Erythrocytes as Carriers of Therapeutic Enzymes


Molecular and Clinical Sciences, St. George’s, University of London, London SW17 0RE, UK
Pharmaceutics 2020, 12(5), 435; https://doi.org/10.3390/pharmaceutics12050435
Submission received: 30 March 2020
/
Revised: 21 April 2020
/
Accepted: 6 May 2020
/
Published: 8 May 2020
(This article belongs to the Special Issue Cell-Based Drug-Delivery Platforms)
Abstract
:Therapeutic enzymes are administered for the treatment of a wide variety of diseases. They exert their effects through binding with a high affinity and specificity to disease-causing substrates to catalyze their conversion to a non-noxious product, to induce an advantageous physiological change. However, the metabolic and clinical efficacies of parenterally or intramuscularly administered therapeutic enzymes are very often limited by short circulatory half-lives and hypersensitive and immunogenic reactions. Over the past five decades, the erythrocyte carrier has been extensively studied as a strategy for overcoming these limitations and increasing therapeutic efficacy. This review examines the rationale for the different therapeutic strategies that have been applied to erythrocyte-mediated enzyme therapy. These strategies include their application as circulating bioreactors, targeting the monocyte–macrophage system, the coupling of enzymes to the surface of the erythrocyte and the engineering of CD34+ hematopoietic precursor cells for the expression of therapeutic enzymes. An overview of the diverse biomedical applications for which they have been investigated is also provided, including the detoxification of exogenous chemicals, thrombolytic therapy, enzyme replacement therapy for metabolic diseases and antitumor therapy.
1. Introduction
Therapeutic enzymes are biocatalyst drugs that bind to target substrates with a high affinity and specificity, catalyzing their conversion into their relevant products. The past five decades have seen the development of therapeutic enzymes for treating a wide range of medical conditions, including inherited enzyme deficiency disorders, acute poisoning, digestive disorders, cancer and cardiovascular diseases. Chemical modifications of the native enzyme (e.g., conjugation with polyethylene glycol) are often employed in the manufacturing process to increase protein stability, decrease immunogenicity, reduce renal ultrafiltration and in some cases, to enable targeting of the enzyme to the appropriate cellular compartment [1]. However, despite these strategies, the metabolic and clinical efficacy of parenterally or intramuscularly administered therapeutic enzymes is still limited. This is principally due to short circulatory half-lives, hypersensitivity reactions and immunogenicity. Immune responses are influenced by several factors including the biophysical characteristics of the protein, the route of delivery, the degree of exposure and the use of immunosuppressive agents during administration. Host factors may also play a part, for example patients with congenital enzyme deficiencies may fail to recognize a therapeutic protein as “self” and may be more likely to mount an immune response during the administration of therapeutic replacement enzyme [2]. The consequences of an immune reaction to a therapeutic enzyme range from a transient appearance of antibodies, without any clinical sequel, to severe life-threatening conditions.
The erythrocyte carrier has been extensively studied as a strategy for overcoming these limitations and increasing therapeutic efficacy. For a majority of the therapeutic applications investigated, the ability of the cell to reseal after creating pores in the membrane has been exploited for the purpose of introducing therapeutic agents [3,4,5]. The resealed erythrocyte is biocompatible and, in the human, has a normal in vivo circulating half-life of 19–29 days, and thus raises the potential to extend the half-life of encapsulated enzymes through the avoidance of plasma clearance due to the action of proteases, anti-enzyme antibodies and renal clearance, and through minimizing immune reactions.
The aim of this article is to provide a review of the available literature relating to in vitro, preclinical and clinical studies of the erythrocyte as a vehicle for therapeutic enzymes.
2. Therapeutic Strategies
Four main therapeutic strategies have been applied to the erythrocyte carrier in terms of a vehicle for therapeutic enzymes. In the first, enzyme-loaded erythrocytes can be employed as circulating bioreactors whereby blood-based molecules diffuse into the erythrocyte and are degraded. This strategy is relevant to the treatment of congenital enzyme-deficiencies where a pathologically elevated plasma metabolite is able to permeate erythrocyte membrane and undergo metabolism to the normal product [6]. Also relevant to this therapeutic strategy is the depletion of plasma metabolites for the treatment of auxotrophic tumors and detoxification after exposure to toxic chemicals, Figure 1.
The second strategy involves the targeting of encapsulated enzymes to the monocyte–macrophage system of the spleen, liver and bone marrow, through exploiting the natural sites of erythrophagocytosis [7]. The loss of phospholipid asymmetry and the exposure of phosphatidylserine on the surface of the plasma membrane of senescence erythrocytes induces erythrocyte sequestration by macrophages. As erythrocyte degradation occurs in the lysosomal compartment of macrophages, it is proposed that erythrocyte encapsulated enzymes can potentially target macromolecules that accumulate in the lysosomal compartment as a result of congenital enzyme-deficiencies, Figure 2.
The third strategy involves the coupling of therapeutic enzymes to the erythrocyte surface with the aim of improving the therapeutic profile, Figure 3.
The fourth strategy is a relatively new approach. This is based on the engineering of CD34+ hematopoietic precursor cells to express therapeutic enzymes and then their subsequent differentiation until the nucleus is ejected, resulting in the mature reticulocyte, Figure 4. This strategy has potential applications for both the metabolism of plasma metabolites and targeting of the monocyte–macrophage system.
3. Therapeutic Applications
Over the past five decades, the erythrocyte carrier has been extensively investigated as a potential modality for targeting a wide range of therapeutic applications. In relation to carriers of enzymes, these applications can broadly be divided into four key areas of research interest: exogeneous chemical detoxification, thrombolytic therapy, treatments for metabolic diseases and antitumor therapies. Although a majority of these studies have not translated beyond the pre-clinical stage, a few are currently undergoing clinical development. The following provides a discussion of the applications investigated under these four research areas.
3.1. Detoxification of Exogenous Chemicals
The utility of the erythrocyte as a carrier of specific enzymatic antidotes against chemical intoxicants has been investigated by a number of groups. Investigations into this therapeutic application have focused on encapsulating the relevant enzyme within the erythrocyte. Way et al. first established the potential of the erythrocyte carrier as antidote candidate for cyanide intoxication [8,9]. Cyanide is a rapidly lethal poison and exerts its toxic effect by blocking the mitochondrial respiration chain and the formation of intracellular adenosine triphosphate through binding to cytochrome-c oxidase, the terminal enzyme complex of the respiratory chain in complex IV. Sodium thiosulfate is one of the antidotes used to combat intoxication and works by acting as a sulfur donor for the mitochondrial enzyme, thiosulfate sulfotransferase (rhodanase, EC 2.8.1.1) [10]. Sodium thiosulfate does not readily permeate cell membranes and therefore is not able to distribute to sites of thiosulfate sulfotransferase or cyanide localization; this provided the rationale for investigating the erythrocyte carrier as an alternative approach to cyanide antagonism. In vivo studies in mice demonstrated that erythrocytes containing sodium thiosulfate and rhodanase could rapidly metabolize cyanide to the less toxic thiocyanate and antagonize the effects of a lethal dose of potassium cyanide [11,12,13]. Moreover, by replacing sodium thiosulfate with butanethiosulfonate, a more reactive sulfur donor substrate, an enhanced protective effect against cyanide was found [14].
The application of the erythrocyte carrier as an antagonist of the lethal effects of parathion, a once widely used agricultural organophosphorous insecticide was also investigated as an alternative antidote approach to paraoxon intoxication. The toxicity of parathion is attributed to its in vivo metabolism to paraoxon which inhibits acetylcholinesterase, leading to an accumulation of acetylcholine and ultimately altering cholinergic synaptic transmission at neuroeffector junctions, at skeletal myoneural junctions and autonomic ganglia in the central nervous system [15]. Two antidotes for parathion poisoning are atropine, a competitive antagonist of acetylcholine at the muscarinic receptor and pralidoxime, which regenerates acetylcholinesterase [16]. However, neither of these antidotes are able to degrade parathion. In vivo studies in mice investigated the efficacy of erythrocyte encapsulated phosphotriesterase (EC 3.1.8.1) in antagonizing the lethal effects of paraoxon through its hydrolysis to the less toxic 4-nitrophenol and diethylphosphate [17]. The results indicated that although the phosphotriesterase-loaded erythrocytes were more effective than the classic antidotal combination of atropine and pralidoxime, a combination of the loaded erythrocytes with the classic antidot, provided a 1000-fold protection against paraoxon. The same group also investigated the application of erythrocyte encapsulated recombinant paraoxonase as an approach to directly hydrolyze paraoxon; treated mice showed no signs of intoxication at paraoxon dose levels that were lethal when using the classical antidotal combination of atropine and pralidoxime. Moreover, erythrocyte encapsulated paraoxonase, in combination with the classic antidotal combination, provided the highest antidotal efficacy ever reported against any chemical toxicant [18].
Another category of detoxifying enzymes that have been investigated are those associated with the metabolism of ethanol and methanol. Ethanol detoxification requires two enzymic reactions: the oxidation of ethanol to acetaldehyde by alcohol dehydrogenase (EC. 1.1.1.1), followed by the oxidation acetaldehyde to acetate by aldehyde dehydrogenase (EC 1.2.1.5). Chronic alcohol consumption decreases acetaldehyde oxidation, either due to decreased aldehyde dehydrogenase activity or impaired mitochondrial function. The application of the erythrocyte carrier as an alcohol detoxifier was first proposed by Magnani et al. They demonstrated that mice administered acetaldehyde dehydrogenase-loaded erythrocytes intraperitoneally had 35% less blood acetaldehyde compared to controls, one hour after receiving an acute dose of ethanol [19]. Lizano et al. investigated the co-encapsulation of alcohol dehydrogenase and aldehyde dehydrogenase in human erythrocytes, demonstrating their superior ability to metabolize ethanol in vitro, compared to erythrocytes loaded with alcohol dehydrogenase alone [20]. In vivo studies in mice receiving acute doses of ethanol and treated with co-encapsulated enzymes showed blood ethanol to be eliminated at rates of 1.7 mmol/L to 4 mmol/L loaded erythrocytes/hour [21,22]. However, these rates of plasma ethanol clearance were lower by an order of magnitude than those expected from the activities of encapsulated enzymes. By employing a mathematical modeling approach and then conducting a subsequent in vitro study, Alexandrovich et al. were able to theorize and then demonstrate the rate limiting step of external ethanol oxidation. They found this was due to the rate of nicotinamide–adenine dinucleotide (NAD+) generation in erythrocyte glycolysis, rather than the activities of the loaded enzymes. By supplementing the erythrocytes with NAD+ and pyruvate they were able to demonstrate an elimination of 17 mmol ethanol/L loaded erythrocytes/hour [23].
In mammalian species, methanol is metabolized to formaldehyde via alcohol dehydrogenase, followed by the conversion of formaldehyde into formic acid via aldehyde dehydrogenase. Formic acid metabolism is mediated through a tetrahydrofolate-dependent pathway by folate-dependent enzymes. Humans have 60% less liver folate concentrations compared to mice and rats, and for this reason humans are more sensitive to methanol poisoning [24]. Specifically, formic acid inhibits mitochondrial cytochrome c oxidase, leading to cellular hypoxia and metabolic acidosis. Magnani et al. investigated the application of erythrocyte-encapsulated methylotrophic yeast alcohol oxidase (EC 1.1.3.13) as an approach to the detoxification of methanol. In vivo studies showed that two hours following an acute dose of methanol (0.7 g/kg), mice that had received enzyme-loaded erythrocytes had 50% less blood methanol compared to controls, with antagonism persisting for at least one week [25]. On the basis that formate dehydrogenase (EC 1.2.1.2) converts formate into CO2 in the presence of NAD, Muthuvel et al. administered formate dehydrogenase-loaded erythrocytes to methanol-intoxicated folate-deficient rats, which were pre-treated with carbicarb to correct the metabolic acidosis, and demonstrated a marked elimination of formate [26].
A detoxifying enzyme that has been investigated in relation to lead poisoning is δ-aminolevulinic acid dehydratase (E.C. 4.2.1.24). Lead is a potent inhibitor of δ-aminolevulinic acid dehydratase through its displacement of zinc from the enzyme’s active site. The resulting inactivation leads to an accumulation of its substrate, aminolevulinic acid, and this has been shown to have a neuropathogenic effect [27]. The in vitro and in vivo mouse studies of Bustos et al. showed that it was possible to correct the defective δ-aminolevulinic acid dehydratase activity in erythrocytes by encapsulating exogenous human enzyme, thus providing a rationale for this approach for treating lead intoxication [28,29]. In the first clinical application, 100 mL autologous erythrocytes loaded with 25,600 units of human δ-aminolevulinic acid dehydratase were administered to a single patient with a history of chronic lead intoxication, producing a reduction in blood lead concentration and decrease in urinary porphyrin excretion, which was sustained for a period of four years [30].
Biochemical decompression has been proposed as a method for reducing the amount of time required for deep-sea divers to return to the surface. Divers breathing H2/O2 mixtures would be administered hydrogenase (EC 1.12.1.4), thus accelerating decompression through the removal of excess hydrogen from the tissues. The application of erythrocyte encapsulated hydrogenase as an approach to converting dissolved hydrogen gas into non-gaseous forms was investigated by Axley et al. Hydrogenase activities of 7-µmol H2/minute/mL packed human erythrocytes were successfully encapsulated, however the cells were not able to consume hydrogen due to the enzyme’s dependence on NAD for activity. The co-encapsulation of FAD with hydrogenase was shown to overcome this [31].
3.2. Thrombolytic Therapy
The application of erythrocytes as a strategy for thrombolytic therapy has been explored by several groups. Thrombolytics (or fibrinolytics), also known as plasminogen activators, facilitate the removal of pre-existing thrombotic clots through degrading the fibrin meshwork into plasmin. However, their application is limited by their rapid clearance and inactivation by plasminogen activator inhibitor-1, the risk of bleeding caused by dissolution of hemostatic clots and filtration into extravascular tissues, such as the central nervous system, causing toxicity [32]. The rationale for the erythrocyte encapsulation of fibrinolytics was based on addressing these issues with the study of Delahousse et al. who successfully demonstrated the encapsulation of the fibrinolytic enzymes urokinase (EC 3.4.21.31), streptokinase (EC 3.4.99.0) and recombinant tissue plasminogen activator (tPA; EC 3.4.21.68) in human erythrocytes. However, in vitro stability studies using human erythrocytes and in vivo studies in the mouse were not able to support the objective of a sustained half-life and demonstrated a rapid destruction of the encapsulated enzymes [33]. Flynn and coworkers reported that Aspergillus oryzae brinase (EC. 3.4.99) encapsulated into rabbit erythrocytes was able to lyse clotted blood in vitro [34]. The coupling of fibrinolytics to the surface of the erythrocyte carrier was proposed as a preferable alternative to encapsulation [35]. Murciano et al. investigated the conjugation of tPA to erythrocytes as a strategy for thromboprophylaxis, by employing mouse and rat models of venous and arterial thrombosis. Following intravenous injection, the fibrinolytic activity of tPA conjugated to erythrocytes was shown to persist in the circulation at least tenfold longer than that of free tPA. Free tPA was able to lyse pulmonary clots that had lodged before administration, but not those that lodged after injection. Erythrocyte conjugated-tPA, however was more selective in lysing nascent over preexisting pulmonary emboli and arterial clots, an effect that is most likely a result of restricting the diffusion of tPA into fibrin [36]. The utility of this approach in preventing cerebrovascular thrombosis was also examined using mouse and rat models of cerebrovascular thromboembolism and ischemia, with the results indicating that erythrocyte-conjugated tPA was an effective thromboprophylaxis, whereas free tPA was ineffective up to a 10-fold higher dose [37].
Worthy of mention is the more recent studies investigating the erythrocyte carrier as a theranostic nanoplatform system. This system consists of vesicles derived from erythrocytes that encapsulate the NIR fluorophore, indocyanine green and of relevance to this review, the conjugation of tPA to the vesicle surface. These constructs are referred to as NIR erythrocyte-derived transducers (NETS). In vitro studies employing a clot model demonstrated the dual functionality of NETS in NIR imaging and clot lysis [38].
3.3. Enzyme Replacement for Metabolic Diseases
The application of the erythrocyte as a carrier of enzymes for the treatment of inherited metabolic diseases and other metabolic disturbances has generated a large amount of interest from those working in a field where there are enormous unmet needs. The following provides a discussion of the metabolic disorders to which the erythrocyte carrier has been applied. With the exception of phenylketonuria, these therapeutic applications have focused on the use of encapsulated enzyme.
3.3.1. Lysosomal Storage Disorders
Ihler et al. first proposed the erythrocyte carrier as a strategy for targeting enzyme replacement for the treatment of disorders of sphingolipid catabolism, and successfully demonstrated the erythrocyte encapsulation of β-glucosidase (EC.3.2.1.45) and β-galactosidase (EC 3.2.1.23) [3]. Sphingolipidoses are a subgroup of lysosomal storage disorders characterized by an abnormal accumulation of phospholipids that have a sphingosine group. The rationale for the therapeutic strategy is based on the erythrocyte being naturally sequestered by cells of the monocyte–macrophage system and subsequent lysosomal degradation at the subcellular site where the enzyme defect manifests, thereby facilitating the delivery of the encapsulated enzyme to the site of phospholipid accumulation. Deloach and coworkers demonstrated that human erythrocytes-loaded with E. coli β-galactosidase could be phagocytosed by bone marrow macrophages that had been matured in vitro. Electron microscopy revealed that intact or partly degraded erythrocytes were localized in intracellular vacuoles, which were presumed to be phagolysosomes. The activity of the untaken β-galactosidase disappeared with a half-life of 15–30 h, which was consistent with the enzyme being degraded within the lysosomes [39]. Studies comparing the fate of erythrocyte-encapsulated bovine β-glucuronidase (EC 3.2.1.31) and free enzyme administered intravenously to β-glucuronidase-deficient mice demonstrated that erythrocyte encapsulated enzyme was retained four times longer in the circulation, five-fold longer in hepatic tissues and was more efficiently delivered to a number of other tissues [40]. In 1977, Beulter et al. reported the first clinical application of the erythrocyte carrier for the treatment of Gaucher disease (Online Mendelian inheritance in Man (OMIM) # 230800, 230900, 231000). Gaucher disease is caused by mutations in the GBA gene which encodes for the enzyme β-glucocerebrosidase (also referred to as β-glucosidase), which catalyzes the hydrolysis of glucosylceramide into ceramide and glucose. A deficiency in the enzyme leads to an accumulation of glucosylceramide in the lysosomes of macrophages in the liver, spleen and bone marrow. The disease is classified into three clinical forms on the basis of neurological involvement: type I (non-neuronopathic), type II (acute neuronopathic) and type III (subacute neuronopathic) with all three having symptoms of hepatosplenomegaly, anemia and orthopedic complications [41]. In the first clinical application, partially purified glucocerebrosidase isolated from the human placenta was encapsulated into erythrocytes and administered to a single patient with advanced type I Gaucher disease. Although there were no conclusive findings that enzyme infusion had been beneficial, this study was able to demonstrate the safety of repeated administrations of enzyme encapsulation in erythrocytes [42]. Studies investigating the in vitro uptake of glucocerebrosidase by Gaucher patient monocytes demonstrated that enzyme loaded erythrocytes coated with human IgG and agglutinated with anti-human serum were more avidly phagocytosed compared to uncoated loaded erythrocytes. In addition, the uptake of enzyme via IgG-coated erythrocytes, was sufficient to normalize cellular glucocerebrosidase activities for at least 18 h [43]. Bax et al. investigated the encapsulation of mannose-terminated glucocerebrosidase (Alglucerase), a licensed pharmaceutical enzyme preparation available for the treatment of Gaucher’s disease. This enzyme preparation has a plasma half-life of 3.6–10.4 min and thus, regular infusions are required to maintain therapeutic levels. Alglucerase encapsulation was found to be maximized by employing the more concentrated pharmaceutical preparation and doubling the hypo-osmotic dialysis time to 180 min [44,45]. Despite the erythrocyte carrier offering a potential solution to the rapid plasma clearance and the prospect of reducing therapy costs and the number of therapeutic interventions, this approach has not been advanced in the clinical setting. The availability of a number of approved and effective enzyme replacement therapies for Gaucher disease that can be administered in the patient’s home no doubt would have contributed to the lack of incentive in developing the erythrocyte carrier further for this disease.
3.3.2. Hyperammonemia
A number of groups have investigated the use of the erythrocyte carrier as an alternative approach to treating metabolic disturbances characterized by an excess of ammonia in the blood. Ammonia is produced by intestinal bacterial flora or as a metabolic byproduct of amino acid catabolism acid and other compounds which contain nitrogen. Normally, ammonia is efficiently handled by the conversion to urea via the urea cycle in the liver or by the reaction catalyzed by glutamine synthetase (EC 6.3.1.2). Congenital deficiencies of enzymes in the urea cycle or diseases that lead to acute or chronic liver failure can result in hyperammonemia, leading to nervous system disturbances, hepatic encephalopathy, coma and death. The metabolic capability of the erythrocyte carrier in reducing blood ammonia concentrations has been investigated by the encapsulation of two enzymes. In the first approach, l-glutamate dehydrogenase (EC. 1.4.1.3) which catalyzes the formation of l-glutamic acid from α-ketoglutarate and ammonium in the presence of NADPH was investigated. The second approach utilized glutamine synthetase (EC. 6.3.1.2) which catalyzes the formation of l-glutamine from l-glutamic acid and ammonium in the presence of ATP. Sanz et al. explored the former approach by examining the capability of encapsulated glutamate dehydrogenase in removing extracellular ammonia in vitro by incubating human loaded erythrocytes in buffer containing ammonium chloride, α-ketoglutarate and NADPH as substrates. After 24 h of incubation, the ammonia concentration was 15 µmol/L, this representing 15% of the zero time concentration and therefore providing support for this approach [46]. Subsequent in vivo studies were performed in mice where hyperammonemia was induced through the intraperitoneal injection of 33 U of urease/Kg of body weight. One hour after urease administration the blood concentration of ammonia was 1333 µmol/L, and this declined to 135 µmol/L by 25 hours. By administering erythrocyte-encapsulated glutamate dehydrogenase to these mice, the blood concentration of ammonia could be reduced by 50%, two hours after urease injection. The ammonia concentration remained unchanged in control mice that received native washed erythrocytes [47]. Using the second approach, Venediktova et al. examined the capability of erythrocytes loaded with glutamine synthetase to eliminate ammonium from the blood of hyperammonemic mice. Blood ammonium concentrations were shown to decrease by approximately 2-fold, 30 min after injection of the loaded cells and 4-fold after 120 min, compared to ammonium concentration in control mice [48,49]. However, after this time, the blood ammonium concentration decreased approximately at the same rate in both the experimental and the control animals. To provide insight into this limitation, Protasov and coworkers developed mathematical models of the erythrocyte carriers by focusing on the glycolytic pathway reactions and enzymes utilizing ammonium. They concluded that the efficiency and duration of the erythrocyte carrier function was limited by the low membrane permeability for α-ketoglutarate and glutamate. To compensate the effect of this, the authors proposed the co-encapsulation of glutamate dehydrogenase and alanine aminotransferase (EC 2.6.1.2). This approach created a metabolic pathway where glutamic acid produced by the glutamate dehydrogenase reaction was converted into α-ketoglutarate by alanine aminotransferase, with α-ketoglutarate then entering the glutamate dehydrogenase reaction. Therefore, α-ketoglutarate and L-glutamic acid are produced and consumed in a cyclical process and hence the system will independent of their transport. The conclusions of the theoretical study were verified experimentally, demonstrating the removal of ammonium in vitro at the rate of 1.5 mmol/h/ L human loaded erythrocytes and in a in vivo model of hyperammonemia in mice at the rate of 2.0 mmol/h/L loaded erythrocytes [50].
An example of a urea cycle disorder that causes hyperammonemia is arginase-1 deficiency (OMIM # 207800). This autosomal recessive disorder is caused by a mutation in the ARG1 gene. Arginase (EC 3.5.3.1) normally controls the final step in the urea cycle through the hydrolysis of arginine to urea and ornithine. Although a deficiency leads to an accumulation of arginine and ammonia in the blood and cerebrospinal fluid, it is the accumulation of arginine which predominantly contributes to the pathological abnormality, with affected individuals experiencing development delay, growth retardation and epileptic seizures [51]. Current treatments focus on reducing plasma ammonia and arginine concentrations and reducing dietary nitrogen. Adriaenssens et al. explored the capacity of arginase-loaded erythrocytes from patients with arginase deficiency to metabolize extracellular arginine in vitro. They demonstrated that when the enzyme-loaded erythrocytes were incubated in plasma containing 45 to 375 µmol/L arginine, the extracellular concentration of arginine was reduced by 34% to 53% after one hour [52].
The utility of the erythrocyte as a carrier of urease (EC 3.5.1.5) for reducing blood concentrations of urea in patients with chronic renal failure has been a long-term interest of Baysal et al. [53]. The rational for this approach is based on urease catabolizing urea into ammonia and bicarbonate, followed by the catalytic conversion of ammonia and erythrocyte pyruvate into alanine by alanine dehydrogenase (EC 1.4.1.1). The ultimate aim is to negate the requirement for hemodialysis and continuous ambulatory peritoneal dialysis. Urease and alanine dehydrogenase and their pegylated counterparts PEG–urease/PEG–alanine dehydrogenase were successfully encapsulated in human erythrocytes [54,55]. In vivo studies in sheep employing erythrocytes containing PEG–urease and PEG–alanine dehydrogenase at an activity unit ratio of 3:9 demonstrated a urea reduction of 51.6 mg/L/24 h. Compared to unencapsulated PEG–urease/PEG–alanine dehydrogenase, which was able to reduce blood urea concentration by 21.7–61.6 mg/L for two days, the same dose encapsulated in erythrocytes sustained these reductions for six days, thus demonstrating the utility of this system in sustaining enzyme activity [56].
3.3.3. Hyperglycemia
The exploitation of enzyme-loaded erythrocytes in the regulation of diabetes associated hyperglycemia was proposed by Magnani et al. who successfully encapsulated hexokinase (EC 2.7.1.1), the first enzyme in the glycolytic pathway, responsible for catalyzing the phosphorylation of glucose by ATP to glucose-6-phosphate. In vitro studies demonstrated that hexokinase-loaded erythrocytes were able to metabolize twice the amount of extracellular glucose compared to unloaded cells, however, this rate of metabolism was deemed insufficient for maintaining blood glucose within the physiological concentrations [57]. De Flora and co-workers investigated the encapsulation of glucose oxidase (EC 1.1.3.4) isolated from Aspergillus niger, another glucose catabolizing enzyme, which converts glucose to hydrogen peroxide and D-glucono-δ-lactone. In vitro studies revealed that glucose oxidase-loaded human and mouse erythrocytes were able to consume glucose 3–4 times faster than the native, unloaded erythrocyte. However, there was a reported 10% increase in methemoglobin formation and a several-fold increase in the activity of the pentose phosphate pathway, indicating a glutathione peroxidase-mediated draining of reduced glutathione for removal of hydrogen peroxide that was produced [58,59]. In vivo studies in the mouse demonstrated a drastically decreased half-life of loaded cells of one to one and a half hours, compared to 10 days for unloaded erythrocyte carriers, with a selective splenic uptake [59]. On the basis that the encapsulation of hexokinase not only increases the glycolytic rate, but also the production of reducing equivalents (NADPH) in response to an oxidative stress, Rossi et al. proposed the co-encapsulation of hexokinase and glucose oxidase in human and mouse erythrocytes. Using this strategy, the authors were able maintain methemoglobin concentrations within an acceptable range and demonstrate a large increase in glucose consumption [60]. In vivo studies in the diabetic mouse model, C57BL/Ks-db/db/01a, revealed that a single intraperitoneal administration of hexokinase/glucose oxidase-loaded erythrocytes could sustain a near normal blood glucose concentration for seven days, while repeated administrations at 10-day intervals were effective in regulating blood glucose at physiological levels for more than 30 days [60].
3.3.4. Hyperlactatemia
The erythrocyte encapsulation of lactate 2-monooxygenase (EC 1.13.12.4) and L-lactate oxidase (EC 1.1.3.2), two lactate-catabolizing enzymes, as strategies for the treatment for hyperlactatemia was explored by Garin et al. [61]. Lactate 2-mono-oxygenase metabolizes lactate in the presence of oxygen to acetate and carbon dioxide, whereas lactate oxidase metabolizes lactate to pyruvate and hydrogen peroxide. In vitro studies in mouse and human erythrocytes demonstrated that encapsulated lactate 2-monooxygenase had a low affinity for lactate, operating at a low rate in the presence of lactate concentrations found in hyperlactatemia (5–20 mM). Erythrocyte encapsulated lactate oxidase however was able to provide a constant catabolic rate under the same range of blood lactate concentrations, but was limited by the generation of hydrogen peroxide which is toxic to the erythrocytes. The co-encapsulation of both enzymes provided a significant rate of lactate metabolism in vitro, over a range of 1–30 mM lactate and counteracted the production of hydrogen peroxide by increasing the amount of glucose metabolized in the pentose phosphate pathway. The in vivo efficacy of the loaded cells in removing blood lactate in the mouse, however, could not be demonstrated due to the high aerobic capacity and high lactate metabolism of this species [61].
3.3.5. Glucose-6-Phosphate Dehydrogenase Deficiency
Glucose-6-phosphate dehydrogenase (EC 1.1.1.49) deficiency (OMIM # 300908) is caused by mutations in the G6PD gene. The enzyme catalyzes nicotinamide adenine dinucleotide phosphate (NADP) to its reduced form, NADPH, in the pentose phosphate pathway. NADPH protects cells from oxidative damage and because mature erythrocytes cannot generate NADPH by any other pathway, a deficiency in glucose-6-phosphate dehydrogenase increases the vulnerability of erythrocytes to oxidative stress. The common clinical manifestations of the disease are neonatal jaundice and acute or chronic hemolytic anemia triggered by exogenous agents [62]. Gerli and co-workers demonstrated that human glucose-6-phosphate dehydrogenase deficient erythrocytes loaded with enzyme extracted from yeast were able to acquire some capacity to reduce glutathione [63]. In the study of Morelli et al., homogeneous human glucose 6-phosphate dehydrogenase was successfully encapsulated into the erythrocytes of individuals affected by glucose 6-phosphate dehydrogenase deficiency, to levels observed in unaffected erythrocytes, with a consequent normalization of the pentose phosphate pathway [64].
3.3.6. Adenosine Deaminase Deficiency
The application of the erythrocyte carrier as an enzyme replacement for adenosine deaminase deficiency (OMIM # 608958) due to severe combined immunodeficiency was explored by Bax et al., [65,66,67,68,69]. The disorder is caused by mutations in the ADA gene which encodes for adenosine deaminase (EC 3.5.4.4), the enzyme responsible for catalyzing the irreversible deamination of adenosine and 2’-deoxyadenosine in the purine catabolic pathway. The subsequent accumulation of 2’-deoxyadenosine results in its preferential phosphorylation to deoxyadenosine triphosphate (dATP) which accumulates intracellularly to high levels, resulting in an impairment of lymphocyte differentiation and proliferation through the inhibition of ribonucleotide reductase. The disease is characterized by low levels of immunoglobulins, a virtual absence of T-lymphocytes and markedly reduced levels of B-lymphocytes and natural killer cells [70]. The encapsulation of native adenosine deaminase and the licensed pharmaceutical preparation, polyethylene glycol-conjugated adenosine deaminase (pegademase), within human erythrocyte carriers was investigated. The rationale was to prolong the in vivo circulatory half-life of these enzyme preparations and maintain therapeutic blood levels. The efficiencies of native enzyme and pegylated enzyme encapsulation were 50% and 9%, respectively, thus indicating that the polyethylene glycol side-chains were impeding the encapsulation of the pegylated preparation. The biochemical characteristics and the osmotic fragility of loaded cells were not adversely affected by the encapsulation of either enzyme preparation [65,66,68]. In vivo survival studies of pegademase-loaded erythrocytes were conducted by labeling with Chromium [51Cr] and infusing into an ADA-deficient adult patient and demonstrated a mean cell half-life of 16 days. Erythrocyte encapsulated pegademase and native ADA had in vivo circulatory half-lives of 20 and 12.5 days, respectively; these are substantially greater than the plasma half-lives of the unencapsulated preparations, which are less than six days [68]. A clinical evaluation of erythrocyte-encapsulated native adenosine deaminase therapy was conducted over a period of nine years in a single patient who received a total of 225 infusions every 2–3 weeks. Observations included a reduction in the erythrocyte dATP concentration to between 24 and 44 µmol/L, compared to 234 µmol/L on diagnosis and a normalization of serum immunoglobulin levels. Also reported were a reduction in the frequency of respiratory problems and a decline in the forced expiratory volume in one second and vital capacity reduced compared with the four years preceding erythrocyte therapy [69].
3.3.7. Mitochondrial Neurogastrointestinal Encephalomyopathy (MNGIE)
MNGIE (OMIM # 603041) is an ultra-rare, fatal disorder caused by mutations in the nuclear TYMP gene which leads to a deficiency in thymidine phosphorylase activity (EC 2.4.2.4). Thymidine phosphorylase plays a pivotal role in the deoxyribonucleoside salvage metabolic pathway through catalyzing the reversible phosphorylation of thymidine and 2’-deoxyuridine to 2-deoxyribose 1-phosphate and their respective bases, thymine and uracil. In the absence of thymidine phosphorylase activity there is a systemic accumulation of thymidine and 2’-deoxyuridine which generate imbalances within the mitochondrial deoxyribonucleotide pools, causing mitochondrial DNA (mtDNA) point mutations, depletion and deletion abnormalities—and ultimately mitochondrial dysfunction. The disorder is characterized by gastrointestinal dysmotility, cachexia, peripheral neuropathy, ophthalmoplegia, ptosis and leukoencephalopathy [71]. Although there are no licensed therapies for patients with MNGIE, there are a number of experiment treatment approaches under investigation and this is attributed to MNGIE being one of the few mitochondrial disorders where the molecular abnormality is metabolically and physically accessible to manipulation. Erythrocyte encapsulated thymidine phosphorylase (EE-TP) is one of these experimental approaches, where recombinant E. coli enzyme was employed as the source of enzyme [71,72,73,74]. In the first proof of concept study, one dose of EE-TP (1020 units encapsulated within 20.25 × 1010 erythrocytes) was administered to a patient with MNGI.E. Three days post infusion, there was a reported decrease in the urinary excretion of thymidine and 2’-deoxyuridine, to 6% and 13%, respectively of the amounts excreted pre-therapy. Plasma metabolites concentrations were also shown to decrease in parallel [75]. Pre-clinical studies conducted in the mouse and dog revealed no safety issues that would preclude a clinical study of EE-TP, other than the production of anti-thymidine phosphorylase antibodies in a small proportion of animals [76,77]. Based on these encouraging results, a compassionate use program was conducted in four further patients, with participants receiving treatment in accordance with the provisions of Schedule 1 of the Medicines for Human Use (marketing authorizations, etc.) Regulations SI 1994/3144, where schedule 1 provides an exemption from the need for a marketing authorization for a relevant medicinal product, which is supplied on an individual patient basis to fulfil a "special need". A chromium [51Cr]–labeling study of EE-TP was conducted in one patient to evaluate the in vivo survival characteristics of the patient’s enzyme-loaded erythrocytes and this revealed a normal circulating mean cell life and half-life of 108 and 32 days, respectively [78]. The administration of doses ranging between 3.7 to 108 U/kg body weight/4 weeks indicated that doses greater than 47 U/kg/4 weeks resulted a greater reduction or an elimination of plasma and urine metabolites. One patient was reported to gain four kg in weight, three months after initiating therapy and this coincided with a decrease in nausea and vomiting, increased ability to walk longer distances and an increase in the physical and mental components of the SF36 health and well-being survey. A second patient showed significant clinical improvements in bilateral muscle power, gait and balance, sensory ataxia and fine finger function, after 23 months of EE-TP therapy. In addition, a weight gain of 5.8 kg was reported and a fall in plasma creatine kinase activity from 1200 U/L pre-therapy to levels within the normal reference range. Patient reported outcomes included an ability to walk longer distances, climb stairs without assistance, tie shoe laces, feel the sensation of sand, and a return to weight training and guitar playing [78,79,80]. Adverse events for EE-TP that were recorded in this compassionate study included nausea, coughing spasms and erythema of the face and neck in two of the four patients. These were transient in nature, occurring within the first five minutes of EE-TP infusion and did not lead to patient discontinuation of EE-TP therapy. Pre-medication with antihistamine, corticosteroid anti-inflammatory and anti-emetic drugs provided a strategy for successfully managing these reactions [78,79]. Specific antibodies to thymidine phosphorylase were detected in one patient, after the tenth treatment cycle. However, this did not raise any concerns with regard to neutralizing antibodies as the efficacy of encapsulated thymidine phosphorylase in metabolizing the plasma metabolites improved over the 5.5 years of EE-TP administration to this patient, and positive clinical responses to treatment continued to be recorded [81]. The development of specific antibodies is not surprising considering that senescent erythrocytes are naturally sequestered from the vascular compartment by macrophages of the monocyte–macrophage system, where macrophages are able to present peptides to T-lymphocytes [82]. EE-TP has received MHRA clearance for a Phase II, multi-center, multiple-dose, open-label trial without a control to determine the safety, tolerability, pharmacodynamics and efficacy this treatment approach in patients with MNGIE (https://clinicaltrials.gov/ct2/show/NCT03866954) [83]. EE-TP has Orphan designation by the EMA and FDA.
3.3.8. Hyperuricemia
Due to the loss of uricase (EC 1.7.3.3) activity during the evolution of hominids, uric acid is the end product of purine metabolism in humans and other higher primates and is excreted in the urine. Hyperuricemia is caused by abnormally elevated concentrations of blood uric acid and arises through several mechanisms, including reduced renal excretion or increased uric acid production. The concept of utilizing the erythrocyte as a mechanism for degrading extracellular molecules was investigated by Ihler et al. through examining the in vitro metabolism of extracellular uric acid by human erythrocytes-loaded with uricase. Uric acid was shown to be transported into enzyme-loaded cells at the same rate as in native erythrocytes, however, the generation of hydrogen peroxide through the metabolism of uric acid to allantoin raised the concern of inducing oxidative stress damage [84]. To address this, Magnani et al. coupled uricase to the extracellular human erythrocyte membrane by a biotin–avidin–biotin–enzyme bridge, and in doing so, demonstrated a more efficient in vitro metabolism of uric acid, compared to that by encapsulated enzyme [85].
3.3.9. Phenylketonuria
Phenylketonuria (OMIM # 261600) is an autosomal recessive disorder caused by mutations in the PAH gene, leading to a deficiency in the enzyme responsible for the hydroxylation of phenylalanine to tyrosine, phenylalanine hydroxylase (EC 1.14.16.1). If untreated, phenylketonuria can result in impaired postnatal cognitive development resulting from a neurotoxic effect of hyperphenylalaninemia [86]. Another enzyme capable of metabolizing phenylalanine is phenylalanine ammonia lyase (EC 4.3.1.24). This enzyme is not expressed in mammals, but is expressed in plants, some bacteria, yeast and fungi and catalyzes the conversion of phenylalanine to ammonia and trans-cinnamic acid. Its potential application via erythrocyte encapsulation as a therapy for phenylketonuria was first proposed by Sprandel and Zoller, with a short-term in vivo study in mice demonstrating the effectiveness of this strategy [87]. Erythrocytes loaded with recombinant Chromobacterium violaceum phenylalanine hydroxylase, together with the cofactor tetrahydrobiopterin, were shown to reduced blood phenylalanine levels when administered to wild-type mice, but not when administered to the BTBR Pahenu2 phenylketonuria mouse model [88]. In contrast, the repeated administration of erythrocytes loaded with recombinant Anabaena variabilis phenylalanine ammonia lyase (rAvPAL) at 9–10-day intervals for 10 weeks, was able to provide a sustained reduction in blood phenylalanine levels in the BTBR Pahenu2 mouse model and also reverse fur pigmentation. Although, anti-enzyme antibodies were generated, these did not affect the efficacy of the encapsulated enzyme [89]. More recent studies examining the clinical efficacy of erythrocyte-loaded rAvPAL in the early treatment of the BTBR Pahenu2 mouse demonstrated a normalized of blood and brain phenylalanine concentrations and prevented cognitive developmental failure, depletion of brain serotonin, dendritic spine abnormalities and myelin basic protein reduction [90]. Rubius Therapeutics, Inc received FDA approval for a Phase Ib clinical trial of genetically engineered erythrocytes expressing phenylalanine ammonia lyase (RTX-134). This approach involves the genetic engineering of CD34+ cells, collected from a healthy O negative donor, with a lentiviral vector to express enzyme phenylalanine ammonia lyase within the cells. The cells are expanded and differentiated until the nucleus is ejected, resulting in the mature reticulocyte. The trial is reported to be active, but no longer recruiting, and is designed to determine a preliminary dose and to inform on a dosing schedule that is deemed safe, tolerable and potentially effective (https://clinicaltrials.gov/ct2/show/NCT04110496). Recent media coverage has, however reported that the trial will be discontinued due to results from the first patient being uninterpretable. This is thought to be due to the low dose of cells administered and the lack of sensitivity of the flow cytometry assay used to detect circulating cells.
3.4. Antitumor Therapy
The implementation of the erythrocyte carrier in antitumor therapy has focused on the use of encapsulated enzymes and represents one of the most clinically advanced applications is for the treatment of acute lymphoblastic leukemia (ALL). This strategy is based on tumor cells not being able to synthesize adequate amounts of amino acids and therefore depend on extracellular sources. The administration of specific enzymes enables the depletion of plasma amino acids, thus starving the tumor cells of amino acids necessary for DNA, RNA and protein synthesis and leading ultimately to cell death. The antitumor characteristics of L-asparaginase (EC 3.5.1.1) was first investigated by Kidd, who reported lymphoma regression in mice and rats in response to guinea pig plasma [91]. In the early 1970s, native Erwinia chrysanthemi and E. coli-derived l-asparaginase were introduced into chemotherapy protocols for the treatment of ALL. However, its use was associated with toxicity against the liver and pancreas. High immunogenicity, manifesting as hypersensitivity reactions and/or the neutralization of asparaginase activity without any signs of hypersensitivity were also observed. The enzymes had a short circulatory half-life, ranging between 8 and 30 h thus necessitating frequent infusions [92]. Pegylated formulations of E. coli l-asparaginase were developed to overcome these limitations, however these preparations are associated with hepatotoxicity, pancreatitis and thrombosis [93]. Updike et al. were the first to employ the erythrocyte carrier as a strategy to counteract the problems of antigenicity and host proteolytic degradation of asparaginase. In vivo studies demonstrated that circulating asparaginase activity in monkeys injected with enzyme-loaded erythrocytes was two orders of magnitude higher than that in animals injected with native asparaginase [94,95]. In addition, monkeys that received a single injection of erythrocyte-loaded enzyme were able to suppress plasma asparagine concentrations for 20 days compared to 10 days for animals that received the native enzyme [96]. Further preliminary in vivo studies in the mouse and human demonstrated the safety of asparaginase-loaded erythrocytes and their efficacy in reducing plasma asparagine concentrations [96,97,98,99,100].
Erytech Pharma have developed a number of programs to further investigate the safety and efficacy of erythrocyte encapsulated asparaginase for the treatment of several types of cancers. In their Phase I/II study of E. coli erythrocyte-encapsulated l-asparaginase (GRASPA) conducted in adults and children with ALL, they were able to demonstrate an effective depletion of L-asparagine. A single injection of the highest dose (150 IU/kg) was shown to provide similar results to eight intravenous injections of 10,000 IU/m2 of the native enzyme. In addition, compared to the native enzyme, GRASPA exhibited a reduction in the number and severity of allergic reactions and a trend towards less coagulation disorders [101]. A subsequent Phase II trial evaluating the safety and efficacy of GRASPA was conducted in patients 55 years of age and older with Philadelphia chromosome-negative ALL. The primary efficacy endpoint of asparagine depletion <2 µmol/L for at least seven days was achieved in 85% and 71% of patients who received 100 and 150 IU/kg, respectively, but not with those patients who received 50 IU/kg. Although no dose limiting toxicity was observed at the lowest dosage, in the 100 IU/kg and 150 IU/kg cohorts, toxicities were 15% and 36%, respectively. This suggested that the 100 IU/kg dose had the best safety/efficacy profile for these elderly patients [102]. Erythrocyte encapsulated asparaginase was subsequently extended to a Phase I clinical trial for the treatment of patients with pancreatic adenocarcinoma with null/low asparagine synthetase expression. The intervention (ERY–ASP) was well tolerated by patients, and demonstrated no dose limiting toxicities reported [103]. A follow-on Phase IIb study evaluating erythrocyte-encapsulated asparaginase (relabeled as eryaspase) in combination with chemotherapy in second-line advanced pancreatic adenocarcinoma was conducted. This demonstrated significantly prolonged overall survival and progression free survival compared with chemotherapy alone, with a 40% reduction in the risk of death on average over time. The therapy combination was shown to be generally well tolerated and revealed no unexpected safety findings [104]. A Phase III study in patients with ductal adenocarcinoma of the pancreas who have failed one prior line of systemic anticancer therapy for advanced pancreatic cancer and have measurable disease is currently active and recruiting. The primary outcome measure is whether the addition of eryaspase to chemotherapy improves overall survival when compared to chemotherapy alone (https://clinicaltrials.gov/ct2/show/NCT03665441). Following the positive clinical data obtained from the pancreatic cancer studies, Erytech are now expanding their solid tumor studies to include selected metastatic triple-negative breast cancer. The ongoing Phase II/III trial is evaluating eryaspase in combination with gemcitabine and carboplatin chemotherapy, compared to chemotherapy alone as a first-line treatment (https://clinicaltrials.gov/ct2/show/NCT03674242).
Other enzymes, with relevance to tumor starvation which have been encapsulated include methionine-γ-lyase (EC 4.4.1.11) and arginine deiminase (EC 3.5.3.6). The administration of erythrocyte encapsulated Pseudomonas putida methionine-γ-lyase to mice grafted with human gastric adenocarcinoma and glioblastoma was effective in providing a sustained and significant plasma methionine depletion in vivo and a reduction in tumor growth [105]. A significant inhibition of tumor growth was observed in mice bearing orthotopic EMT-6 syngeneic breast carcinoma when infused with erythrocyte-encapsulated methionine-γ-lyase at a dose of 60 U/kg and in combination with anti-mouse PD-1 antibody, compared to the separate entities. Survival time was 35 days for the combined therapy, compared to 23 days for the separate therapies [106]. With regard to erythrocyte encapsulated arginine deiminase, a dose of 10.4 IU/mL administered to CD1 mice was shown to completely deplete plasma arginine over a period of five days compared to the same dose of free enzyme. The depletion was sustained for 24 h, with the plasma levels returning to baseline by 2.5 days [107].
4. Challenges and Limitations of Erythrocyte-Based Enzyme Therapy
The limitation of the erythrocyte enzyme carrier is that its metabolic capacity is restricted to the vascular compartment and monocyte–macrophage system. For the former, a further restriction for the encapsulated enzyme, would be the inability of the enzyme’s substrate to permeate the erythrocyte membrane. Thus, the spectrum of clinical applications to which the erythrocyte enzyme carrier can be applied to is fairly narrow. As with most cell-based therapies, the translation of the erythrocyte carrier into the clinical setting faces many complex process-related and regulatory challenges. The enzyme to be encapsulated or conjugated to the erythrocyte membrane will require manufacture in compliance with current good manufacturing practice (cGMP). Although advancements in the field of recombinant DNA biotechnologies have facilitated the manufacture of therapeutic enzymes, many challenging activities exist, including full characterization of cell banks, process- and product-related impurities and enzyme; validation of analytical procedures; formulation design; and stability studies. The transition of the manufacture of the erythrocyte carrier from a laboratory-based scale, to a scale that is clinically relevant is a critical step in the development pathway. Manufacture can be via a decentralized automated process, such as the Red Cell Loader as employed by Erydel or via a centralized process as used by Erytech Pharma [108,109]. Either way, the process will need to be performed in a closed-circuit system, using single use, sterile disposables. A key regulatory requirement is the manufacture of a reproducible product. Standardizing the loading procedure (and thus dose) when autologous erythrocytes are employed can be difficult to achieve, and may necessitate the consideration of individualized metrics such as patient hematocrit or red blood cell count in the manufacturing process. To minimize substantial differences in the quality of the end product and to produce a consistent encapsulation rate, Erytech Pharma who employ homologous donated erythrocytes, adjust the process operating conditions such as erythrocyte flow rate and osmolality of buffers according to the osmotic fragility of the initial donor erythrocyte pellet [109]. Prior to clinical batch release by a pharmacist, the product will need to conform to product specifications. For erythrocyte carriers manufactured from homologous blood, the 72-h shelf-life specified by Erytech provides sufficient time to conduct the necessary tests [109]. However, erythrocyte carriers manufactured from blood collected from autologous blood require a much shorter shelf-life (30 min) thus providing a major hurdle to testing. The MHRA approved Phase II trial of EE-TP was able to navigate this issue by proposing a retrospective testing of two of the release parameters. The impact of manufacturing process and chemical modification (where enzymes are conjugated to the erythrocyte membrane) on the function, viability and biocompatibility of the erythrocyte will also need to be established to ensure safety of the medicinal product.
5. Conclusions
The erythrocyte carrier has the advantage of counteracting some of the key issues associated with the administration of free enzymes by virtual of shielding the enzyme from the immune system and plasma proteases, and thereby enhance the circulatory half-life and minimize immunogenic reactions. The feasibility of erythrocyte-mediated enzyme therapy to deplete pathological molecules in the circulation has been investigated by many workers using animal models and humans. This paper provides an overview of the immense range of applications that have been investigated, covering the early studies of the 1970s up to the more recent biomedical applications that have overcome the process and regulatory challenges that face cell therapy development and have reached the clinic; these are summarized in Table 1. Although there are currently no licensed erythrocyte-mediated enzyme therapeutics, this is very likely to change over the next few years, with several products undergoing advanced clinical development for conditions where there are unmet needs.
Funding
This author is supported by grants awarded by the Medical Research Council (Grant Number: K025406) https://mrc.ukri.org/ and the Lily Foundation https://www.thelilyfoundation.org.uk/.
Conflicts of Interest
The author declares no conflict of interest.
References
- Baldo, B.A. Enzymes Approved for Human Therapy: Indications, Mechanisms and Adverse Effects. BioDrugs 2015, 29, 31–55. [Google Scholar] [CrossRef]
- Purcell, R.T.; Lockey, R.F. Immunologic Responses to Therapeutic Biologic Agents. J. Investig. Allergol. Clin. Immunol. 2008, 18, 335–342. [Google Scholar] [PubMed]
- Ihler, G.M.; Glew, R.H.; Schnure, F.W. Enzyme Loading of Erythrocytes. Proc. Natl. Acad. Sci. USA 1973, 70, 2663–2666. [Google Scholar] [CrossRef]
- Hoffman, J.F. Physiological Characteristics of Human Red Blood Cell Ghosts. J. Gen. Physiol. 1958, 42, 9–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffman, J.F. On Red Blood Cells, Hemolysis and Resealed Ghosts. Adv. Exp. Med. Biol. 1992, 326, 1–15. [Google Scholar]
- Sprandel, U. Erythrocytes as Carrier for Therapeutic Enzymes--an Approach towards Enzyme Therapy of Inborn Errors of Metabolism. Bibl. Haematol. 1985, 51, 7–14. [Google Scholar]
- Bratosin, D.; Mazurier, J.; Tissier, J.P.; Estaquier, J.; Huart, J.J.; Ameisen, J.C.; Aminoff, D.; Montreuil, J. Cellular and Molecular Mechanisms of Senescent Erythrocyte Phagocytosis by Macrophages. A Review. Biochimie 1998, 80, 173–195. [Google Scholar] [CrossRef]
- Way, J.L.; Leung, P.; Ray, L.; Sander, C. Erythrocyte Encapsulated Thiosulfate Sulfurtransferase. Bibl. Haematol. 1985, 75–81. [Google Scholar]
- Leung, P.; Ray, L.E.; Sander, C.; Way, J.L.; Sylvester, D.M.; Way, J.L. Encapsulation of Thiosulfate: Cyanide Sulfurtransferase by Mouse Erythrocytes. Toxicol. Appl. Pharmacol. 1986, 83, 101–107. [Google Scholar] [CrossRef]
- Hall, A.H.; Rumack, B.H. Hydroxycobalamin/sodium Thiosulfate as a Cyanide Antidote. J. Emerg. Med. 1987, 5, 115–121. [Google Scholar] [CrossRef]
- Leung, P.; Cannon, E.P.; Petrikovics, I.; Hawkins, A.; Way, J.L. In Vivo Studies on Rhodanese Encapsulation in Mouse Carrier Erythrocytes. Toxicol. Appl. Pharmacol. 1991, 110, 268–274. [Google Scholar] [CrossRef]
- Way, J.L.; Cannon, E.P.; Leung, P.; Hawkins-Zitzer, A.; Pei, L.; Petrikovics, I. Antagonism of the Lethal Effects of Cyanide with Resealed Erythrocytes Containing Rhodanese and Thiosulfate. Adv. Exp. Med. Biol. 1992, 326, 159–161. [Google Scholar] [PubMed]
- Cannon, E.P.; Leung, P.; Hawkins, A.; Petrikovics, I.; Deloach, J.; Way, J.L. Antagonism of Cyanide Intoxication with Murine Carrier Erythrocytes Containing Bovine Rhodanese and Sodium Thiosulfate. J. Toxicol. Environ. Health 1994, 41, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Petrikovics, I. Cyanide Antagonism with Carrier Erythrocytes and Organic Thiosulfonates. Fundam. Appl. Toxicol. 1995, 24, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Jan, Y.H.; Richardson, J.R.; Baker, A.A.; Mishin, V.; Heck, D.E.; Laskin, D.L.; Laskin, J.D. Novel Approaches to Mitigating Parathion Toxicity: Targeting Cytochrome P450–mediated Metabolism with Menadione. Ann. N. Y. Acad. Sci. 2016, 1378, 80–86. [Google Scholar] [CrossRef] [Green Version]
- Chung, S.P.; Roh, H.K. Antidote for Organophosphate Insecticide Poisoning: Atropine and Pralidoxime. J. Korean Med Assoc. 2013, 56, 1057–1066. [Google Scholar] [CrossRef] [Green Version]
- Pei, L. Antagonism of the Lethal Effects of Paraoxon by Carrier Erythrocytes Containing Phosphotriesterase. Fundam. Appl. Toxicol. 1995, 28, 209–214. [Google Scholar] [CrossRef]
- Way, J.L.; Pei, L.; Petrikovics, I.; McGuinn, D.; Tamulinas, C.; Hu, Q.Z.; Cannon, E.P.; Zitzer, A. Organophosphorus Antagonism by Resealed Erythrocytes Containing Recombinant Paraoxonase. In Erythrocytes as Drug Carriers in Medicine; Springer: Boston, MA, USA, 1997; pp. 89–92. [Google Scholar]
- Magnani, M.; Laguerre, M.; Rossi, L.; Bianchi, M.; Ninfali, P.; Mangani, F.; Ropars, C. In Vivo Accelerated Acetaldehyde Metabolism Using Acetaldehyde Dehydrogenase-Loaded Erythrocytes. Alcohol Alcohol. 1990, 25, 627–637. [Google Scholar] [CrossRef]
- Lizano, C.; Sanz, S.; Luque, J.; Pinilla, M. In Vitro Study of Alcohol Dehydrogenase and Acetaldehyde Dehydrogenase Encapsulated into Human Erythrocytes by an Electroporation Procedure. Biochim. Biophys. Acta Gen. Subj. 1998, 1425, 328–336. [Google Scholar] [CrossRef]
- Lizano, C.; Pérez, M.T.; Pinilla, M. Mouse Erythrocytes as Carriers for Coencapsulated Alcohol and Aldehyde Dehydrogenase Obtained by Electroporation - In Vivo Survival Rate in Circulation, Organ Distribution and Ethanol Degradation. Life Sci. 2001, 68, 2001–2016. [Google Scholar] [CrossRef]
- Kaminsky, Y.G.; Kosenko, E.A.; Alexandrovich, Y.G.; Ataullakhanov, F.I. Experiments on Alcocytes Containing Enzyme Nanoparticles for Reducing Toxic Blood Concentration of Ethanol. Bull. Exp. Biol. Med. 2012, 153, 170–172. [Google Scholar] [CrossRef] [PubMed]
- Alexandrovich, Y.G.; Kosenko, E.A.; Sinauridze, E.I.; Obydennyi, S.I.; Kireev, I.I.; Ataullakhanov, F.I.; Kaminsky, Y.G. Rapid Elimination of Blood Alcohol Using Erythrocytes: Mathematical Modeling and In Vitro Study. Biomed Res. Int. 2017, 2017, 5849593. [Google Scholar] [CrossRef] [PubMed]
- Johlin, F.C.; Fortman, C.S.; Nghiem, D.D.; Tephly, T.R. Studies on the Role of Folic Acid and Folate-Dependent Enzymes in Human Methanol Poisoning. Mol. Pharmacol. 1987, 31, 557–561. [Google Scholar] [PubMed]
- Magnani, M.; Fazi, A.; Mangani, F.; Rossi, L.; Mancini, U. Methanol Detoxification by Enzyme-Loaded Erythrocytes. Biotechnol. Appl. Biochem. 1993, 18, 217–226. [Google Scholar]
- Muthuvel, A.; Rajamani, R.; Manikandan, S.; Sheeladevi, R. Detoxification of Formate by Formate Dehydrogenase-Loaded Erythrocytes and Carbicarb in Folate-Deficient Methanol-Intoxicated Rats. Clin. Chim. Acta 2006, 367, 162–169. [Google Scholar] [CrossRef]
- Wani, A.L.; Ara, A.; Usmani, J.A. Lead Toxicity: A Review. Interdiscip. Toxicol. 2015, 8, 55–64. [Google Scholar] [CrossRef] [Green Version]
- Bustos, N.L.; Stella, A.M.; Wider, E.A.; Alcira, A.M. Enzyme Replacement Therapy in Porphyrias-III: Potential Use of Erythrocyte Ghosts as Carriers of δ-Aminolaevulinate Dehydratase. Int. J. Biochem. 1983, 15, 447–452. [Google Scholar] [CrossRef]
- Bustos, N.L.; Batlle, A.M. Enzyme Replacement Therapy in Porphyrias--V. In Vivo Correction of Delta-Aminolaevulinate Dehydratase Defective in Erythrocytes in Lead Intoxicated Animals by Enzyme-Loaded Red Blood Cell Ghosts. Drug Des. Deliv. 1989, 5, 125–131. [Google Scholar]
- Batlle, A.M.; Bustos, N.L.; Stella, A.M.; Wider, E.A.; Conti, H.A.; Mendez, A. Enzyme Replacement Therapy in Porphyrias--IV. First Successful Human Clinical Trial of Delta-Aminolevulinate Dehydratase-Loaded Erythrocyte Ghosts. Int. J. Biochem. 1983, 15, 1261–1265. [Google Scholar] [CrossRef]
- Axley, M.J.; Dad, L.K.; Harabin, A.L. Hydrogenase Encapsulation into Red Blood Cells and Regeneration of Electron Acceptor. Biotechnol. Appl. Biochem. 1996, 24, 95–100. [Google Scholar]
- Adivitiya; Khasa, Y.P. The Evolution of Recombinant Thrombolytics: Current Status and Future Directions. Bioengineered 2017, 8, 331–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delahousse, B.; Kravtzoff, R.; Ropars, C. Use of Erythrocytes as a New Route of Administration of Fibrinolytic Agents. In Erythrocytes as Drug Carriers in Medicine; Springer: Boston, MA, USA, 1997; pp. 35–42. [Google Scholar]
- Flynn, G.; Hackett, T.J.; McHale, L.; McHale, A.P. Encapsulation of the Thrombolytic Enzyme, Brinase, in Photosensitized Erythrocytes: A Novel Thrombolytic System Based on Photodynamic Activation. J. Photochem. Photobiol. B Biol. 1994, 26, 193–196. [Google Scholar] [CrossRef]
- Muzykantov, V.R.; Sakharov, D.V.; Smirnov, M.D.; Samokhin, G.P.; Smirnov, V.N. Immunotargeting of Erythrocytes-Bound Streptokinase Provides Local Lysis of a Fibrin Clot. BBA Gen. Subj. 1986, 884, 355–362. [Google Scholar] [CrossRef]
- Murciano, J.C.; Medinilla, S.; Eslin, D.; Atochina, E.; Cines, D.B.; Muzykantov, V.R. Prophylactic Fibrinolysis through Selective Dissolution of Nascent Clots by tPA-Carrying Erythrocytes. Nat. Biotechnol. 2003, 21, 891–896. [Google Scholar] [CrossRef] [PubMed]
- Danielyan, K.; Ganguly, K.; Ding, B.-S.; Atochin, D.; Zaitsev, S.; Murciano, J.-C.; Huang, P.L.; Kasner, S.E.; Cines, D.B.; Muzykantov, V.R. Cerebrovascular Thromboprophylaxis in Mice by Erythrocyte-Coupled Tissue-Type Plasminogen Activator. Circulation 2008, 118, 1442–1449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vankayala, R.; Corber, S.R.; Mac, J.T.; Rao, M.P.; Shafie, M.; Anvari, B. Erythrocyte-Derived Nanoparticles as a Theranostic Agent for Near-Infrared Fluorescence Imaging and Thrombolysis of Blood Clots. Macromol. Biosci. 2018, 18, 1700379. [Google Scholar] [CrossRef]
- Deloach, J.R.; Widnell, C.C.; Ihler, G.M. Phagocytosis of Enzyme-Containing Carrier Erythrocytes by Macrophages. J. Appl. Biochem. 1979, 1, 95–103. [Google Scholar]
- Thorpe, S.R.; Fiddler, M.B.; Desnick, R.J. Enzyme Therapy. V. In Vivo Fate of Erythrocyte-Entrapped β-Glucuronidase in β- Glucuronidase-Deficient Mice. Pediatr. Res. 1975, 9, 918–923. [Google Scholar] [CrossRef]
- Stirnemann, J.Ô.; Belmatoug, N.; Camou, F.; Serratrice, C.; Froissart, R.; Caillaud, C.; Levade, T.; Astudillo, L.; Serratrice, J.; Brassier, A.; et al. A Review of Gaucher Disease Pathophysiology, Clinical Presentation and Treatments. Int. J. Mol. Sci. 2017, 18, 441. [Google Scholar] [CrossRef]
- Beutler, E.; Dale, G.L.; Guinto, E.; Kuhl, W. Enzyme Replacement Therapy in Gaucher’s Disease: Preliminary Clinical Trial of a New Enzyme Preparation. Proc. Natl. Acad. Sci. USA 1977, 74, 4620–4623. [Google Scholar] [CrossRef] [Green Version]
- Dale, G.L.; Kuhl, W.; Beutler, E. Incorporation of Glucocerebrosidase into Gaucher’s Disease Monocytes in Vitro. Proc. Natl. Acad. Sci. USA 1979, 76, 473–475. [Google Scholar] [CrossRef] [Green Version]
- Bax, B.E.; Bain, M.D.; Ward, C.P.; Fensom, A.H.; Chalmers, R.A. The Entrapment of Mannose-Terminated Glucocerebrosidase (Alglucerase) in Human Carrier Erythrocytes. In Biochemical Society Transactions; Springer: Boston, MA, USA, 1996; Volume 24, p. 441S. [Google Scholar]
- Bax, B.E.; Bain, M.D.; Ward, C.P.; Fensom, A.H.; Chalmers, R.A. The Entrapment of Mannose-Terminated Glucocerebrosidase (Alglucerase) in Human Carrier Erythrocytes. In Erythrocytes as Drug Carriers in Medicine; Springer: Boston, MA, USA, 1997; pp. 59–62. [Google Scholar]
- Sanz, S.; Lizano, C.; Garín, M.I.; Luque, J.; Pinilla, M. Biochemical Properties of Alcohol Dehydrogenase and Glutamate Dehydrogenase Encapsulated into Human Erythrocytes by a Hypotonic-Dialysis Procedure. In Erythrocytes as Drug Carriers in Medicine; Springer: Boston, MA, USA, 1997; pp. 101–108. [Google Scholar]
- Sanz, S.; Lizano, C.; Luque, J.; Pinilla, M. In Vitro and in Vivo Study of Glutamate Dehydrogenase Encapsulated into Mouse Erythrocytes by a Hypotonic Dialysis Procedure. Life Sci. 1999, 65, 2781–2789. [Google Scholar] [CrossRef]
- Venediktova, N.I.; Kosenko, E.A.; Kaminsky, Y.G. Studies on Ammocytes: Development, Metabolic Characteristics, and Detoxication of Ammonium. Bull. Exp. Biol. Med. 2008, 146, 730–732. [Google Scholar] [CrossRef] [PubMed]
- Kosenko, E.A.; Venediktova, N.I.; Kudryavtsev, A.A.; Ataullakhanov, F.I.; Kaminsky, Y.G.; Felipo, V.; Montoliu, C. Encapsulation of Glutamine Synthetase in Mouse Erythrocytes: A New Procedure for Ammonia Detoxification. Biochem. Cell Biol. 2008, 86, 469–476. [Google Scholar] [CrossRef] [PubMed]
- Protasov, E.S.; Borsakova, D.V.; Alexandrovich, Y.G.; Korotkov, A.V.; Kosenko, E.A.; Butylin, A.A.; Ataullakhanov, F.I.; Sinauridze, E.I. Erythrocytes as Bioreactors to Decrease Excess Ammonium Concentration in Blood. Sci. Rep. 2019, 9, 1455. [Google Scholar] [CrossRef]
- Ah Mew, N.; Simpson, K.L.; Gropman, A.L.; Lanpher, B.C.; Chapman, K.A.; Summar, M.L. Urea Cycle Disorders Overview; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Adriaenssens, K.; Karcher, D.; Lowenthal, A.; Terheggen, H.G. Use of Enzyme Loaded Erythrocytes in in Vitro Correction of Arginase Deficient Erythrocytes in Familial Hyperargininemia. Clin. Chem. 1976, 22, 323–326. [Google Scholar] [CrossRef]
- Baysal, Ş.H.; Uslan, A.H. Encapsulation of Urease and PEG-Urease in Erythrocyte. Artif. Cells Blood Substit. Immobil. Biotechnol. 2000, 28, 263–271. [Google Scholar] [CrossRef] [Green Version]
- Hamarat Baysal, S.; Uslan, A.H. Encapsulation of PEG-urease/PEG-AlaDH Enzyme System in Erythrocyte. Artif. Cells Blood Substit. Immobil. Biotechnol. 2001, 29, 405–412. [Google Scholar] [CrossRef]
- Baysal, S.H.; Uslan, A.H. In Vitro Study of urease/AlaDH Enzyme System Encapsulated into Human Erythrocytes and Research into Its Medical Applications. Artif. Cells Blood Substit. Immobil. Biotechnol. 2002, 30, 71–77. [Google Scholar] [CrossRef]
- Baysal, S.H.; Uslan, A.H.; Pala, H.H.; Tunçoku, O. Encapsulation of PEG-urease/PEG-AlaDH within Sheep Erythrocytes and Determination of the System’s Activity in Lowering Blood Levels of Urea in Animal Models. Artif. Cells Blood Substit. Immobil. Biotechnol. 2007, 35, 391–403. [Google Scholar] [CrossRef]
- Magnani, M.; Rossi, L.; Bianchi, M.; Fornaini, G.; Benatti, U.; Guida, L.; Zocchi, E.; De Flora, A. Improved Metabolic Properties of Hexokinase-Overloaded Human Erythrocytes. Biochim. Biophys. Acta 1988, 972, 1–8. [Google Scholar]
- De Flora, A.; Guida, L.; Zocchi, E.; Tonetti, M.; Benatti, U. Construction of Glucose Oxidase-Loaded human Erythrocytes: A Model of Oxidative Cytotoxicity. Available online: https://www.ncbi.nlm.nih.gov/pubmed/3804702 (accessed on 27 March 2020).
- Zocchi, E.; Benatti, U.; Guida, L.; Tonetti, M.; Damonte, G.L.; De Flora, A. Encapsulation of Glucose Oxidase in Mouse Erythrocytes: An Experimental Model of Oxidant-Induced Cytotoxicity and a Means for Splenic Targeting of. In Red Blood Cells as Carriers for Drugs; Ropars, C., Chassaigne, M., Nicolau, C., Eds.; Pergamon Press: Oxford, UK, 1987. [Google Scholar]
- Rossi, L.; Bianchi, M.; Magnani, M. Increased Glucose Metabolism by Enzyme-loaded Erythrocytes in Vitro and in Vivo Normalization of Hyperglycemia in Diabetic Mice. Biotechnol. Appl. Biochem. 1992, 15, 207–216. [Google Scholar] [PubMed]
- Garin, M.; Rossi, L.; Luque, J.; Magnani, M. Lactate Catabolism by Enzyme-Loaded Red Blood Cells. Biotechnol. Appl. Biochem. 1995, 22, 295–303. [Google Scholar] [PubMed]
- Belfield, K.D.; Tichy, E.M. Review and Drug Therapy Implications of Glucose-6-Phosphate Dehydrogenase Deficiency. Am. J. Health-Syst. Pharm. 2018, 75, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Gerli, G.C.; Agostoni, A.; Fiorelli, G. G-6PD Loading of G-6PD-Deficient Erythrocytes. Experientia 1978, 34, 431–432. [Google Scholar] [CrossRef]
- Morelli, A.; Benatti, U.; Salamino, F.; Sparatore, B.; Michetti, M.; Melloni, E.; Pontremoli, S.; De Flora, A. In Vitro Correction of Erythrocyte Glucose 6-Phosphate Dehydrogenase (G6PD) Deficiency. Arch. Biochem. Biophys. 1979, 197, 543–550. [Google Scholar] [CrossRef]
- Bax, B.E.; Fairbanks, L.D.; Bain, M.D.; Simmonds, H.A.; Chalmers, R.A. The Entrapment of Polyethylene Glycol-Bound Adenosine Deaminase (Pegademase) in Human Carrier Erythrocytes. In Biochemical Society Transactions; Springer: Boston, MA, USA, 1996; Volume 24, p. 442S. [Google Scholar]
- Bax, B.E.; Fairbanks, L.D.; Bain, M.D.; Simmonds, H.A.; Chalmers, R.A. The Entrapment of Polyethylene Glycol-Conjugated Adenosine Deaminase (Pegademase) and Native Adenosine Deaminase in Human Carrier Erythrocytes. In Erythrocytes as Drug Carriers in Medicine; Springer: Boston, MA, USA, 1997; pp. 31–34. [Google Scholar]
- Bax, B.E.; Bain, M.D.; Fairbanks, L.D.; Anne Simmonds, H.; David Webster, A.; Chalmers, R.A. Carrier Erythrocyte Entrapped Adenosine Deaminase Therapy in Adenosine Deaminase Deficiency. Adv. Exp. Med. Biol. 2000, 486, 47–50. [Google Scholar]
- Bax, B.E.; Bain, M.D.; Fairbanks, L.D.; Webster, A.D.B.; Chalmers, R.A. In Vitro and in Vivo Studies with Human Carrier Erythrocytes Loaded with Polyethylene Glycol-Conjugated and Native Adenosine Deaminase. Br. J. Haematol. 2000, 109, 549–554. [Google Scholar] [CrossRef]
- Bax, B.E.; Bain, M.D.; Fairbanks, L.D.; Webster, A.D.B.; Ind, P.W.; Hershfield, M.S.; Chalmers, R.A. A 9-Yr Evaluation of Carrier Erythrocyte Encapsulated Adenosine Deaminase (ADA) Therapy in a Patient with Adult-Type ADA Deficiency. Eur. J. Haematol. 2007, 79, 338–348. [Google Scholar] [CrossRef]
- Flinn, A.M.; Gennery, A.R. Adenosine Deaminase Deficiency: A Review. Orphanet J. Rare Dis. 2018, 13, 65. [Google Scholar] [CrossRef]
- Bax, B.E. Mitochondrial Neurogastrointestinal Encephalomyopathy: Approaches to Diagnosis and Treatment. J. Transl. Genet. Genom. 2020, 4, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Godfrin, Y.; Horand, F.; Franco, R.; Dufour, E.; Kosenko, E.; Bax, B.E.; Banz, A.; Skorokhod, O.A.; Lanao, J.M.; Vitvitsky, V.; et al. International Seminar on the Red Blood Cells as Vehicles for Drugs. Expert Opin. Biol. Ther. 2012, 12, 127–133. [Google Scholar] [CrossRef] [Green Version]
- Godfrin, Y.; Bax, B.E. Enzyme bioreactors as drugs. Drugs Future 2012, 37, 263–272. [Google Scholar] [CrossRef]
- Pacitti, D.; Levene, M.; Garone, C.; Nirmalananthan, N.; Bax, B.E. Mitochondrial Neurogastrointestinal Encephalomyopathy: Into the Fourth Decade, What We Have Learned So Far. Front. Genet. 2018, 9, 669. [Google Scholar] [CrossRef] [Green Version]
- Moran, N.F.; Bain, M.D.; Muqit, M.M.K.; Bax, B.E. Carrier Erythrocyte Entrapped Thymidine Phosphorylase Therapy for Mngie. Neurology 2008, 71, 686–688. [Google Scholar] [CrossRef]
- Levene, M.; Coleman, D.G.; Kilpatrick, H.C.; Fairbanks, L.D.; Gangadharan, B.; Gasson, C.; Bax, B.E. Preclinical Toxicity Evaluation of Erythrocyte-Encapsulated Thymidine Phosphorylase in BALB/c Mice and Beagle Dogs: An Enzyme-Replacement Therapy for Mitochondrial Neurogastrointestinal Encephalomyopathy. Toxicol. Sci. 2013, 131, 311–324. [Google Scholar] [CrossRef]
- Gasson, C.; Levene, M.; Bax, B.E. The Development and Validation of an Immunoassay for the Measurement of Anti-Thymidine Phosphorylase Antibodies in Mouse and Dog Sera. J. Pharm. Biomed. Anal. 2013, 72, 16–24. [Google Scholar] [CrossRef]
- Levene, M.; Bain, M.; Moran, N.; Nirmalananthan, N.; Poulton, J.; Scarpelli, M.; Filosto, M.; Mandel, H.; MacKinnon, A.; Fairbanks, L.; et al. Safety and Efficacy of Erythrocyte Encapsulated Thymidine Phosphorylase in Mitochondrial Neurogastrointestinal Encephalomyopathy. J. Clin. Med. 2019, 8, 457. [Google Scholar] [CrossRef] [Green Version]
- Bax, B.E.; Bain, M.D.; Scarpelli, M.; Filosto, M.; Tonin, P.; Moran, N. Clinical and Biochemical Improvements in a Patient with MNGIE Following Enzyme Replacement. Neurology 2013, 81, 1269–1271. [Google Scholar] [CrossRef] [Green Version]
- Kipper, K.; Hecht, M.; Antunes, N.J.; Fairbanks, L.D.; Levene, M.; Uçar, S.K.; Schaefer, A.; Blakely, E.L.; Bax, B.E. Quantification of Plasma and Urine Thymidine and 2 ’ -Deoxyuridine by LC-MS / MS for the Pharmacodynamic Evaluation of Erythrocyte Encapsulated Thymidine Phosphorylase in Patients with Mitochondrial Neurogastrointestinal Encephalomyopathy. J. Clin. Med. 2020, 9, 788. [Google Scholar] [CrossRef] [Green Version]
- Levene, M.; Pacitti, D.; Gasson, C.; Hall, J.; Sellos-Moura, M.; Bax, B.E. Validation of an Immunoassay for Anti-Thymidine Phosphorylase Antibodies in Patients with MNGIE Treated with Enzyme Replacement Therapy. Mol. Ther. Methods Clin. Dev. 2018, 11, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Murray, A.M.; Pearson, I.F.S.; Fairbanks, L.D.; Chalmers, R.A.; Bain, M.D.; Bax, B.E. The Mouse Immune Response to Carrier Erythrocyte Entrapped Antigens. Vaccine 2006, 24, 6129–6139. [Google Scholar] [CrossRef]
- Bax, B.E.; Levene, M.; Bain, M.D.; Fairbanks, L.D.; Filosto, M.; Kalkan Uçar, S.; Klopstock, T.; Kornblum, C.; Mandel, H.; Rahman, S.; et al. Erythrocyte Encapsulated Thymidine Phosphorylase for the Treatment of Patients with Mitochondrial Neurogastrointestinal Encephalomyopathy: Study Protocol for a Multi-Centre, Multiple Dose, Open Label Trial. J. Clin. Med. 2019, 8, 1096. [Google Scholar] [CrossRef] [Green Version]
- Ihler, G.; Lantzy, A.; Purpura, J.; Glew, R.H. Enzymatic Degradation of Uric Acid by Uricase-Loaded Human Erythrocytes. J. Clin. Investig. 1975, 56, 595–602. [Google Scholar] [CrossRef]
- Magnani, M.; Mancini, U.; Bianchi, M.; Fazi, A. Comparison of Uricase-Bound and Uricase-Loaded Erythrocytes as Bioreactors for Uric Acid Degradation. Adv. Exp. Med. Biol. 1992, 326, 189–194. [Google Scholar]
- Williams, R.A.; Mamotte, C.D.S.; Burnett, J.R. Phenylketonuria: An Inborn Error of Phenylalanine Metabolism. Clin. Biochem. Rev. 2008, 29, 31–41. [Google Scholar]
- Sprandel, U.; Zöllner, N. Biochemical Studies of Phenylalanine Ammonia-Lyase Encapsulated in Erythrocytes. Biochem. Soc. Trans. 1990, 18, 654–655. [Google Scholar] [CrossRef] [Green Version]
- Yew, N.S.; Dufour, E.; Przybylska, M.; Putelat, J.; Crawley, C.; Foster, M.; Gentry, S.; Reczek, D.; Kloss, A.; Meyzaud, A.; et al. Erythrocytes Encapsulated with Phenylalanine Hydroxylase Exhibit Improved Pharmacokinetics and Lowered Plasma Phenylalanine Levels in Normal Mice. Mol. Genet. Metab. 2013, 109, 339–344. [Google Scholar] [CrossRef]
- Rossi, L.; Pierigè, F.; Carducci, C.; Gabucci, C.; Pascucci, T.; Canonico, B.; Bell, S.M.; Fitzpatrick, P.A.; Leuzzi, V.; Magnani, M. Erythrocyte-Mediated Delivery of Phenylalanine Ammonia Lyase for the Treatment of Phenylketonuria in BTBR-Pahenu2mice. J. Control. Release 2014, 194, 37–44. [Google Scholar] [CrossRef] [Green Version]
- Pascucci, T.; Rossi, L.; Colamartino, M.; Gabucci, C.; Carducci, C.; Valzania, A.; Sasso, V.; Bigini, N.; Pierigè, F.; Viscomi, M.T.; et al. A New Therapy Prevents Intellectual Disability in Mouse with Phenylketonuria. Mol. Genet. Metab. 2018, 124, 39–49. [Google Scholar] [CrossRef]
- Kidd, J.G. Regression of Transplanted Lymphomas Induced in Vivo by Means of Normal Guinea Pig Serum. I. Course of Transplanted Cancers of Various Kinds in Mice and Rats given Guinea Pig Serum, Horse Serum, or Rabbit Serum. J. Exp. Med. 1953, 98, 565–582. [Google Scholar] [CrossRef]
- Wriston, J.C.; Yellin, T.O. L-Asparaginase: A Review. In Advances in Enzymology and Related Areas of Molecular Biology; Wiley: Hoboken, NJ, USA, 2006; Volume 39, pp. 185–248. [Google Scholar]
- Heo, Y.A.; Syed, Y.Y.; Keam, S.J. Pegaspargase: A Review in Acute Lymphoblastic Leukaemia. Drugs 2019, 79, 767–777. [Google Scholar] [CrossRef] [Green Version]
- Updike, S.J.; Wakamiya, R.T.; Lightfoot, E.N. Asparaginase Entrapped in Red Blood Cells: Action and Survival. Science 1976, 193, 681–683. [Google Scholar] [CrossRef]
- Updike, S.J.; Wakamiya, R.T. Infusion of Red Blood Cell-Loaded Asparaginase in Monkey. Immunologic, Metabolic, and Toxicologic Consequences. J. Lab. Clin. Med. 1983, 101, 679–691. [Google Scholar]
- Kravtzoff, R.; Colombat, P.; Desbois, I.; Linassier, C.; Muh, J.P.; Philip, T.; Blay, J.Y.; Gardenbas, M.; Poumier-Gaschard, P.; Lamagnere, J.P.; et al. Tolerance Evaluation of L-Asparaginase Loaded in Red Blood Cells. Eur. J. Clin. Pharmacol. 1996, 51, 221–225. [Google Scholar] [CrossRef]
- Kravtzoff, R.; Desbois, I.; Lamagnere, J.P.; Muh, J.P.; Valat, C.; Chassaigne, M.; Colombat, P.; Ropars, C. Improved Pharmacodynamics of L-Asparaginase-Loaded in Human Red Blood Cells. Eur. J. Clin. Pharmacol. 1996, 49, 465–470. [Google Scholar] [CrossRef]
- Garin, M.I.; Kravtzoff, R.; Chestier, N.; Sanz, S.; Pinilla, M.; Luque, J.; Ropars, C. Density Gradient Separation of L-Asparaginase-Loaded Human Erythrocytes. Biochem. Mol. Biol. Int. 1994, 33, 807–814. [Google Scholar]
- Ktavtzoff, R.; Desbois, I.; Doinel, C.; Colombat, P.; Lamagnere, J.P.; Chassaigne, M.; Ropars, C. Immunological Response to L-Asparaginase Loaded into Red Blood Cells. Adv. Exp. Med. Biol. 1992, 326, 175–182. [Google Scholar]
- Kravtzoff, R.; Ropars, C.; Laguerre, M.; Muh, J.P.; Chassaigne, M. Erythrocytes as Carriers for L-Asparaginase. Methodological and Mouse In-vivo Studies. J. Pharm. Pharmacol. 1990, 42, 473–476. [Google Scholar] [CrossRef]
- Halfon-Domenech, C.; Thomas, X.; Chabaud, S.; Baruchel, A.; Gueyffier, F.; Mazingue, F.; Auvrignon, A.; Corm, S.; Dombret, H.; Chevallier, P.; et al. L-Asparaginase Loaded Red Blood Cells in Refractory or Relapsing Acute Lymphoblastic Leukaemia in Children and Adults: Results of the GRASPALL 2005-01 Randomized Trial. Br. J. Haematol. 2011, 153, 58–65. [Google Scholar] [CrossRef]
- Hunault-Berger, M.; Leguay, T.; Huguet, F.; Leprêtre, S.; Deconinck, E.; Ojeda-Uribe, M.; Bonmati, C.; Escoffre-Barbe, M.; Bories, P.; Himberlin, C.; et al. Group for Research on Adult Acute Lymphoblastic Leukemia (GRAALL). A Phase 2 Study of L-Asparaginase Encapsulated in Erythrocytes in Elderly Patients with Philadelphia Chromosome Negative Acute Lymphoblastic Leukemia: The GRASPALL/GRAALL-SA2-2008 Study. Am. J. Hematol. 2015, 90, 811–818. [Google Scholar] [CrossRef] [Green Version]
- Bachet, J.-B.; Gay, F.; Maréchal, R.; Galais, M.-P.; Adenis, A.; MsC, D.S.; Cros, J.; Demetter, P.; Svrcek, M.; Bardier-Dupas, A.; et al. Asparagine Synthetase Expression and Phase I Study With L-Asparaginase Encapsulated in Red Blood Cells in Patients With Pancreatic Adenocarcinoma. Pancreas 2015, 44, 1141–1147. [Google Scholar] [CrossRef]
- Hammel, P.; Fabienne, P.; Mineur, L.; Metges, J.P.; Andre, T.; De La Fouchardiere, C.; Louvet, C.; El Hajbi, F.; Faroux, R.; Guimbaud, R.; et al. Erythrocyte-Encapsulated Asparaginase (Eryaspase) Combined with Chemotherapy in Second-Line Treatment of Advanced Pancreatic Cancer: An Open-Label, Randomized Phase IIb Trial. Eur. J. Cancer 2020, 124, 91–101. [Google Scholar] [CrossRef] [Green Version]
- Gay, F.; Aguera, K.; Sénéchal, K.; Tainturier, A.; Berlier, W.; Maucort-Boulch, D.; Honnorat, J.; Horand, F.; Godfrin, Y.; Bourgeaux, V. Methionine Tumor Starvation by Erythrocyte-Encapsulated Methionine Gamma-Lyase Activity Controlled with per Os Vitamin B6. Cancer Med. 2017, 6, 1437–1452. [Google Scholar] [CrossRef]
- Sénéchal, K.; Maubant, S.; Leblanc, M.; Ciré, S.; Gallix, F.; Andrivon, A.; Duchamp, O.; Viviani, F.; Horand, F.; Scheer, A.; et al. Abstract 2258: Erymethionase (Methionine-Gamma-Lyase Encapsulated into Red Blood Cells) Potentiates Anti-PD-1 Therapy in TNBC Syngeneic Mouse Model. In Cancer Research; American Association for Cancer Research (AACR): Philadelphia, PA, USA, 2019; Volume 79, p. 2258. [Google Scholar]
- Gay, F.; Aguera, K.; Senechal, K.; Bes, J.; Chevrier, A.-M.; Gallix, F.; Guicher, C.; Lorenzi, P.; Bourgeaux, V.; Berlier, W.; et al. Abstract 4812: Arginine Deiminase Loaded in Erythrocytes: A Promising Formulation for L-Arginine Deprivation Therapy in Cancers. In Cancer Research; American Association for Cancer Research (AACR): Philadelphia, PA, USA, 2016; Volume 76, p. 4812. [Google Scholar]
- Magnani, M.; Rossi, L.; D’ascenzo, M.; Panzani, I.; Bigi, L.; Zanella, A. Erythrocyte Engineering for Drug Delivery and Targeting. Biotechnol. Appl. Biochem. 1998, 28, 1–6. [Google Scholar]
- Bourgeaux, V.; Lanao, J.M.; Bax, B.E.; Godfrin, Y. Drug-Loaded Erythrocytes: On the Road toward Marketing Approval. Drug Des. Dev. Ther. 2016, 10, 665–676. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Strategy for the removal of pathological metabolites from the circulation. Pathological metabolites in the blood enter the erythrocyte carrier, to undergo metabolism by the encapsulated enzyme (E).
Figure 1.
Strategy for the removal of pathological metabolites from the circulation. Pathological metabolites in the blood enter the erythrocyte carrier, to undergo metabolism by the encapsulated enzyme (E).

Figure 2.
Strategy for targeting enzymes to the monocyte–macrophage system of the spleen, liver and bone marrow. Senescent erythrocyte carriers loaded with enzymes are removed from the circulation by macrophages. The resulting phagosome fuses with macromolecule-laden lysosomes, resulting in the release of enzyme and breakdown of macromolecules.
Figure 2.
Strategy for targeting enzymes to the monocyte–macrophage system of the spleen, liver and bone marrow. Senescent erythrocyte carriers loaded with enzymes are removed from the circulation by macrophages. The resulting phagosome fuses with macromolecule-laden lysosomes, resulting in the release of enzyme and breakdown of macromolecules.

Figure 3.
Therapeutic strategy for therapeutic enzymes coupled to the erythrocyte membrane. Pathological plasma metabolites are catalyzed by the conjugated enzyme to their corresponding non-pathological product.
Figure 3.
Therapeutic strategy for therapeutic enzymes coupled to the erythrocyte membrane. Pathological plasma metabolites are catalyzed by the conjugated enzyme to their corresponding non-pathological product.

Figure 4.
Strategy for therapeutic enzymes produced through the transfection of CD34+ cells with lentiviral vectors containing constructs encoding for the enzyme of interest. Cells are expanded and differentiated until the nucleus is ejected, resulting in the mature reticulocyte expressing the recombinant enzyme.
Figure 4.
Strategy for therapeutic enzymes produced through the transfection of CD34+ cells with lentiviral vectors containing constructs encoding for the enzyme of interest. Cells are expanded and differentiated until the nucleus is ejected, resulting in the mature reticulocyte expressing the recombinant enzyme.


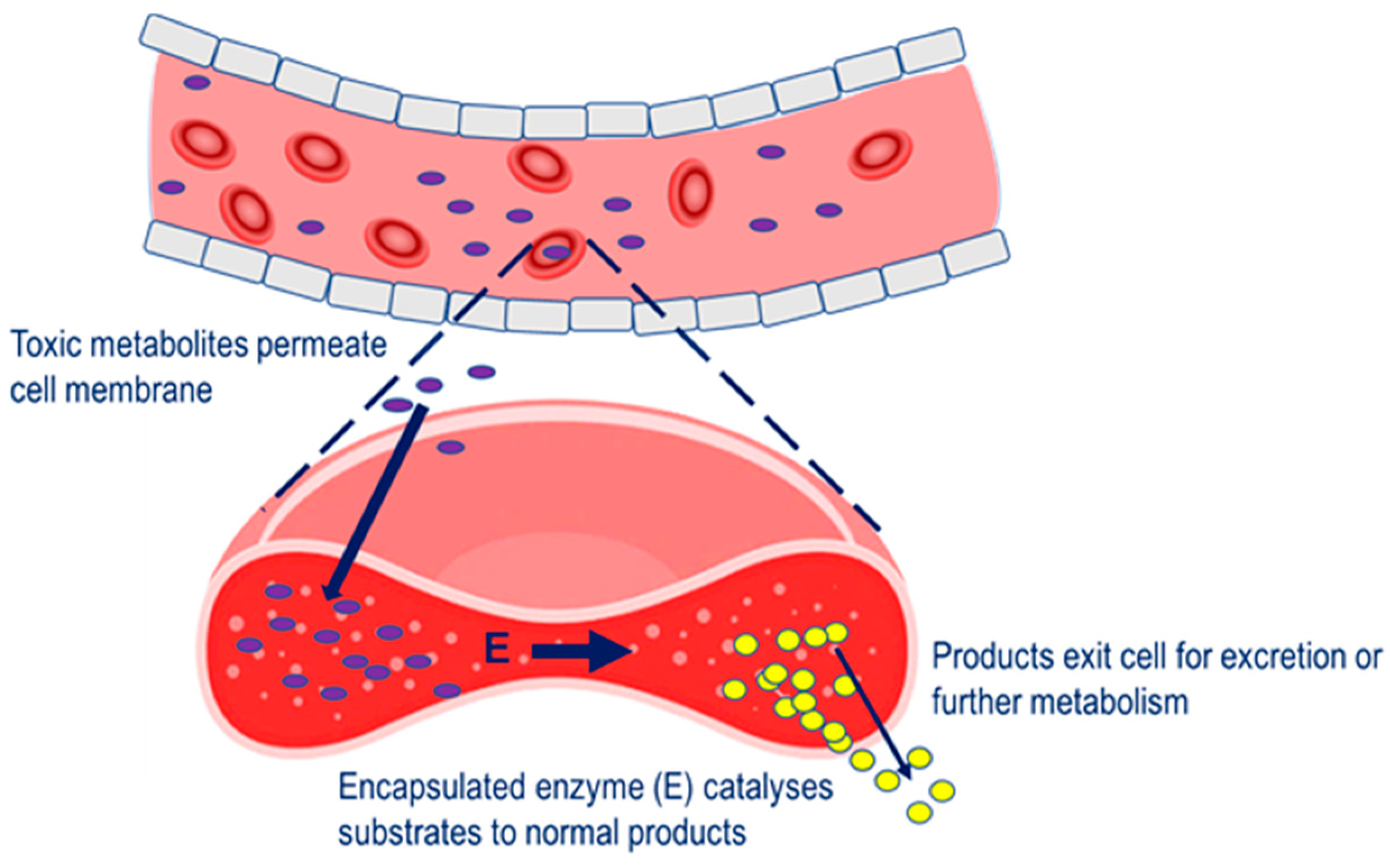{kind=link}
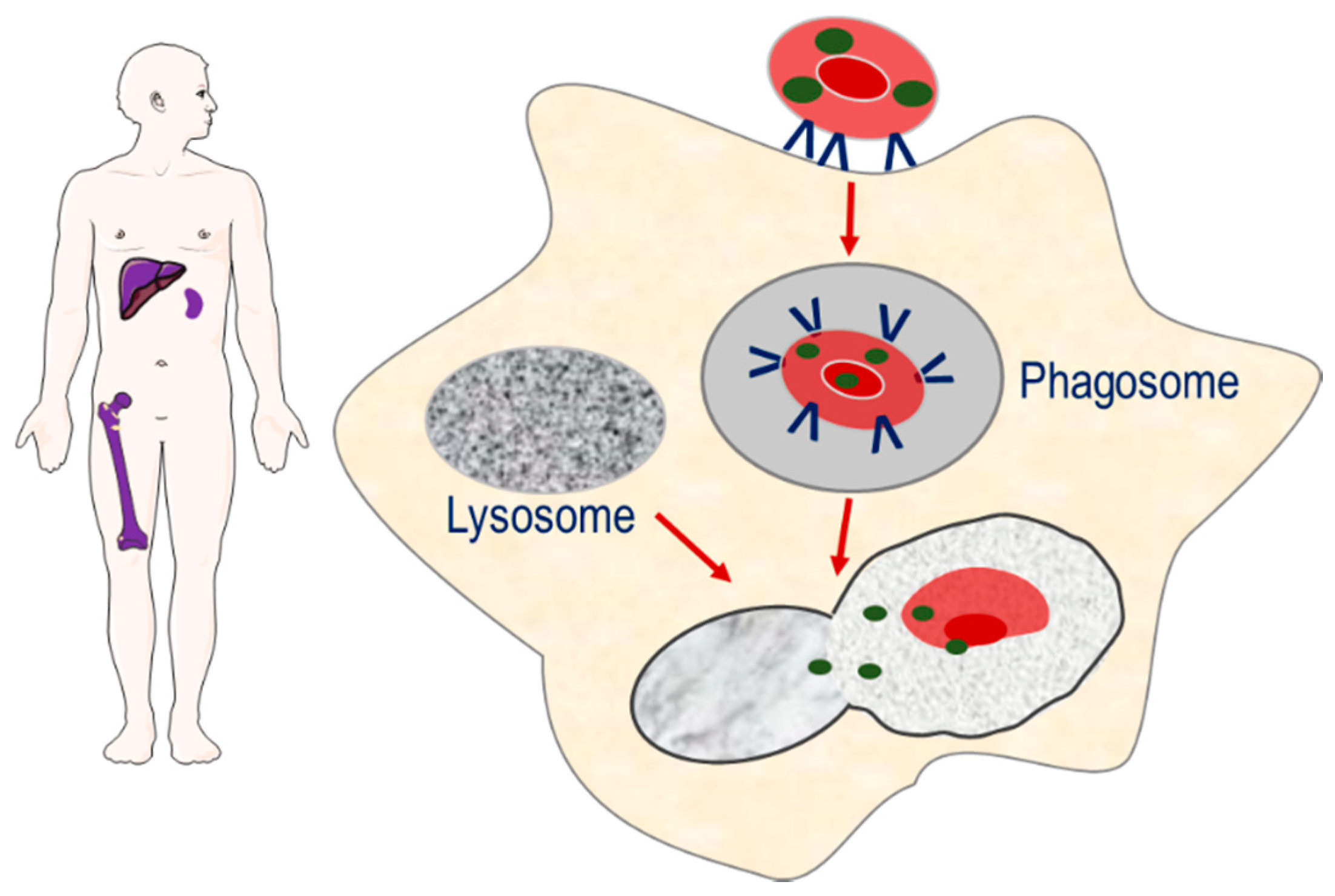{kind=link}
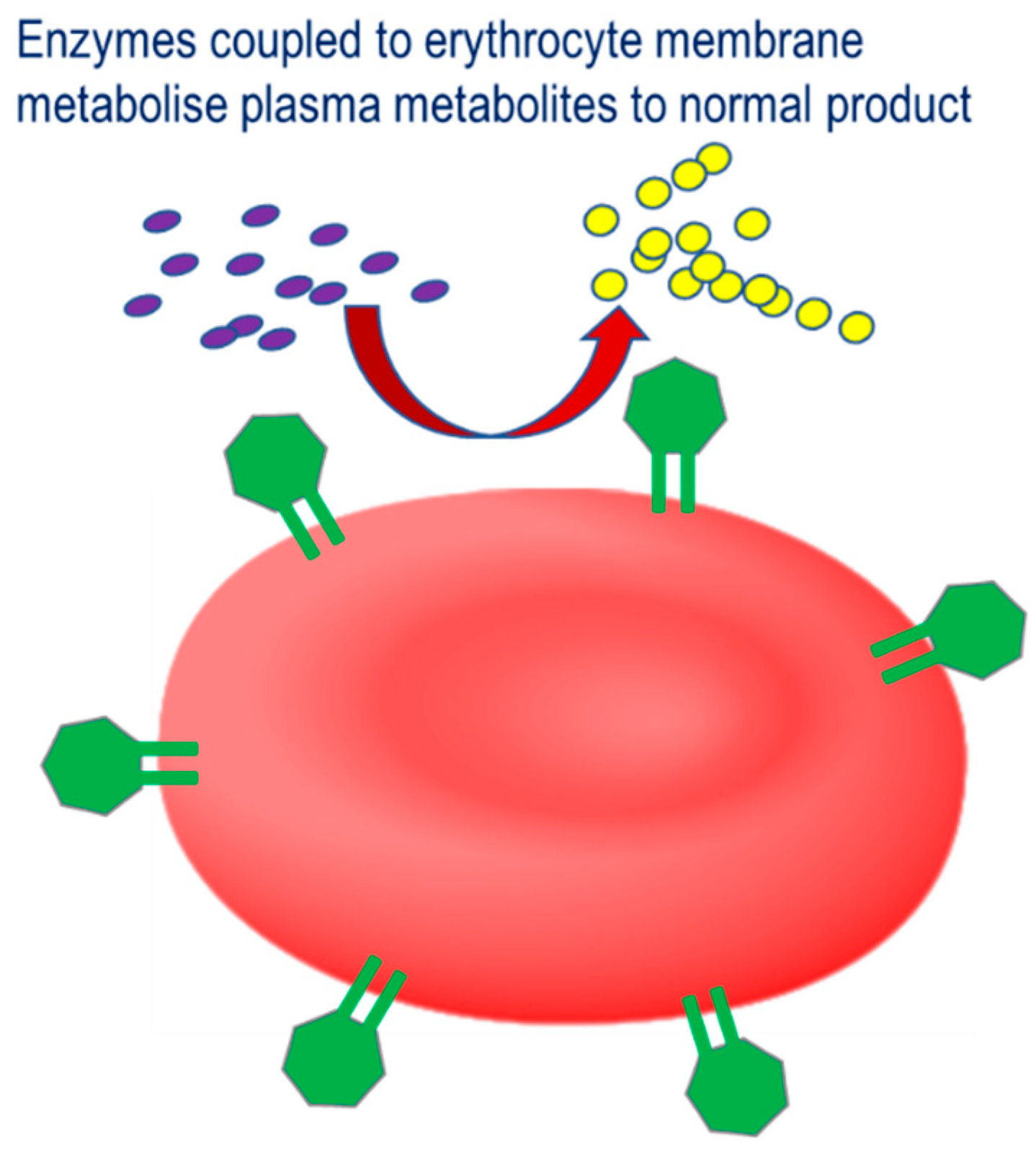{kind=link}
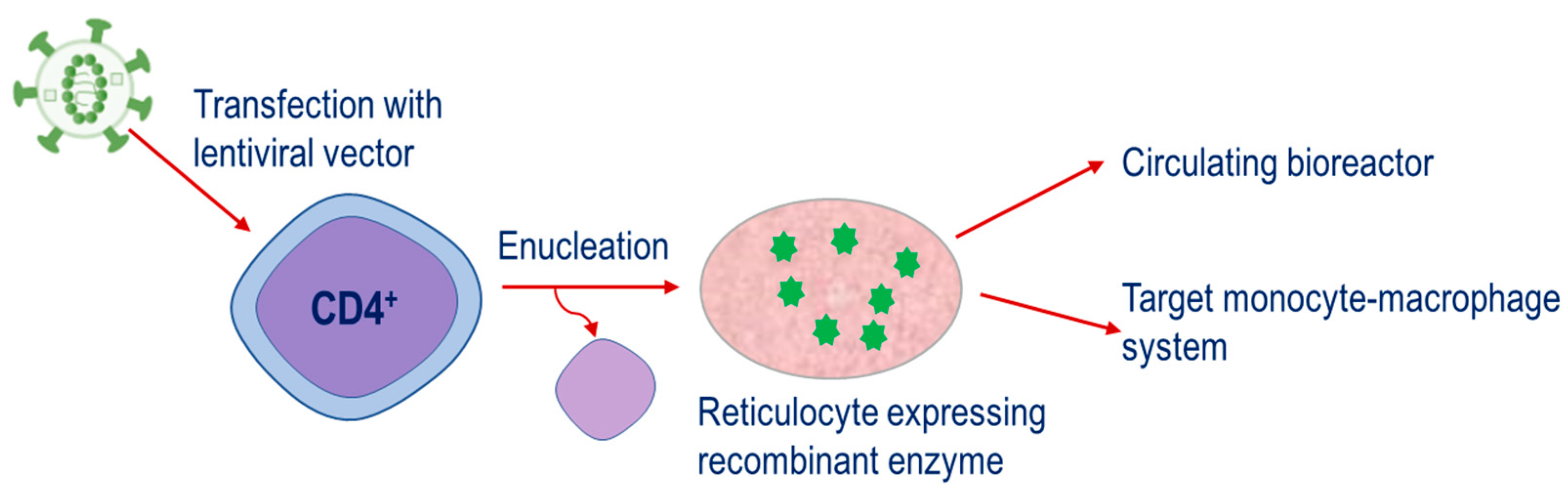{kind=link}
Table 1.
Summary of in vitro, in vivo and clinical applications of erythrocyte-mediated enzyme therapy.
Table 1.
Summary of in vitro, in vivo and clinical applications of erythrocyte-mediated enzyme therapy.
| Therapeutic Application | Therapeutic Target/Disorder | Encapsulated/Conjugated Enzyme | Investigations |
|---|---|---|---|
| Detoxification of exogenous chemicals | Cyanide [8,9,10,11,12,13] | Rhodanase | Mouse in vivo |
| Paraoxon [14,15] | Phosphotriesterase | Mouse in vivo | |
| Paraoxonase | Mouse in vivo | ||
| Ethanol [16,17,18,19,20] | Acetaldehyde dehydrogenase | Mouse in vivo | |
| Alcohol dehydrogenase/aldehyde dehydrogenase | Mouse in vitro/in vivo Human in silico/in vitro | ||
| Methanol [21,22] | Alcohol oxidase | Mouse in vivo Mouse in vivo | |
| Formate dehydrogenase | |||
| Lead [23,24,25] | δ-aminolevulinic acid dehydratase | Mouse in vitro/in vivo Human clinical | |
| Hydrogen gas [26] | Hydrogenase | Human in vitro | |
| Thrombolytic therapy | Plasminogen [27,28,29,30,31] | Urokinase | Human in vitro Mouse in vivo |
| Streptokinase | Human in vitro Mouse in vivo | ||
| tPA | Human in vitro Mouse/rat in vivo | ||
| Brinase | Rabbit in vitro | ||
| Treatment of metabolic disease | Sphingolipids (Lysosomal storage disorders) [32,33,34,35,36,37] | β-glucosidase | Human in vitro |
| β-galactosidase | Human in vitro | ||
| β-glucuronidase | Mouse in vivo | ||
| β-glucocerebrosidase | Human in vitro/clinical | ||
| Alglucerase | Human in vitro | ||
| Ammonia (hyperammonemia) [38,39,40,41,42] | l-glutamate dehydrogenase | Human in vitro Mouse in vivo | |
| Glutamine synthetase | Mouse in vivo | ||
| Glutamate dehydrogenase/alanine aminotransferase | In silico Human in vitro Mouse in vivo | ||
| Arginine and ammonia (arginase-1 deficiency) [43] | Arginase | Human in vitro | |
| Urea and ammonia (chronic renal failure) [44,45,46,47] | Urease/alanine dehydrogenase | Human in vitro Sheep in vivo | |
| Glucose (hyperglycemia) [48,49,50,51] | Hexokinase | Human in vitro | |
| Glucose oxidase | Human in vitro Mouse in vitro/in vivo | ||
| Hexokinase/glucose oxidase | Human in vitro Mouse in vitro/in vivo | ||
| Lactate (hyperlactatemia) [52] | Lactate 2-mono-oxygenase | Mouse/human in vitro | |
| Lactate oxidase | Mouse/human in vitro | ||
| Lactate 2-mono-oxygenase/ lactate oxidase | Mouse in vitro/in vivo Human in vitro | ||
| NADP (Glucose-6-phosphate dehydrogenase deficiency) [3,54] | Glucose-6-phosphate dehydrogenase | Human in vitro | |
| Adenosine and 2‘-deoxyadenosine (Adenosine deaminase deficiency) [55,56,57,58,59] | Adenosine deaminase Pegademase | Human in vitro/in vivo Human in vitro/clinical | |
| Thymidine and 2’-deoxyuridine (MNGIE) [60,61,62,63,64,65,66,67,68,69,70,71,72] | Thymidine phosphorylase | Mouse/dog in vivo Human Phase II | |
| Uric acid (hyperuricemia) [73,74] | Uricase | Human in vitro Mouse in vivo | |
| Phenylalanine (phenylketonuria) [75,76,77,78] | Phenylalanine ammonia lyase | Mouse in vitro/in vivo | |
| Phenylalanine hydroxylase | Mouse in vivo | ||
| RTX-134 | Human Phase Ib | ||
| Antitumor therapy | Asparagine (acute lymphoblastic leukemia) [79,80,81,82,83,84,85,86,87,88,89,90] | Asparaginase GRASPA | Monkey in vivo Human Phase I/II |
| Asparagine (pancreatic adenocarcinoma) [91,92] | ERY-ASP Eryaspase | Human Phase I Human Phase IIb/ Phase III | |
| (triple negative breast cancer) | Eryaspase | Human Phase II/III | |
| Methionine (gastric adenocarcinoma, glioblastoma, breast carcinoma) [93,94] | Methionine-γ-lyase | Mouse in vivo | |
| Arginine [95] | Arginine deiminase | Mouse in vivo |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Bax, B.E. Erythrocytes as Carriers of Therapeutic Enzymes. Pharmaceutics 2020, 12, 435. https://doi.org/10.3390/pharmaceutics12050435
AMA Style
Bax BE. Erythrocytes as Carriers of Therapeutic Enzymes. Pharmaceutics. 2020; 12(5):435. https://doi.org/10.3390/pharmaceutics12050435
Chicago/Turabian StyleBax, Bridget E. 2020. "Erythrocytes as Carriers of Therapeutic Enzymes" Pharmaceutics 12, no. 5: 435. https://doi.org/10.3390/pharmaceutics12050435
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.