLIAMENT AND OF THE COUNCIL of 16 April 2014 on Clinical Trials on Medicinal Products for Human Use, and Repealing Directive 2001/20/EC”, Official Journal of the European Union, Luxembourg, EU. 2014. Available online: https://ec.europa.eu/health/sites/health/files/files/eudralex/vol-%201/reg_2014_536/reg_2014_536_en.pdf (accessed on 15 April 2020).
- Boráň, T.; Menezes-Ferreira, M.; Reischl, I.; Celis, P.; Ferry, N.; Gänsbacher, B.; Krafft, H.; Lipucci Di Paola, M.; Sladowski, D.; Salmikangas, P. Clinical Development and Commercialization of Advanced Therapy Medicinal Products (ATMPs) in the EU: How are the product pipeline and regulatory framework evolving? Hum. Gene Ther. Clin. Dev. 2017. [Google Scholar] [CrossRef] [PubMed]
- Meij, P.; Canals, J.M.; Lowery, M.; Scott, M. Advanced Therapy Medicinal Products; LERU: Leuven, Belgium, 2019. [Google Scholar]
- De Wilde, S.; Veltrop-Duits, L.; Hoozemans-Strik, M.; Ras, T.; Blom-Veenman, J.; Guchelaar, H.-J.; Zandvliet, M.; Meij, P. Hurdles in clinical implementation of academic advanced therapy medicinal products: A national evaluation. Cytotherapy 2016, 18, 797–805. [Google Scholar] [CrossRef] [PubMed]
- Huff, R.; Hoofwijk, T.J.; Turner, J.R. The role of regulatory agencies in new drug development: A global perspective. Regulatory 2014, 6, 20–22. [Google Scholar]
- Salmikangas, P. Design and optimisation of a quality target product profile for ATMPs. Regul. Rapp. 2019, 16, 4–7. [Google Scholar]
- Thrasher, A.J.; Williams, D.A. Evolving Gene Therapy in Primary Immunodeficiency. Mol. Ther. 2017, 25, 1132–1141. [Google Scholar] [CrossRef] [PubMed]
- Booth, C.; Romano, R.; Roncarolo, M.G.; Thrasher, A.J. Gene therapy for primary immunodeficiency. Hum. Mol. Genet. 2019, 28, R15–R23. [Google Scholar] [CrossRef] [Green Version]
- Staal, F.J.T.; Aiuti, A.; Cavazzana, M. Autologous Stem-Cell-Based Gene Therapy for Inherited Disorders: State of the Art and Perspectives. Front. Pediatr. 2019, 7. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Disease | Gene | Vector | Clinical Study | Phase | Participants | Location | Study Type | Status | Outcome/References | |
|---|---|---|---|---|---|---|---|---|---|---|
| ADA-SCID | ADA | RV | NCT01279720 | Phase 1/2 | 8 | EU | interventional | completed | Positive benefit-risk profile [28,29,30,31,32,33,34,35,36] | |
| NCT00018018 | Phase 1 | 8 | USA | interventional | completed | |||||
| NCT00794508 | Phase 2 | 10 | USA | interventional | completed | |||||
| NCT00599781 | Phase 1/2 | 8 | EU | interventional | completed | |||||
| Strimvelis (RV) | NCT00598481 | Phase 2 | 18 | EU | interventional | completed | Immune reconstitution [33,37] | |||
| NCT03232203 | 10 | EU | observational | recruiting | ||||||
| NCT03478670 | 50 | EU | observational | enrolling | ||||||
| SIN LV | NCT01852071 | Phase 1/2 | 20 | USA | interventional | completed | Immune reconstitution, well tolerated [36,38,39] | |||
| NCT02999984 | Phase 1/2 | 10 | USA | interventional | completed | |||||
| NCT01380990 | Phase 1/2 | 36 | EU | interventional | completed | |||||
| NCT04140539 | Phase 2/3 | 3 | USA | interventional | recruiting | |||||
| NCT03645460 | n.a. | 10 | China | interventional | recruiting | |||||
| NCT04049084 | 70 | USA/EU | observational | enrolling | ||||||
| Artemis-SCID | DCLRE1C | SIN LV | NCT03538899 | Phase 1/2 | 15 | USA | interventional | recruiting | Immune reconstitution [40] | |
| Chronic granulomatous disease | Gp91 phox | RV | NCT00927134 | Phase 1/2 | 2 | EU | interventional | completed | Sustained engraftment, insertional mutagenesis, [41,42] | |
| NCT00564759 | Phase 1/2 | 2 | EU | interventional | unknown | |||||
| SIN | NCT01906541 | Phase 1/2 | 5 | EU | interventional | unknown | ||||
| NCT00778882 | Phase 1/2 | 2 | Korea | interventional | active | |||||
| SIN LV | NCT01855685 | Phase 1/2 | 3 | EU | interventional | active | [16] | |||
| NCT02757911 | Phase 1/2 | 3 | EU | interventional | active | |||||
| NCT02234934 | Phase 1/2 | 10 | USA | interventional | active | |||||
| NCT03645486 | n.a. | 10 | China | Interventional | active | |||||
| Leukocyte Adhesion Deficiency-I | CD18 | RV | NCT00023010 | Phase 1 | 2 | USA | observational | completed | ||
| SIN LV | NCT03812263 | Phase 1/2 | 9 | USA | interventional | recruiting | ||||
| NCT03825783 | Phase 1 | 2 | EU | interventional | recruiting | |||||
| Wiskott–Aldrich Syndrome | WAS | SIN LV | NCT01347242 | Phase 1/2 | 6 | EU | interventional | completed | Successful engraftment, immune reconstitution, no adverse reactions [15,43,44] | |
| NCT01347346 | Phase 1/2 | 5 | EU | interventional | completed | |||||
| NCT02333760 | Phase 1/2 | 10 | EU | interventional | active | |||||
| NCT03837483 | Phase 2 | 6 | EU | interventional | active | |||||
| NCT01410825 | Phase 1/2 | 5 | USA | interventional | active | |||||
| NCT01515462 | Phase 1/2 | 8 | EU | interventional | active | |||||
| X linked-SCID | IL2RG | RV | NCT00028236 | Phase 1 | 3 | USA | interventional | completed | Sustained immune correction, risk acute leukemia [6,45,46,47,48] | |
| SIN | NCT01175239 | n.a. | 1 | EU | interventional | unknown | ||||
| NCT01410019 | Phase 1/2 | 5 | EU | interventional | unknown | |||||
| SIN | NCT01129544 | Phase 1/2 | 8 | USA | interventional | active | ||||
| SIN LV | NCT03315078 | Phase 1/2 | 13 | USA | interventional | recruiting | Multilineage engraftment, immune reconstitution [49] | |||
| NCT03311503 | Phase 1/2 | 10 | USA | interventional | recruiting | |||||
| NCT01512888 | Phase 1/2 | 28 | USA | interventional | recruiting | |||||
| NCT01306019 | Phase 1/2 | 30 | USA | interventional | recruiting | |||||
| NCT03601286 | Phase 1 | 5 | EU | interventional | recruiting | |||||
| NCT04286815 | n.a. | 10 | China | interventional | recruiting | |||||
| NCT03217617 | Phase 1/2 | 10 | China | interventional | recruiting | |||||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garcia-Perez, L.; Ordas, A.; Canté-Barrett, K.; Meij, P.; Pike-Overzet, K.; Lankester, A.; Staal, F.J.T. Preclinical Development of Autologous Hematopoietic Stem Cell-Based Gene Therapy for Immune Deficiencies: A Journey from Mouse Cage to Bed Side. Pharmaceutics 2020, 12, 549. https://doi.org/10.3390/pharmaceutics12060549
Garcia-Perez L, Ordas A, Canté-Barrett K, Meij P, Pike-Overzet K, Lankester A, Staal FJT. Preclinical Development of Autologous Hematopoietic Stem Cell-Based Gene Therapy for Immune Deficiencies: A Journey from Mouse Cage to Bed Side. Pharmaceutics. 2020; 12(6):549. https://doi.org/10.3390/pharmaceutics12060549
Chicago/Turabian StyleGarcia-Perez, Laura, Anita Ordas, Kirsten Canté-Barrett, Pauline Meij, Karin Pike-Overzet, Arjan Lankester, and Frank J. T. Staal. 2020. "Preclinical Development of Autologous Hematopoietic Stem Cell-Based Gene Therapy for Immune Deficiencies: A Journey from Mouse Cage to Bed Side" Pharmaceutics 12, no. 6: 549. https://doi.org/10.3390/pharmaceutics12060549