Vascular Drug Delivery Using Carrier Red Blood Cells: Focus on RBC Surface Loading and Pharmacokinetics

 , , and
, , and
Abstract
:
1. Introduction
2. Principles of RBC Drug Delivery
2.1. Encapsulation of Drugs into Carrier RBC
2.2. Surface Loading of RBCs
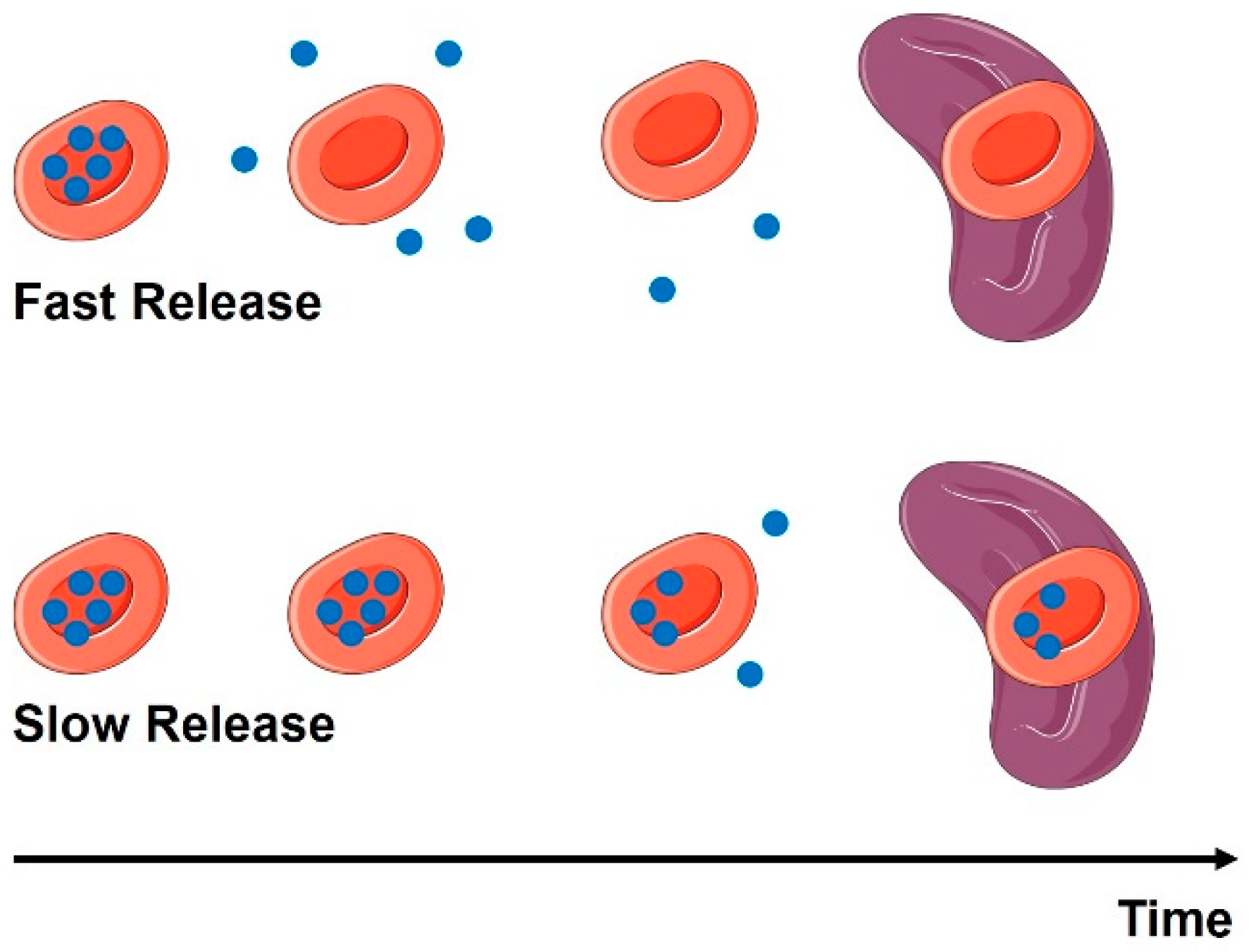
2.3. RBC Loading: Effects on Cargoes
2.4. Novel Strategies
3. Pharmacokinetics of RBC-Associated Drugs
3.1. Pharmacokinetics—A Brief Primer
3.1.1. Absorption and Routes of Administration
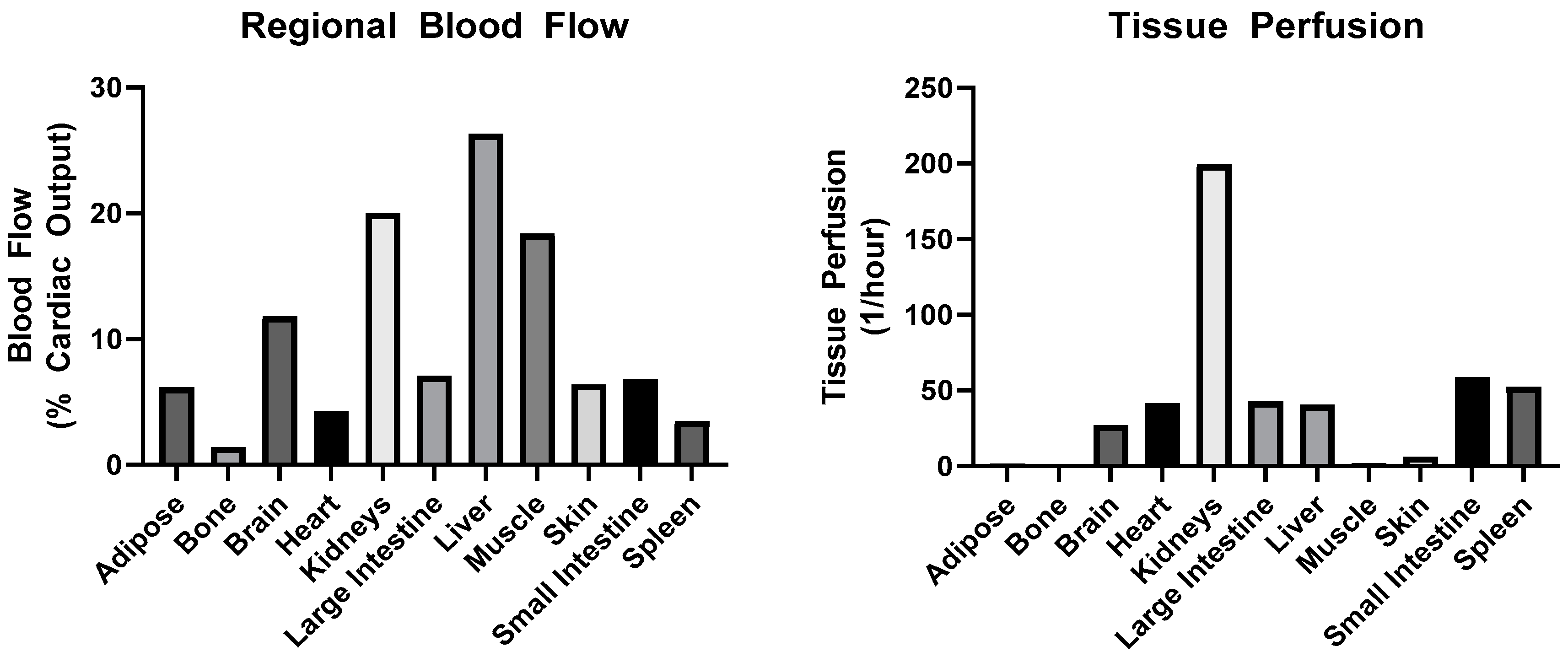
3.1.2. Distribution
3.1.3. Metabolism/Elimination
3.1.4. Pharmacokinetics in Drug Development
3.1.5. ADME of RBC-Associated Drugs
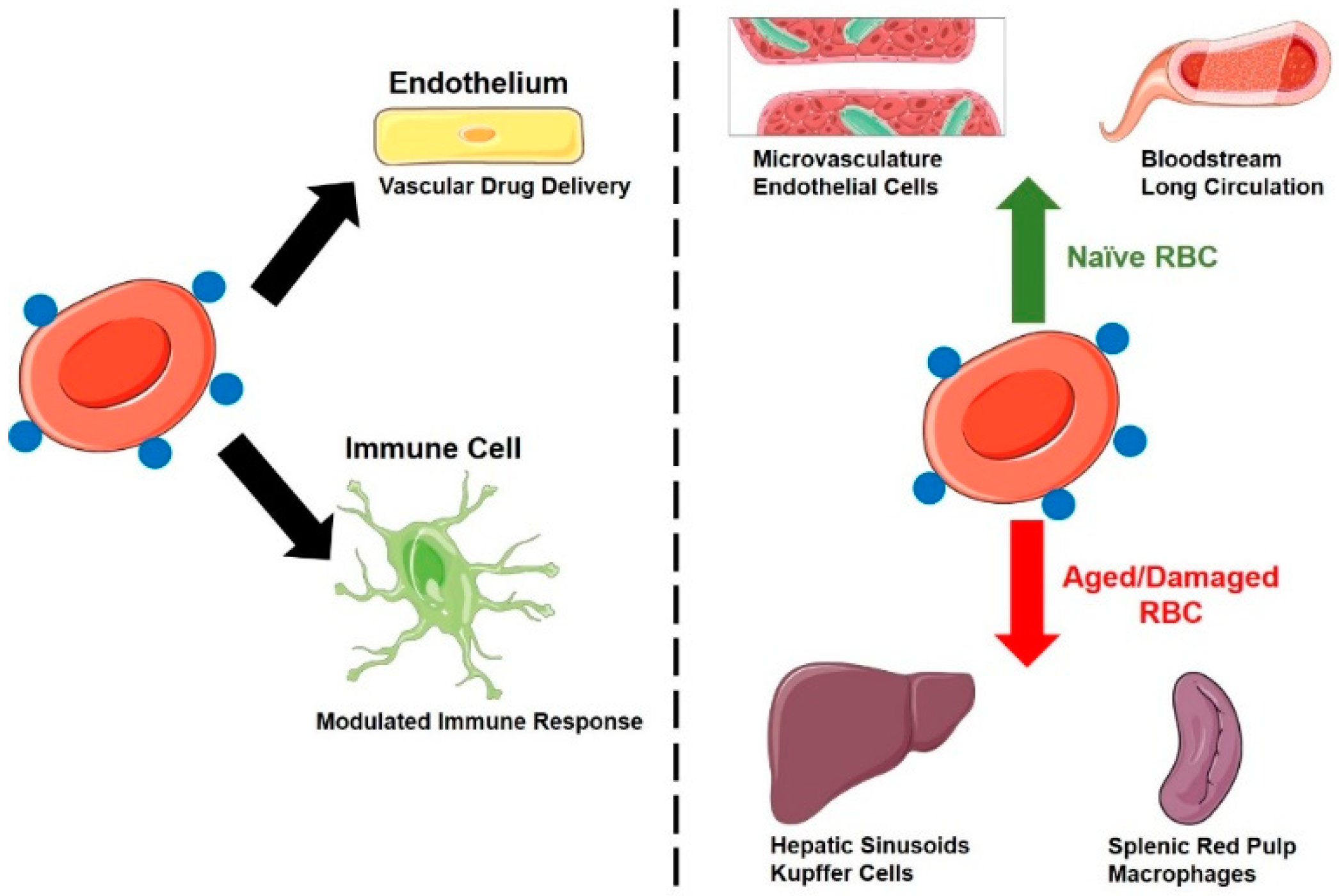
3.1.6. Unique Aspects of RBC PK
3.2. PK of RBC-Associated Drugs—Key Considerations
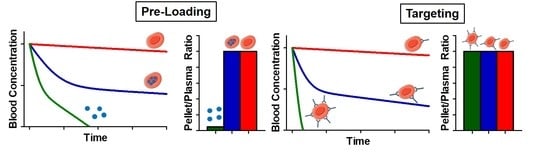
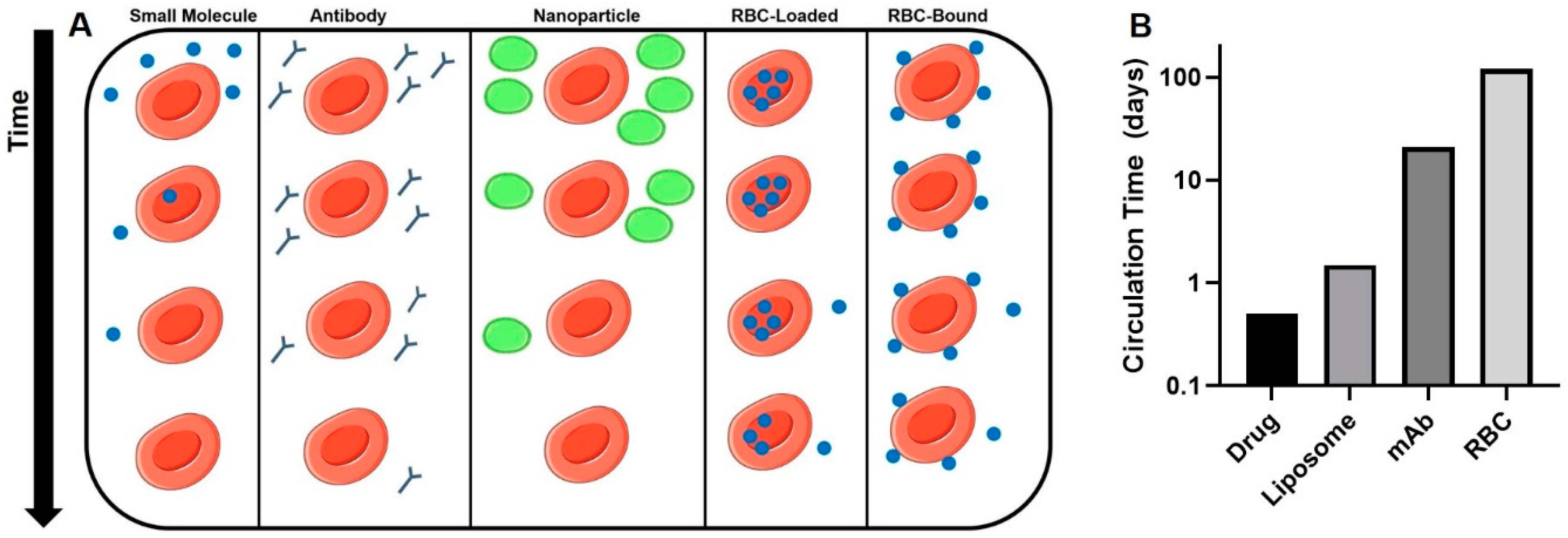
3.2.1. Drugs Loaded inside the RBC—Effects on RBC Circulation
3.2.2. Drugs Loaded Inside the RBC—Effects on Drug Pharmacokinetics
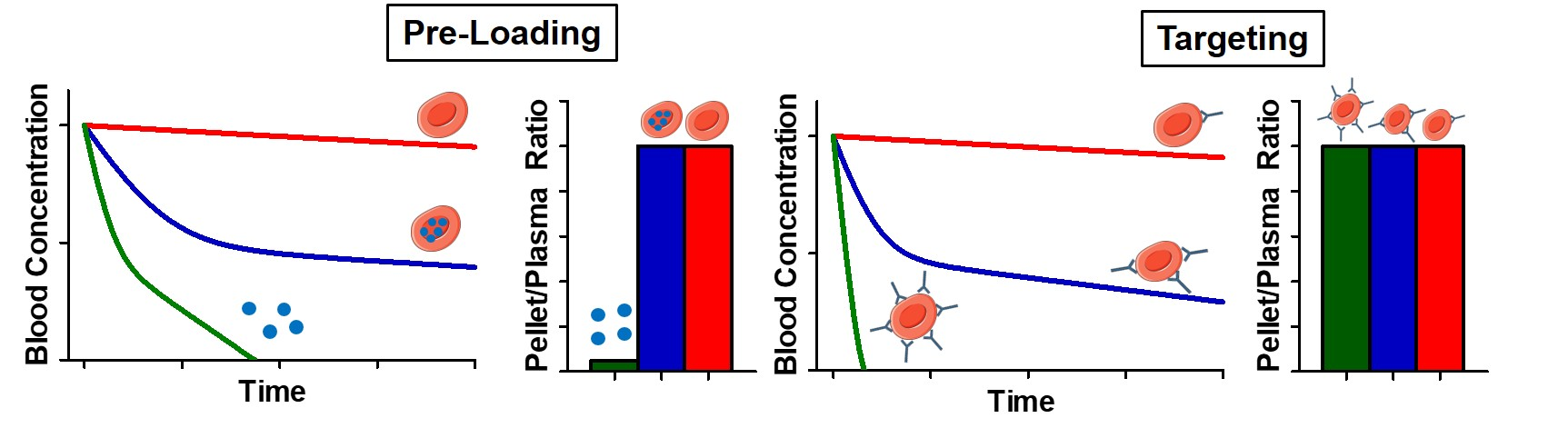
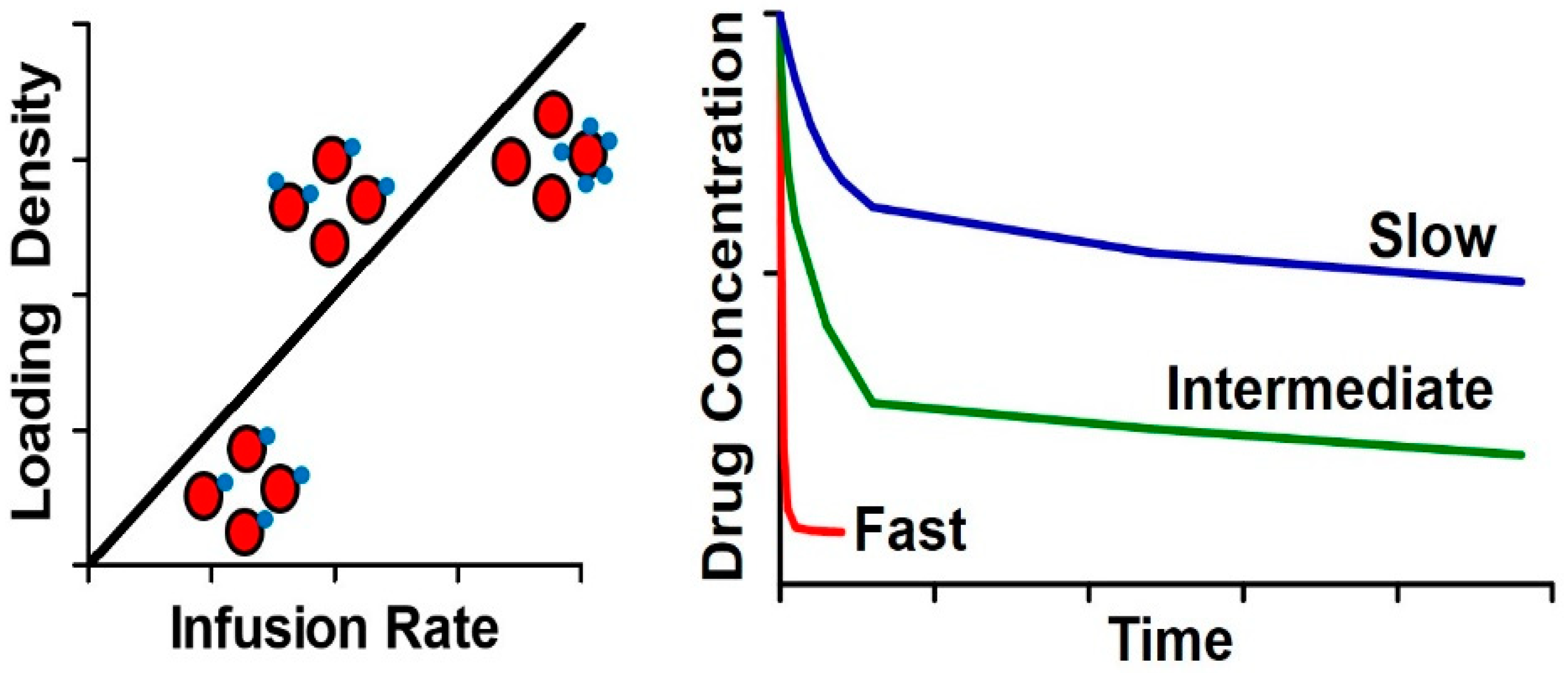
3.2.3. Surface Loading of RBC—Impact of Dose and Coupling Strategy
3.2.4. Surface Loading of RBC—Impact of Affinity
3.2.5. Surface Loading—Ex Vivo or In Vivo?
4. Conclusions and Other Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Ihler, G.M.; Glew, R.H.; Schnure, F.W. Enzyme loading of erythrocytes. Proc. Natl. Acad. Sci. USA 1973, 70, 2663–2666. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Kundrat, L.; Pishesha, N.; Bilate, A.; Theile, C.; Maruyama, T.; Dougan, S.K.; Ploegh, H.L.; Lodish, H.F. Engineered red blood cells as carriers for systemic delivery of a wide array of functional probes. Proc. Natl. Acad. Sci. USA 2014, 111, 10131–10136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pishesha, N.; Bilate, A.M.; Wibowo, M.C.; Huang, N.J.; Li, Z.; Deshycka, R.; Bousbaine, D.; Li, H.; Patterson, H.C.; Dougan, S.K.; et al. Engineered erythrocytes covalently linked to antigenic peptides can protect against autoimmune disease. Proc. Natl. Acad. Sci. USA 2017, 114, 3157–3162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimm, A.J.; Kontos, S.; Diaceri, G.; Quaglia-Thermes, X.; Hubbell, J.A. Memory of tolerance and induction of regulatory T cells by erythrocyte-targeted antigens. Sci. Rep. 2015, 5, 15907. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.; Ukidve, A.; Gao, Y.; Kim, J.; Mitragotri, S. Erythrocyte leveraged chemotherapy (ELeCt): Nanoparticle assembly on erythrocyte surface to combat lung metastasis. Sci. Adv. 2019, 5, eaax9250. [Google Scholar] [CrossRef] [Green Version]
- Brenner, J.S.; Pan, D.C.; Myerson, J.W.; Marcos-Contreras, O.A.; Villa, C.H.; Patel, P.; Hekierski, H.; Chatterjee, S.; Tao, J.Q.; Parhiz, H.; et al. Red blood cell-hitchhiking boosts delivery of nanocarriers to chosen organs by orders of magnitude. Nat. Commun. 2018, 9, 2684. [Google Scholar] [CrossRef]
- Anselmo, A.C.; Kumar, S.; Gupta, V.; Pearce, A.M.; Ragusa, A.; Muzykantov, V.; Mitragotri, S. Exploiting shape, cellular-hitchhiking and antibodies to target nanoparticles to lung endothelium: Synergy between physical, chemical and biological approaches. Biomaterials 2015, 68, 1–8. [Google Scholar] [CrossRef]
- He, H.; Ye, J.; Wang, Y.; Liu, Q.; Chung, H.S.; Kwon, Y.M.; Shin, M.C.; Lee, K.; Yang, V.C. Cell-penetrating peptides meditated encapsulation of protein therapeutics into intact red blood cells and its application. J. Control. Release 2014, 176, 123–132. [Google Scholar] [CrossRef] [Green Version]
- Favretto, M.E.; Cluitmans, J.C.; Bosman, G.J.; Brock, R. Human erythrocytes as drug carriers: Loading efficiency and side effects of hypotonic dialysis, chlorpromazine treatment and fusion with liposomes. J. Control. Release 2013, 170, 343–351. [Google Scholar] [CrossRef]
- Mosca, A.; Paleari, R.; Russo, V.; Rosti, E.; Nano, R.; Boicelli, A.; Villa, S.; Zanella, A. IHP entrapment into human erythrocytes: Comparison between hypotonic dialysis and DMSO osmotic pulse. Adv. Exp. Med. Biol. 1992, 326, 19–26. [Google Scholar] [CrossRef]
- Bourgeaux, V.; Lanao, J.M.; Bax, B.E.; Godfrin, Y. Drug-loaded erythrocytes: On the road toward marketing approval. Drug Des. Devel. Ther. 2016, 10, 665–676. [Google Scholar] [CrossRef] [Green Version]
- Magnani, M.; Rossi, L.; D’Ascenzo, M.; Panzani, I.; Bigi, L.; Zanella, A. Erythrocyte engineering for drug delivery and targeting. Biotechnol. Appl. Biochem. 1998, 28, 1–6. [Google Scholar] [PubMed]
- Muzykantov, V.R.; Sakharov, D.V.; Smirnov, M.D.; Domogatsky, S.P.; Samokhin, G.P. Targeting of enzyme immobilized on erythrocyte membrane to collagen-coated surface. FEBS Lett. 1985, 182, 62–66. [Google Scholar] [CrossRef] [Green Version]
- Samokhin, G.P.; Smirnov, M.D.; Muzykantov, V.R.; Domogatsky, S.P.; Smirnov, V.N. Red blood cell targeting to collagen-coated surfaces. FEBS Lett. 1983, 154, 257–261. [Google Scholar] [CrossRef] [Green Version]
- Kontos, S.; Hubbell, J.A. Improving Protein Pharmacokinetics by Engineering Erythrocyte Affinity. Mol. Pharmaceut. 2010, 7, 2141–2147. [Google Scholar] [CrossRef] [PubMed]
- Mqadmi, A.; Abramowitz, S.; Zheng, X.; Yazdanbakhsh, K. Reduced red blood cell destruction by antibody fragments. Immunohematology 2006, 22, 11–14. [Google Scholar] [PubMed]
- Zaitsev, S.; Kowalska, M.A.; Neyman, M.; Carnemolla, R.; Tliba, S.; Ding, B.S.; Stonestrom, A.; Spitzer, D.; Atkinson, J.P.; Poncz, M.; et al. Targeting recombinant thrombomodulin fusion protein to red blood cells provides multifaceted thromboprophylaxis. Blood 2012, 119, 4779–4785. [Google Scholar] [CrossRef] [Green Version]
- Zaitsev, S.; Spitzer, D.; Murciano, J.C.; Ding, B.S.; Tliba, S.; Kowalska, M.A.; Bdeir, K.; Kuo, A.; Stepanova, V.; Atkinson, J.P.; et al. Targeting of a mutant plasminogen activator to circulating red blood cells for prophylactic fibrinolysis. J. Pharmacol. Exp. Ther. 2010, 332, 1022–1031. [Google Scholar] [CrossRef] [Green Version]
- Ji, W.; Smith, P.N.; Koepsel, R.R.; Andersen, J.D.; Baker, S.L.; Zhang, L.; Carmali, S.; Myerson, J.W.; Muzykantov, V.; Russell, A.J. Erythrocytes as carriers of immunoglobulin-based therapeutics. Acta Biomater. 2020, 101, 422–435. [Google Scholar] [CrossRef]
- Carnemolla, R.; Villa, C.H.; Greineder, C.F.; Zaitsev, S.; Patel, K.R.; Kowalska, M.A.; Atochin, D.N.; Cines, D.B.; Siegel, D.L.; Esmon, C.T.; et al. Targeting thrombomodulin to circulating red blood cells augments its protective effects in models of endotoxemia and ischemia-reperfusion injury. FASEB J. 2017, 31, 761–770. [Google Scholar] [CrossRef] [Green Version]
- Villa, C.H.; Cines, D.B.; Siegel, D.L.; Muzykantov, V. Erythrocytes as Carriers for Drug Delivery in Blood Transfusion and Beyond. Transfus. Med. Rev. 2017, 31, 26–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greineder, C.F.; Howard, M.D.; Carnemolla, R.; Cines, D.B.; Muzykantov, V.R. Advanced drug delivery systems for antithrombotic agents. Blood 2013, 122, 1565–1575. [Google Scholar] [CrossRef] [PubMed]
- Muzykantov, V.R.; Sakharov, D.V.; Smirnov, M.D.; Samokhin, G.P.; Smirnov, V.N. Immunotargeting of erythrocyte-bound streptokinase provides local lysis of a fibrin clot. Biochim. Biophys. Acta 1986, 884, 355–362. [Google Scholar] [CrossRef]
- Smirnov, V.N.; Domogatsky, S.P.; Dolgov, V.V.; Hvatov, V.B.; Klibanov, A.L.; Koteliansky, V.E.; Muzykantov, V.R.; Repin, V.S.; Samokhin, G.P.; Shekhonin, B.V.; et al. Carrier-directed targeting of liposomes and erythrocytes to denuded areas of vessel wall. Proc. Natl. Acad. Sci. USA 1986, 83, 6603–6607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muzykantov, V.R.; Sakharov, D.V.; Domogatsky, S.P.; Goncharov, N.V.; Danilov, S.M. Directed targeting of immunoerythrocytes provides local protection of endothelial cells from damage by hydrogen peroxide. Am. J. Pathol. 1987, 128, 276–285. [Google Scholar] [PubMed]
- Glukhova, M.A.; Domogatsky, S.P.; Kabakov, A.E.; Muzykantov, V.R.; Ornatsky, O.I.; Sakharov, D.V.; Frid, M.G.; Smirnov, V.N. Red blood cell targeting to smooth muscle cells. FEBS Lett. 1986, 198, 155–158. [Google Scholar] [CrossRef] [Green Version]
- Magnani, M.; Rossi, L. Approaches to erythrocyte-mediated drug delivery. Expert Opin. Drug Deliv. 2014, 11, 677–687. [Google Scholar] [CrossRef]
- Mukthavaram, R.; Shi, G.; Kesari, S.; Simberg, D. Targeting and depletion of circulating leukocytes and cancer cells by lipophilic antibody-modified erythrocytes. J. Control. Release 2014, 183, 146–153. [Google Scholar] [CrossRef] [Green Version]
- Shi, G.; Mukthavaram, R.; Kesari, S.; Simberg, D. Distearoyl anchor-painted erythrocytes with prolonged ligand retention and circulation properties in vivo. Adv. Healthc Mater. 2014, 3, 142–148. [Google Scholar] [CrossRef] [Green Version]
- Danielyan, K.; Ganguly, K.; Ding, B.S.; Atochin, D.; Zaitsev, S.; Murciano, J.C.; Huang, P.L.; Kasner, S.E.; Cines, D.B.; Muzykantov, V.R. Cerebrovascular thromboprophylaxis in mice by erythrocyte-coupled tissue-type plasminogen activator. Circulation 2008, 118, 1442–1449. [Google Scholar] [CrossRef] [Green Version]
- Stein, S.C.; Ganguly, K.; Belfield, C.M.; Xu, X.; Swanson, E.W.; Chen, X.H.; Browne, K.D.; Johnson, V.E.; Smith, D.H.; LeBold, D.G.; et al. Erythrocyte-bound tissue plasminogen activator is neuroprotective in experimental traumatic brain injury. J. Neurotrauma. 2009, 26, 1585–1592. [Google Scholar] [CrossRef]
- Armstead, W.M.; Ganguly, K.; Kiessling, J.W.; Chen, X.H.; Smith, D.H.; Higazi, A.A.; Cines, D.B.; Bdeir, K.; Zaitsev, S.; Muzykantov, V.R. Red blood cells-coupled tPA prevents impairment of cerebral vasodilatory responses and tissue injury in pediatric cerebral hypoxia/ischemia through inhibition of ERK MAPK activation. J. Cereb. Blood Flow. Metab. 2009, 29, 1463–1474. [Google Scholar] [CrossRef] [Green Version]
- Pisapia, J.M.; Xu, X.; Kelly, J.; Yeung, J.; Carrion, G.; Tong, H.; Meghan, S.; El-Falaky, O.M.; Grady, M.S.; Smith, D.H.; et al. Microthrombosis after experimental subarachnoid hemorrhage: Time course and effect of red blood cell-bound thrombin-activated pro-urokinase and clazosentan. Exp. Neurol. 2012, 233, 357–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gersh, K.C.; Zaitsev, S.; Muzykantov, V.; Cines, D.B.; Weisel, J.W. The spatial dynamics of fibrin clot dissolution catalyzed by erythrocyte-bound vs. free fibrinolytics. J. Thromb. Haemost. 2010, 8, 1066–1074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gersh, K.C.; Zaitsev, S.; Cines, D.B.; Muzykantov, V.; Weisel, J.W. Flow-dependent channel formation in clots by an erythrocyte-bound fibrinolytic agent. Blood 2011, 117, 4964–4967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murciano, J.C.; Medinilla, S.; Eslin, D.; Atochina, E.; Cines, D.B.; Muzykantov, V.R. Prophylactic fibrinolysis through selective dissolution of nascent clots by tPA-carrying erythrocytes. Nat. Biotechnol. 2003, 21, 891–896. [Google Scholar] [CrossRef] [PubMed]
- Ganguly, K.; Goel, M.S.; Krasik, T.; Bdeir, K.; Diamond, S.L.; Cines, D.B.; Muzykantov, V.R.; Murciano, J.C. Fibrin affinity of erythrocyte-coupled tissue-type plasminogen activators endures hemodynamic forces and enhances fibrinolysis in vivo. J. Pharmacol. Exp. Ther. 2006, 316, 1130–1136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armstead, W.M.; Ganguly, K.; Riley, J.; Kiessling, J.W.; Cines, D.B.; Higazi, A.A.; Zaitsev, S.; Muzykantov, V.R. Red blood cell-coupled tissue plasminogen activator prevents impairment of cerebral vasodilatory responses through inhibition of c-Jun-N-terminal kinase and potentiation of p38 mitogen-activated protein kinase after cerebral photothrombosis in the newborn pig. Pediatr. Crit. Care Med. 2011, 12, e369–e375. [Google Scholar] [CrossRef] [Green Version]
- Zaitsev, S.; Danielyan, K.; Murciano, J.C.; Ganguly, K.; Krasik, T.; Taylor, R.P.; Pincus, S.; Jones, S.; Cines, D.B.; Muzykantov, V.R. Human complement receptor type 1-directed loading of tissue plasminogen activator on circulating erythrocytes for prophylactic fibrinolysis. Blood 2006, 108, 1895–1902. [Google Scholar] [CrossRef] [Green Version]
- Zaitsev, S.; Spitzer, D.; Murciano, J.C.; Ding, B.S.; Tliba, S.; Kowalska, M.A.; Marcos-Contreras, O.A.; Kuo, A.; Stepanova, V.; Atkinson, J.P.; et al. Sustained thromboprophylaxis mediated by an RBC-targeted pro-urokinase zymogen activated at the site of clot formation. Blood 2010, 115, 5241–5248. [Google Scholar] [CrossRef] [Green Version]
- Kontos, S.; Kourtis, I.C.; Dane, K.Y.; Hubbell, J.A. Engineering antigens for in situ erythrocyte binding induces T-cell deletion. Proc. Natl. Acad. Sci. USA 2013, 110, E60–E68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorentz, K.M.; Kontos, S.; Diaceri, G.; Henry, H.; Hubbell, J.A. Engineered binding to erythrocytes induces immunological tolerance to E. coli asparaginase. Sci. Adv. 2015, 1, e1500112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murciano, J.C.; Higazi, A.A.; Cines, D.B.; Muzykantov, V.R. Soluble urokinase receptor conjugated to carrier red blood cells binds latent pro-urokinase and alters its functional profile. J. Control. Release 2009, 139, 190–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armstead, W.M.; Ganguly, K.; Kiessling, J.W.; Riley, J.; Chen, X.H.; Smith, D.H.; Stein, S.C.; Higazi, A.A.; Cines, D.B.; Bdeir, K.; et al. Signaling, delivery and age as emerging issues in the benefit/risk ratio outcome of tPA For treatment of CNS ischemic disorders. J. Neurochem. 2010, 113, 303–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atukorale, P.U.; Yang, Y.S.; Bekdemir, A.; Carney, R.P.; Silva, P.J.; Watson, N.; Stellacci, F.; Irvine, D.J. Influence of the glycocalyx and plasma membrane composition on amphiphilic gold nanoparticle association with erythrocytes. Nanoscale 2015, 7, 11420–11432. [Google Scholar] [CrossRef] [PubMed]
- Ganguly, K.; Murciano, J.C.; Westrick, R.; Leferovich, J.; Cines, D.B.; Muzykantov, V.R. The glycocalyx protects erythrocyte-bound tissue-type plasminogen activator from enzymatic inhibition. J. Pharmacol. Exp. Ther. 2007, 321, 158–164. [Google Scholar] [CrossRef] [Green Version]
- Murad, K.L.; Mahany, K.L.; Brugnara, C.; Kuypers, F.A.; Eaton, J.W.; Scott, M.D. Structural and functional consequences of antigenic modulation of red blood cells with methoxypoly(ethylene glycol). Blood 1999, 93, 2121–2127. [Google Scholar] [CrossRef]
- Chapanian, R.; Constantinescu, I.; Brooks, D.E.; Scott, M.D.; Kizhakkedathu, J.N. In vivo circulation, clearance, and biodistribution of polyglycerol grafted functional red blood cells. Biomaterials 2012, 33, 3047–3057. [Google Scholar] [CrossRef]
- Blackall, D.P.; Armstrong, J.K.; Meiselman, H.J.; Fisher, T.C. Polyethylene glycol-coated red blood cells fail to bind glycophorin A-specific antibodies and are impervious to invasion by the Plasmodium falciparum malaria parasite. Blood 2001, 97, 551–556. [Google Scholar] [CrossRef] [Green Version]
- Le, Y.; Toyofuku, W.M.; Scott, M.D. Immunogenicity of murine mPEG-red blood cells and the risk of anti-PEG antibodies in human blood donors. Exp. Hematol. 2017, 47, 36–47.e32. [Google Scholar] [CrossRef]
- Chapanian, R.; Constantinescu, I.; Rossi, N.A.; Medvedev, N.; Brooks, D.E.; Scott, M.D.; Kizhakkedathu, J.N. Influence of polymer architecture on antigens camouflage, CD47 protection and complement mediated lysis of surface grafted red blood cells. Biomaterials 2012, 33, 7871–7883. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Fan, M.; Chen, Y.; Liu, Z.; Shao, C.; Jin, B.; Wang, X.; Hui, L.; Wang, S.; Liao, Z.; et al. Surface-anchored framework for generating RhD-epitope stealth red blood cells. Sci. Adv. 2020, 6, eaaw9679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klei, T.R.L.; Meinderts, S.M.; van den Berg, T.K.; van Bruggen, R. From the Cradle to the Grave: The Role of Macrophages in erythropoiesis and erythrophagocytosis. Front. Immunol. 2017, 8, 292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Z.; Ukidve, A.; Kim, J.; Mitragotri, S. Targeting Strategies for Tissue-Specific Drug Delivery. Cell 2020, 181, 151–167. [Google Scholar] [CrossRef] [PubMed]
- Anderson, H.L.; Brodsky, I.E.; Mangalmurti, N.S. The Evolving Erythrocyte: Red Blood Cells as Modulators of Innate Immunity. J. Immunol. 2018, 201, 1343–1351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minasyan, H. Mechanisms and pathways for the clearance of bacteria from blood circulation in health and disease. Pathophysiology 2016, 23, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Halma, C.; Breedveld, F.C.; Daha, M.R.; Blok, D.; Evers-Schouten, J.H.; Hermans, J.; Pauwels, E.K.; van Es, L.A. Elimination of soluble 123I-labeled aggregates of IgG in patients with systemic lupus erythematosus. Effect of serum IgG and numbers of erythrocyte complement receptor type 1. Arthritis Rheum 1991, 34, 442–452. [Google Scholar] [CrossRef]
- Anselmo, A.C.; Gupta, V.; Zern, B.J.; Pan, D.; Zakrewsky, M.; Muzykantov, V.; Mitragotri, S. Delivering Nanoparticles to Lungs while Avoiding Liver and Spleen through Adsorption on Red Blood Cells. ACS Nano 2013, 7, 11129–11137. [Google Scholar] [CrossRef] [Green Version]
- Chambers, E.; Mitragotri, S. Long circulating nanoparticles via adhesion on red blood cells: Mechanism and extended circulation. Exp. Biol. Med. 2007, 232, 958–966. [Google Scholar]
- Therapeutics, R. Rubius Therapeutics. Available online: https://www.rubiustx.com/our-science/#therapeutic-modalities (accessed on 10 April 2020).
- Zhang, X.; Shamael, M.L.; Dastagir, R.; Nixon, M.; Lee, A.; Schmidt, A.; Khamhoung, A.; Douglas, C.; Moore, C.; Pawar, S.; et al. An Engineered Allogeneic Artificial Antigen-Presenting Red Cell Therapeutic, RTX-321, Promotes Antigen-Specific T Cell Expansion and Anti-Tumor Activity. In Proceedings of the Keystone Symposia 2020-Emerging Cellular Therapies: Cancer and Beyond, Banff, AB, Canada, 8–12 February 2020. [Google Scholar]
- Anne-Sophie Dugast, S.M.; Hoover, M.; Hong, E.; Leonard, S.C.; Bollampalli, A.; McLaughlin, D.C.; Mellen, J.; Nissen, T.S.; Carpenter, C.L.; Wickham, T.J.; et al. RTX-240, an Allogeneic Red Cell Therapeutic Expressing 4-1BBL and IL-15TP, Exhibits Potent In Vitro and In Vivo Activity and a Favorable Safety Profile. In Proceedings of the AACR 2019, Atlanta, GA, USA, 29 March–3 April 2019. [Google Scholar]
- Anne-Sophie, D.; Maegan Hoover, E.H.; Bollampalli, A.; McLaughlin, D.C.; Bhate, O.; Lyford, T.J.; Nissen, T.S.; Carpenter, C.L.; Wickham, T.J.; Melançon, L.; et al. RTX-224, an Allogeneic Red Cell Therapeutic Expressing IL-12 and 4-1BBL, Exhibits Potent In Vitro and In Vivo Activity and a Favorable Safety Profile. In Proceedings of the AACR 2019, Atlanta, GA, USA, 29 March–3 April 2019. [Google Scholar]
- Asher, D.R.; Cerny, A.M.; Finberg, R.W. The erythrocyte viral trap: Transgenic expression of viral receptor on erythrocytes attenuates coxsackievirus B infection. Proc. Natl. Acad. Sci. USA 2005, 102, 12897–12902. [Google Scholar] [CrossRef] [Green Version]
- Kuhl, H. Pharmacology of estrogens and progestogens: Influence of different routes of administration. Climacteric 2005, 8, 3–63. [Google Scholar] [CrossRef] [PubMed]
- Moeller, E.H.; Jorgensen, L. Alternative routes of administration for systemic delivery of protein pharmaceuticals. Drug Discov. Today Technol. 2008, 5, e89–e94. [Google Scholar] [CrossRef] [PubMed]
- Richter, W.F.; Jacobsen, B. Subcutaneous absorption of biotherapeutics: Knowns and unknowns. Drug Metab. Dispos 2014, 42, 1881–1889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patton, J.S.; Fishburn, C.S.; Weers, J.G. The Lungs as a Portal of Entry for Systemic Drug Delivery. Proc. Am. Thorac. Soc. 2004, 1, 338–344. [Google Scholar] [CrossRef]
- Grond, S.; Radbruch, L.; Lehmann, K.A. Clinical pharmacokinetics of transdermal opioids: Focus on transdermal fentanyl. Clin. Pharm. 2000, 38, 59–89. [Google Scholar] [CrossRef]
- Brown, R.P.; Delp, M.D.; Lindstedt, S.L.; Rhomberg, L.R.; Beliles, R.P. Physiological parameter values for physiologically based pharmacokinetic models. Toxicol. Ind. Health 1997, 13, 407–484. [Google Scholar] [CrossRef]
- International Transporter, C.; Giacomini, K.M.; Huang, S.M.; Tweedie, D.J.; Benet, L.Z.; Brouwer, K.L.; Chu, X.; Dahlin, A.; Evers, R.; Fischer, V.; et al. Membrane transporters in drug development. Nat. Rev. Drug Discov. 2010, 9, 215–236. [Google Scholar] [CrossRef]
- Sarin, H. Physiologic upper limits of pore size of different blood capillary types and another perspective on the dual pore theory of microvascular permeability. J. Angiogenes Res. 2010, 2, 14. [Google Scholar] [CrossRef] [Green Version]
- Thuenauer, R.; Muller, S.K.; Romer, W. Pathways of protein and lipid receptor-mediated transcytosis in drug delivery. Expert Opin. Drug Deliv. 2017, 14, 341–351. [Google Scholar] [CrossRef]
- Shi, S.; Li, Y. Interplay of Drug-Metabolizing Enzymes and Transporters in Drug Absorption and Disposition. Curr. Drug Metab. 2014, 15, 915–941. [Google Scholar] [CrossRef]
- Glassman, P.M.; Muzykantov, V.R. Pharmacokinetic and Pharmacodynamic Properties of Drug Delivery Systems. J. Pharmacol. Exp. Ther. 2019, 370, 570–580. [Google Scholar] [CrossRef] [PubMed]
- Kola, I.; Landis, J. Can the pharmaceutical industry reduce attrition rates? Nat. Rev. Drug Discov. 2004, 3, 711–715. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Fadiran, E.O.; Jones, C.D.; Lesko, L.; Huang, S.M.; Higgins, K.; Hu, C.; Machado, S.; Maldonado, S.; Williams, R.; et al. Population pharmacokinetics. A regulatory perspective. Clin. Pharm. 1999, 37, 41–58. [Google Scholar] [CrossRef]
- Ogasawara, K.; Alexander, G.C. Use of Population Pharmacokinetic Analyses among FDA-Approved Biologics. Clin. Pharmacol. Drug Dev. 2019, 8, 914–921. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.P.; Guan, Y.S.; Jin, X.R.; Jiang, S.S.; Lu, Z.J.; Wu, Y.; Li, Y.; Li, M.; Luo, F. Development of novel 5-fluorouracil carrier erythrocyte with pharmacokinetics and potent antitumor activity in mice bearing malignant ascites. J. Gastroenterol. Hepatol. 2010, 25, 985–990. [Google Scholar] [CrossRef]
- Bax, B.E.; Bain, M.D.; Fairbanks, L.D.; Webster, A.D.; Chalmers, R.A. In vitro and in vivo studies with human carrier erythrocytes loaded with polyethylene glycol-conjugated and native adenosine deaminase. Br. J. Haematol. 2000, 109, 549–554. [Google Scholar] [CrossRef]
- Lizano, C.; Perez, M.T.; Pinilla, M. Mouse erythrocytes as carriers for coencapsulated alcohol and aldehyde dehydrogenase obtained by electroporation-In vivo survival rate in circulation, organ distribution and ethanol degradation. Life Sci. 2001, 68, 2001–2016. [Google Scholar] [CrossRef]
- Briones, E.; Colino, C.I.; Millan, C.G.; Lanao, J.M. Increasing the selectivity of amikacin in rat peritoneal macrophages using carrier erythrocytes. Eur. J. Pharm. Sci. 2009, 38, 320–324. [Google Scholar] [CrossRef]
- Millan, C.G.; Castaneda, A.Z.; Lopez, F.G.; Marinero, M.L.S.; Lanao, J.M. Pharmacokinetics and biodistribution of amikacin encapsulated in carrier erythrocytes. J. Antimicrob. Chemoth. 2008, 61, 375–381. [Google Scholar] [CrossRef]
- Alvarez, F.J.; Herraez, A.; Murciano, J.C.; Jordan, J.A.; Diez, J.C.; Tejedor, M.C. In vivo survival and organ uptake of loaded carrier rat erythrocytes. J. Biochem. Tokyo 1996, 120, 286–291. [Google Scholar] [CrossRef] [Green Version]
- Skorokhod, O.A.; Garmaeva, T.T.; Vitvitsky, V.M.; Isaev, V.G.; Parovichnikova, E.N.; Savchenko, V.G.; Ataullakhanov, F.I. Pharmacokinetics of erythrocyte-bound daunorubicin in patients with acute leukemia. Med. Sci. Monitor 2004, 10, Pi55–Pi64. [Google Scholar]
- Annese, V.; Latiano, A.; Rossi, L.; Bossa, F.; Damonte, G.; Dallapiccola, B.; Serafini, S.; Pierige, F.; Andriulli, A.; Magnani, M. The polymorphism of multi-drug resistance 1 gene (MDR1) does not influence the pharmacokinetics of dexamethasone loaded into autologous erythrocytes of patients with inflammatory bowel disease. Eur. Rev. Med. Pharmacol. Sci. 2006, 10, 27–31. [Google Scholar] [PubMed]
- Annese, V.; Latiano, A.; Rossi, L.; Lombardi, G.; Dallapiccola, B.; Serafini, S.; Damonte, G.; Andriulli, A.; Magnani, M. Erythrocytes-mediated delivery of dexamethasone in steroid-dependent IBD patients-a pilot uncontrolled study. Am. J. Gastroenterol. 2005, 100, 1370–1375. [Google Scholar] [CrossRef] [PubMed]
- Ogiso, T.; Iwaki, M.; Ohtori, A. Encapsulation of dexamethasone in rabbit erythrocytes, the disposition in circulation and anti-inflammatory effect. J. Pharmacobiodyn. 1985, 8, 1032–1040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossi, L.; Castro, M.; D’Orio, F.; Damonte, G.; Serafini, S.; Bigi, L.; Panzani, I.; Novelli, G.; Dallapiccola, B.; Panunzi, S.; et al. Low doses of dexamethasone constantly delivered by autologous erythrocytes slow the progression of lung disease in cystic fibrosis patients. Blood Cells Mol. Dis. 2004, 33, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Rossi, L.; Serafini, S.; Cenerini, L.; Picardi, F.; Bigi, L.; Panzani, I.; Magnani, M. Erythrocyte-mediated delivery of dexamethasone in patients with chronic obstructive pulmonary disease. Biotechnol. Appl. Biochem. 2001, 33, 85–89. [Google Scholar] [CrossRef]
- Skorokhod, O.; Kulikova, E.V.; Galkina, N.M.; Medvedev, P.V.; Zybunova, E.E.; Vitvitsky, V.M.; Pivnik, A.V.; Ataullakhanov, F.I. Doxorubicin pharmacokinetics in lymphoma patients treated with doxorubicin-loaded eythrocytes. Haematologica 2007, 92, 570–571. [Google Scholar] [CrossRef] [Green Version]
- Garin, M.I.; Lopez, R.M.; Luque, J. Pharmacokinetic properties and in-vivo biological activity of recombinant human erythropoietin encapsulated in red blood cells. Cytokine 1997, 9, 66–71. [Google Scholar] [CrossRef]
- Sinauridze, E.I.; Vuimo, T.A.; Kulikova, E.V.; Shmyrev, I.I.; Ataullakhanov, F.I. A new drug form of blood coagulation factor IX: Red blood cell-entrapped factor IX. Med. Sci. Monit. 2010, 16, PI19–PI26. [Google Scholar]
- Eichler, H.G.; Rameis, H.; Bauer, K.; Korn, A.; Bacher, S.; Gasic, S. Survival of gentamicin-loaded carrier erythrocytes in healthy human volunteers. Eur. J. Clin. Investig. 1986, 16, 39–42. [Google Scholar] [CrossRef]
- Deloach, J.R.; Wagner, G.G.; Corrier, D.E. Pharmacokinetics of Imidocarb Dipropionate Encapsulated in Carrier Erythrocytes and Use of Carrier Cells for Babesiosis Chemotherapy. J. Control. Release 1989, 9, 243–248. [Google Scholar] [CrossRef]
- Bhikshapathi, D.V.R.N.; Krishna, A.S.; Ramesh, M.; Rajesham, V.V.; Suresh, G.; Sri, S.J. Erythrocytes as Carriers of Indinavir: Preparation, Characterization, in vitro and in vivo Pharmacokinetic Evaluation in Rats. Int. J. Pharm. Sci. Nano 2017, 10, 3573–3581. [Google Scholar]
- Kravtzoff, R.; Ropars, C.; Laguerre, M.; Muh, J.P.; Chassaigne, M. Erythrocytes as Carriers for L-Asparaginase-Methodological and Mouse Invivo Studies. J. Pharm. Pharmacol. 1990, 42, 473–476. [Google Scholar] [CrossRef] [PubMed]
- Kwon, Y.M.; Chung, H.S.; Moon, C.; Yockman, J.; Park, Y.J.; Gitlin, S.D.; David, A.E.; Yang, V.C. L-Asparaginase encapsulated intact erythrocytes for treatment of acute lymphoblastic leukemia (ALL). J. Control. Release 2009, 139, 182–189. [Google Scholar] [CrossRef] [Green Version]
- Yuan, S.H.; Ge, W.H.; Huo, J.; Wang, X.H. Slow release properties and liver-targeting characteristics of methotrexate erythrocyte carriers. Fundam. Clin. Pharmacol. 2009, 23, 189–196. [Google Scholar] [CrossRef]
- Yew, N.S.; Dufour, E.; Przybylska, M.; Putelat, J.; Crawley, C.; Foster, M.; Gentry, S.; Reczek, D.; Kloss, A.; Meyzaud, A.; et al. Erythrocytes encapsulated with phenylalanine hydroxylase exhibit improved pharmacokinetics and lowered plasma phenylalanine levels in normal mice. Mol. Genet. Metab. 2013, 109, 339–344. [Google Scholar] [CrossRef]
- Shavi, G.V.; Doijad, R.C.; Deshpande, P.B.; Manvi, F.V.; Meka, S.R.; Udupa, N.; Omprakash, R.; Dhirendra, K. Erythrocytes as Carrier for Prednisolone: In Vitro and in Vivo Evaluation. Pak. J. Pharm. Sci. 2010, 23, 194–200. [Google Scholar]
- Leung, P.; Cannon, E.P.; Petrikovics, I.; Hawkins, A.; Way, J.L. In vivo studies on rhodanese encapsulation in mouse carrier erythrocytes. Toxicol. Appl. Pharmacol. 1991, 110, 268–274. [Google Scholar] [CrossRef]
- Ganguly, K.; Krasik, T.; Medinilla, S.; Bdeir, K.; Cines, D.B.; Muzykantov, V.R.; Murciano, J.C. Blood clearance and activity of erythrocyte-coupled fibrinolytics. J. Pharmacol. Exp. Ther. 2005, 312, 1106–1113. [Google Scholar] [CrossRef]
- Ito, Y.; Ogiso, T.; Iwaki, M.; Atago, H. Encapsulation of human urokinase in rabbit erythrocytes and its disposition in the circulation system in rabbits. J. Pharm. 1987, 10, 550–556. [Google Scholar] [CrossRef] [Green Version]
- Khandelwal, S.; van Rooijen, N.; Saxena, R.K. Reduced expression of CD47 during murine red blood cell (RBC) senescence and its role in RBC clearance from the circulation. Transfusion 2007, 47, 1725–1732. [Google Scholar] [CrossRef] [PubMed]
- Gabizon, A.; Shmeeda, H.; Barenholz, Y. Pharmacokinetics of pegylated liposomal Doxorubicin: Review of animal and human studies. Clin. Pharm. 2003, 42, 419–436. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wang, E.Q.; Balthasar, J.P. Monoclonal antibody pharmacokinetics and pharmacodynamics. Clin. Pharmacol. Ther. 2008, 84, 548–558. [Google Scholar] [CrossRef] [PubMed]
- Beck, A.; Goetsch, L.; Dumontet, C.; Corvaia, N. Strategies and challenges for the next generation of antibody-drug conjugates. Nat. Rev. Drug Discov. 2017, 16, 315–337. [Google Scholar] [CrossRef] [PubMed]
- Havel, H.; Finch, G.; Strode, P.; Wolfgang, M.; Zale, S.; Bobe, I.; Youssoufian, H.; Peterson, M.; Liu, M. Nanomedicines: From Bench to Bedside and Beyond. AAPS J. 2016, 18, 1373–1378. [Google Scholar] [CrossRef] [PubMed]
- Hamidi, M.; Tajerzadeh, H. Carrier erythrocytes: An overview. Drug Deliv. 2003, 10, 9–20. [Google Scholar] [CrossRef]
- Muzykantov, V.R.; Murciano, J.C.; Taylor, R.P.; Atochina, E.N.; Herraez, A. Regulation of the complement-mediated elimination of red blood cells modified with biotin and streptavidin. Anal. Biochem. 1996, 241, 109–119. [Google Scholar] [CrossRef]
- Muzykantov, V.R.; Murciano, J.C. Attachment of antibody to biotinylated red blood cells: Immuno-red blood cells display high affinity to immobilized antigen and normal biodistribution in rats. Biotechnol. Appl. Bioc. 1996, 24, 41–45. [Google Scholar]
- Deng, R.; Balthasar, J.P. Comparison of the effects of antibody-coated liposomes, IVIG, and anti-RBC immunotherapy in a murine model of passive chronic immune thrombocytopenia. Blood 2007, 109, 2470–2476. [Google Scholar] [CrossRef] [Green Version]
- Villa, C.H.; Pan, D.C.; Johnston, I.H.; Greineder, C.F.; Walsh, L.R.; Hood, E.D.; Cines, D.B.; Poncz, M.; Siegel, D.L.; Muzykantov, V.R. Biocompatible coupling of therapeutic fusion proteins to human erythrocytes. Blood Adv. 2018, 2, 165–176. [Google Scholar] [CrossRef] [Green Version]
- Wibroe, P.P.; Anselmo, A.C.; Nilsson, P.H.; Sarode, A.; Gupta, V.; Urbanics, R.; Szebeni, J.; Hunter, A.C.; Mitragotri, S.; Mollnes, T.E.; et al. Bypassing adverse injection reactions to nanoparticles through shape modification and attachment to erythrocytes. Nat. Nanotechnol. 2017, 12, 589–594. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Company | Drug | Disease | Trial Identifier |
|---|---|---|---|
| EryDel | Dexamethasone | Ataxia Telangiectasia | NCT03563053 |
| Erytech | L-Asparaginase | Triple-Negative Breast Cancer | NCT03674242 |
| Acute Lymphoblastic Leukemia | NCT03267030 | ||
| Pancreatic Ductal Adenocarcinoma | NCT03665441 | ||
| Anokion | KAN-101 | Celiac Disease | NCT04248855 |
| Rubius | Phenylalanine Ammonia Lyase | Phenylkentonuria | NCT04110496 |
| Coupling Strategy | Drug | Indication | References |
|---|---|---|---|
| Streptavidin-Biotin | tPA | Pulmonary Embolism | [36,37] |
| Arterial Thrombosis | [36] | ||
| Thrombotic Stroke | [30,38] | ||
| Traumatic Brain Injury | [31] | ||
| Cerebral Hypoxia | [32] | ||
| Antibody (or Fragment) Binding | tPA | Pulmonary Embolism | [39] |
| Arterial Thrombosis | [39] | ||
| Pulmonary Embolism | [18] | ||
| Reteplase | Venous Thrombosis | [40] | |
| scuPA-T | Cerebral Thrombosis | [33] | |
| Thrombomodulin | Vascular Thrombosis | [17] | |
| Endotoxemia | [20] | ||
| Cerebral Ischemia/Reperfusion | [20] | ||
| Peptide Binding | Protein Antigens | Immune Tolerance Induction | [41,42] |
| Passive Adsorption | NP-Reteplase | Pulmonary Embolism | [6] |
| NP-Doxorubicin | Lung Metastasis | [5] |
| Parameter | Definition |
|---|---|
| Area Under the Curve (AUC) | Primary metric of overall drug exposure |
| Terminal Half-Life (t1/2) | Time for drug concentrations to reduce by 50% during the terminal slope (λz) |
| Maximum Blood Concentration (Cmax) | Highest observed blood concentration |
| Time of Cmax (tmax) | Time post-dosing where Cmax occurs |
| Bioavailability (F) | Fraction of administered dose that reaches the systemic circulation |
| Clearance (CL) | Volume cleared of drug per unit time |
| Mean Residence Time (MRT) | Average time that a drug molecule stays in the body |
| Volume of Distribution (Vss) | Relationship between the amount of drug in the body and the blood concentration |
| Route | Typical tmax | Barriers | Advantages | Clinical Use |
|---|---|---|---|---|
| Oral | Variable a | Harsh GI environment Efflux transporters First-pass metabolism | Safe and painless Patient convenience | Small molecule |
| Subcutaneous | Hours–days [67] | Immune system | Patient convenience | Peptides |
| No first-pass | Proteins | |||
| Inhaled | Seconds–minutes [68] | Airway branching | Local delivery | Small molecule |
| Muco-ciliary clearance | Rapid absorption | |||
| Immune system | No first-pass | |||
| Transdermal | Hours–days [69] | Dense layers of skin and fat | Prolonged delivery | Small molecule |
| Immune system | No first-pass |
| Drug Class | Mechanisms | Barriers |
|---|---|---|
| Small Molecule | Diffusion | Plasma protein binding |
| Uptake transporters [71] | Efflux transporters [71] | |
| Peptides/Proteins | Diffusion (Low MW) | Vascular permeability [72] |
| Bulk fluid flow Receptor-mediated transcytosis [73] | ||
| Drug Delivery Systems | Bulk fluid flow Receptor-mediated transcytosis [73] | Vascular permeability [72] |
| Erythrocytes | N/A | Vascular permeability [72] |
| Drug Class | Mechanisms | Primary Tissues |
|---|---|---|
| Small Molecule | Renal filtration | Kidney |
| Active tubular secretion | Kidney | |
| Metabolism [74] | Liver, GI, etc. | |
| Peptides/Proteins | Renal filtration (<60 kDa) | Kidney |
| Non-specific catabolism | Liver, spleen, etc. | |
| Receptor-mediated clearance | Target tissue | |
| Drug Delivery Systems [75] | Immune cell uptake | Liver, spleen |
| Receptor-mediated clearance | Target tissue | |
| Erythrocytes | Macrophage uptake | Spleen, liver |
| Drug | Species | Condition | PK Changes | Pharmacologic Effect (Relative to Free Drug) | References |
|---|---|---|---|---|---|
| 5-Fluoruracil (5-FU) | Mouse | Malignant Ascites | 2-fold increase in AUC0-inf in ascites fluid | 70% survival at 20 days vs. 20% in malignant ascites model | [79] |
| Adenosine Deaminase (ADA) | Human | ADA Deficiency | 2–4-fold increase in ADA t1/2 57-day lifespan of loaded RBC | [80] | |
| Alcohol Dehydrogenase (ADH) Aldehyde Dehydrogenase (ALDH) | Mouse | Healthy | 4.5-day RBC t1/2 | 43% reduction in blood ethanol concentrations vs. empty RBC | [81] |
| Amikacin | Rat | Healthy | 2-fold increase in AUC0-inf in plasma Large increases in liver/spleen AUC0-inf | [82,83] | |
| Carbonic Anhydrase | Rat | Healthy | Similar circulation time as carrier RBC 9-day t1/2 of loaded RBC | [84] | |
| Daunorubicin | Human | Acute Leukemia | ~2-fold increase in blood t1/2 | [85] | |
| Dexamethasone | Human Human Human Rabbit | Inflammatory Bowel Disease Chronic Obstructive Pulmonary Disease Cystic Fibrosis Healthy | Plasma concentrations detectable 28 days post-infusion Plasma concentrations detectable for at least 1 week post-infusion Relatively constant plasma concentrations for at least 10 days ~60-fold increase in plasma t1/2 | 50% reduction in ESR and CRP relative to standard of care Reduction in ‘as-needed’ use of corticosteroids and β-agonists Improved FEV1 and 51% reduction in antibiotic use Reduction in histamine response | [86,87,88,89,90] |
| Doxorubicin | Human | Lymphoma | 2-7-fold increase in plasma t1/2 | [91] | |
| Erythropoietin | Mouse | Healthy | ~5-fold increase in blood AUC 5.6 day RBC t1/2 | ~2-fold increase in 59Fe incorporation into circulating RBC | [92] |
| Factor IX | Human | Healthy | ~8-fold increase in blood t12 | [93] | |
| Gentamicin | Human | Healthy | 22 day blood t1/2 | [94] | |
| Imidocarb | Mouse | Parasitemia | Significantly increased blood concentrations | ~25% reduction in peak parasitemia | [95] |
| Indinavir | Rat | Healthy | 9-fold increase in plasma AUC0-inf | [96] | |
| L-Asparaginase | Mouse Mouse Mouse | Healthy Healthy Acute Lymphoblastic Leukemia | ~3-fold increase in blood t1/2 9–10.6 day RBC t1/2 16-fold increase in blood t1/2 2.4–4 day blood t1/2 | 4–5-fold increase in duration of maximal asparagine lowering >10-fold increase in duration of total asparagine suppression Reduced ADA formation 44% increase in survival time vs. untreated | [42,97,98] |
| Maltose-Binding Protein | Mouse | Healthy | ~3-fold increase in blood t1/2 | [15] | |
| Methotrexate | Mouse | Healthy | 3.5-fold increase in plasma t1/2 ~2-fold increase in liver and spleen uptake | [99] | |
| Phenylalanine Hydroxylase | Mouse | Naive | Detectable drug in blood for at least 10 days post-injection vs. <6 h | ~50% reduction in blood Phe vs. 25% | [100] |
| Prednisolone | Rat | Healthy | High drug uptake in liver | [101] | |
| Polystyrene Nanoparticles | Mouse | Healthy | 2–3-fold increase in blood exposure ~5-fold increase in lung uptake >50% decrease in spleen uptake No effect on RBC survival (t1/2 = 33.5 h) | [58] | |
| Reteplase | Mouse | Acute Thrombosis | Blood t1/2 of ~10 h vs. minutes No impact on RBC circulation time | ~3-fold delay in time to arterial occlusion Complete prevention of venous occlusion | [18] |
| Rhodanese | Mouse | Healthy | 230-fold increase in t1/2 | 40% reduction in blood cyanide following IV injection | [102] |
| Tissue Plasminogen Activator | Mouse Rat | Acute Thrombosis Acute Thrombosis | ~10-fold increase in blood exposure No changes in RBC survival >10-fold increase in blood AUC Minimal effects on RBC circulation | ~50% lysis of pulmonary emboli Significant reduction in mortality from thromboembolic stroke ~80% lysis of pulmonary emboli ~80% of blood flow recovery in carotid artery | [30,31,32,36,38,39,103] |
| Thrombomodulin | Mouse | Acute Thrombosis Ischemic Stroke Endotoxemia | 10% of drug present in blood 2 days post-injection vs. 1 h No changes in RBC survival | Complete protection against jugular vein thrombosis ~50% reduction in infarct volume and neurological deficit >50-fold improved potency at reduction of pro-inflammatory cytokines | [17,20] |
| Urokinase | Rabbit Mouse | Healthy Acute Thrombosis | Significant increase in blood exposure 14-fold increase in blood concentration at 30 min No changes in RBC survival | 4–5-fold increase in blood flow following carotid artery thrombosis ~3-fold increase in blood flow following venous thrombosis | [33,40,104] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Glassman, P.M.; Villa, C.H.; Ukidve, A.; Zhao, Z.; Smith, P.; Mitragotri, S.; Russell, A.J.; Brenner, J.S.; Muzykantov, V.R. Vascular Drug Delivery Using Carrier Red Blood Cells: Focus on RBC Surface Loading and Pharmacokinetics. Pharmaceutics 2020, 12, 440. https://doi.org/10.3390/pharmaceutics12050440
Glassman PM, Villa CH, Ukidve A, Zhao Z, Smith P, Mitragotri S, Russell AJ, Brenner JS, Muzykantov VR. Vascular Drug Delivery Using Carrier Red Blood Cells: Focus on RBC Surface Loading and Pharmacokinetics. Pharmaceutics. 2020; 12(5):440. https://doi.org/10.3390/pharmaceutics12050440
Chicago/Turabian StyleGlassman, Patrick M., Carlos H. Villa, Anvay Ukidve, Zongmin Zhao, Paige Smith, Samir Mitragotri, Alan J. Russell, Jacob S. Brenner, and Vladimir R. Muzykantov. 2020. "Vascular Drug Delivery Using Carrier Red Blood Cells: Focus on RBC Surface Loading and Pharmacokinetics" Pharmaceutics 12, no. 5: 440. https://doi.org/10.3390/pharmaceutics12050440