4D Micro-Computed X-ray Tomography as a Tool to Determine Critical Process and Product Information of Spin Freeze-Dried Unit Doses

 , ,
, ,  and
and 
Abstract
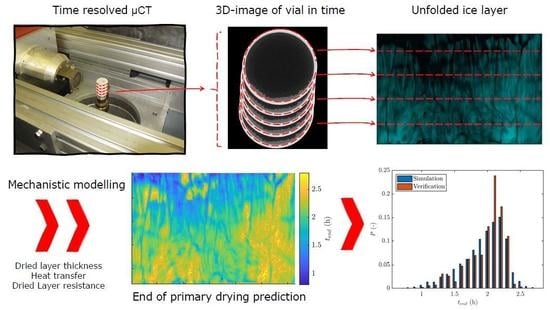
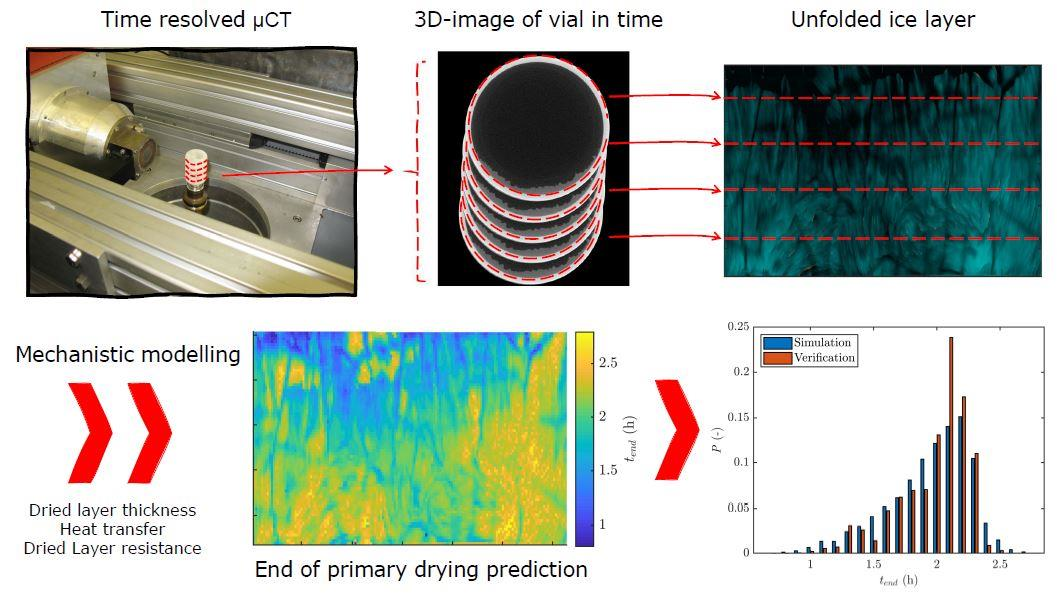
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Determination of Collapse Temperature
2.3. Spin-Freezing
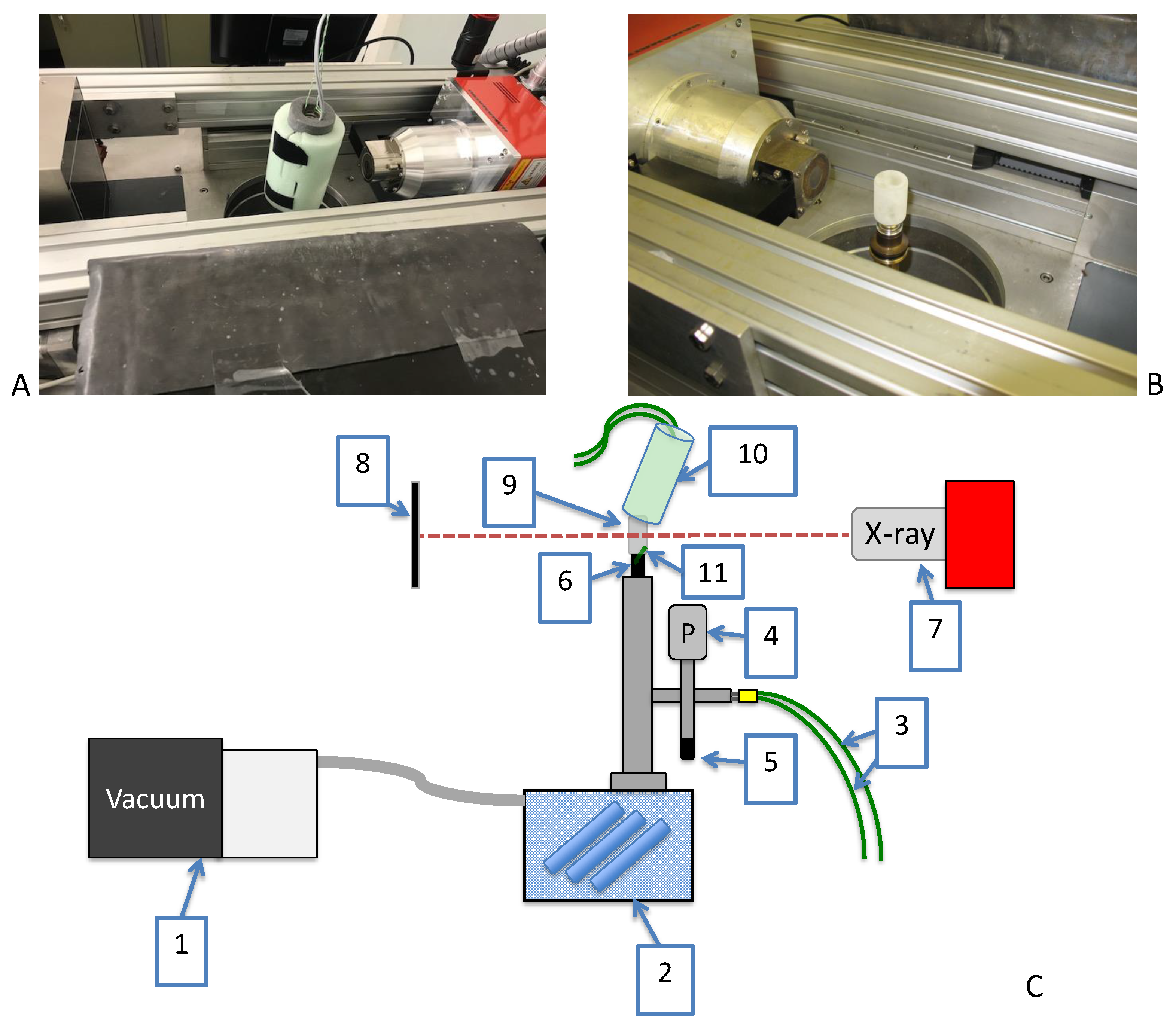
2.4. In-Situ Micro-CT Set-Up
2.4.1. EMCT Scanner
2.4.2. Freeze-Drying Set-Up
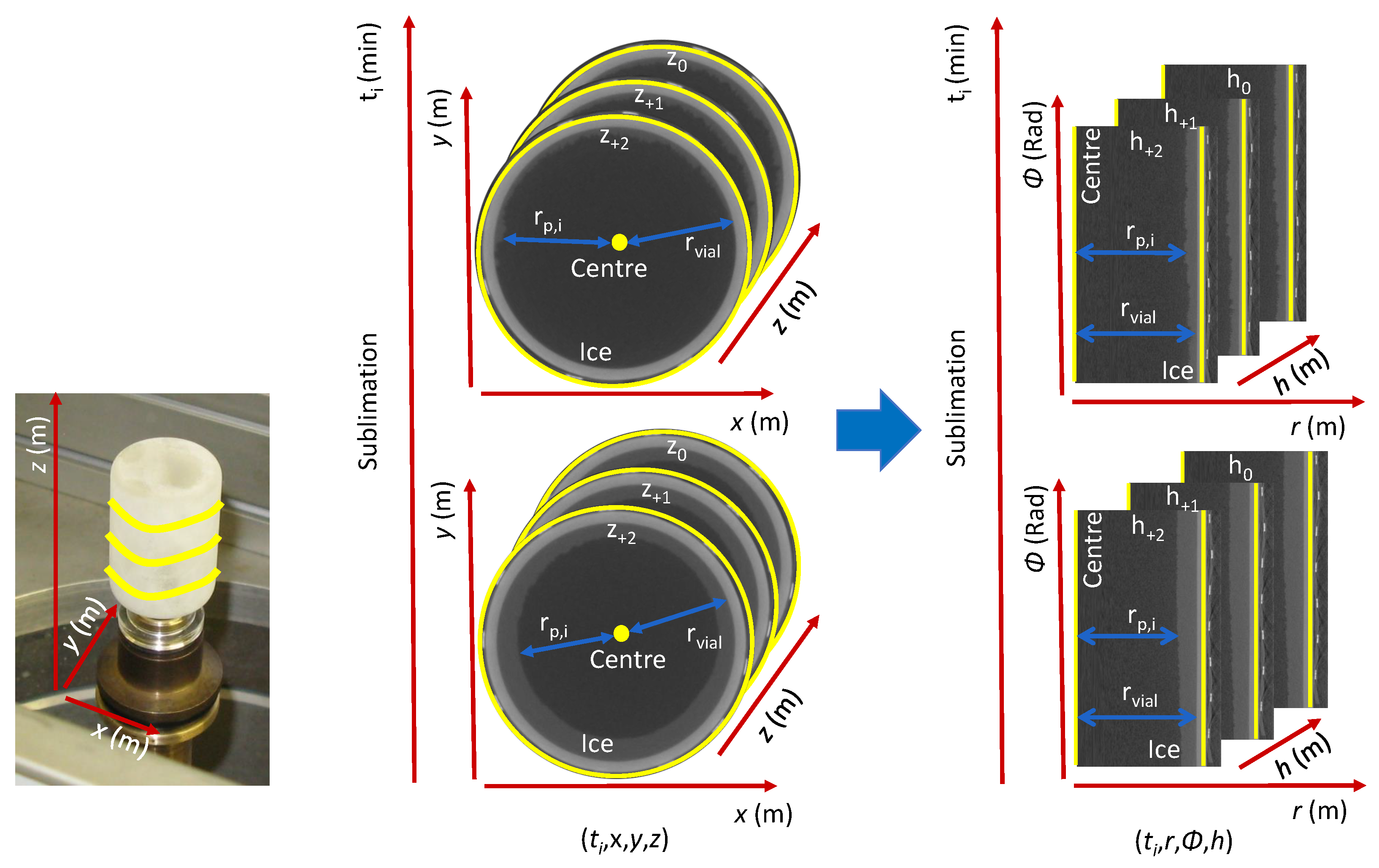
2.4.3. Data Structure
2.5. Direction of Sublimation
2.6. Determination of Dried Product Mass Transfer Resistance
2.7. Simulation of the Primary Drying Time Distribution
2.8. Verification Experiment
3. Results and Discussion
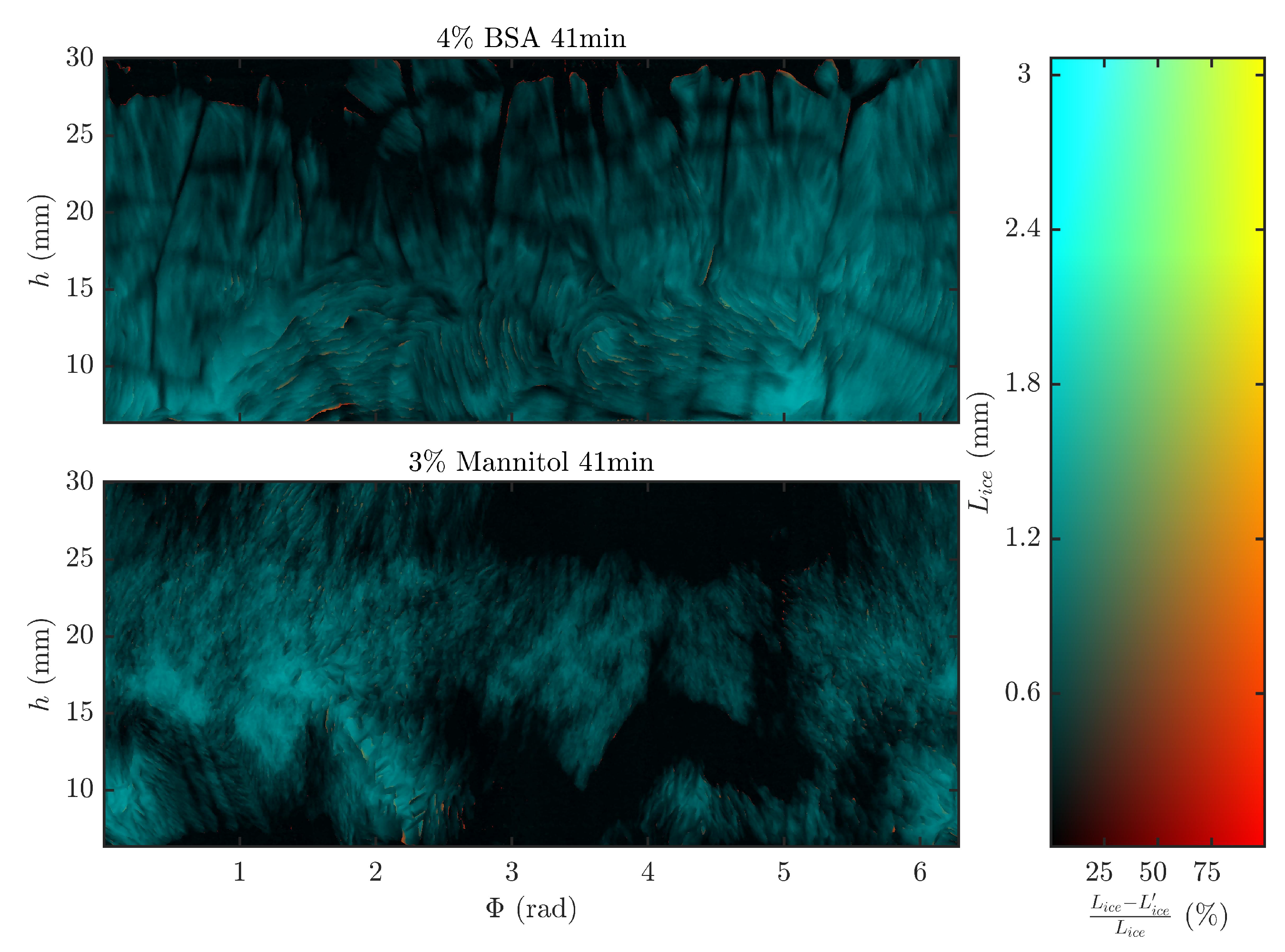
3.1. Direction of Sublimation
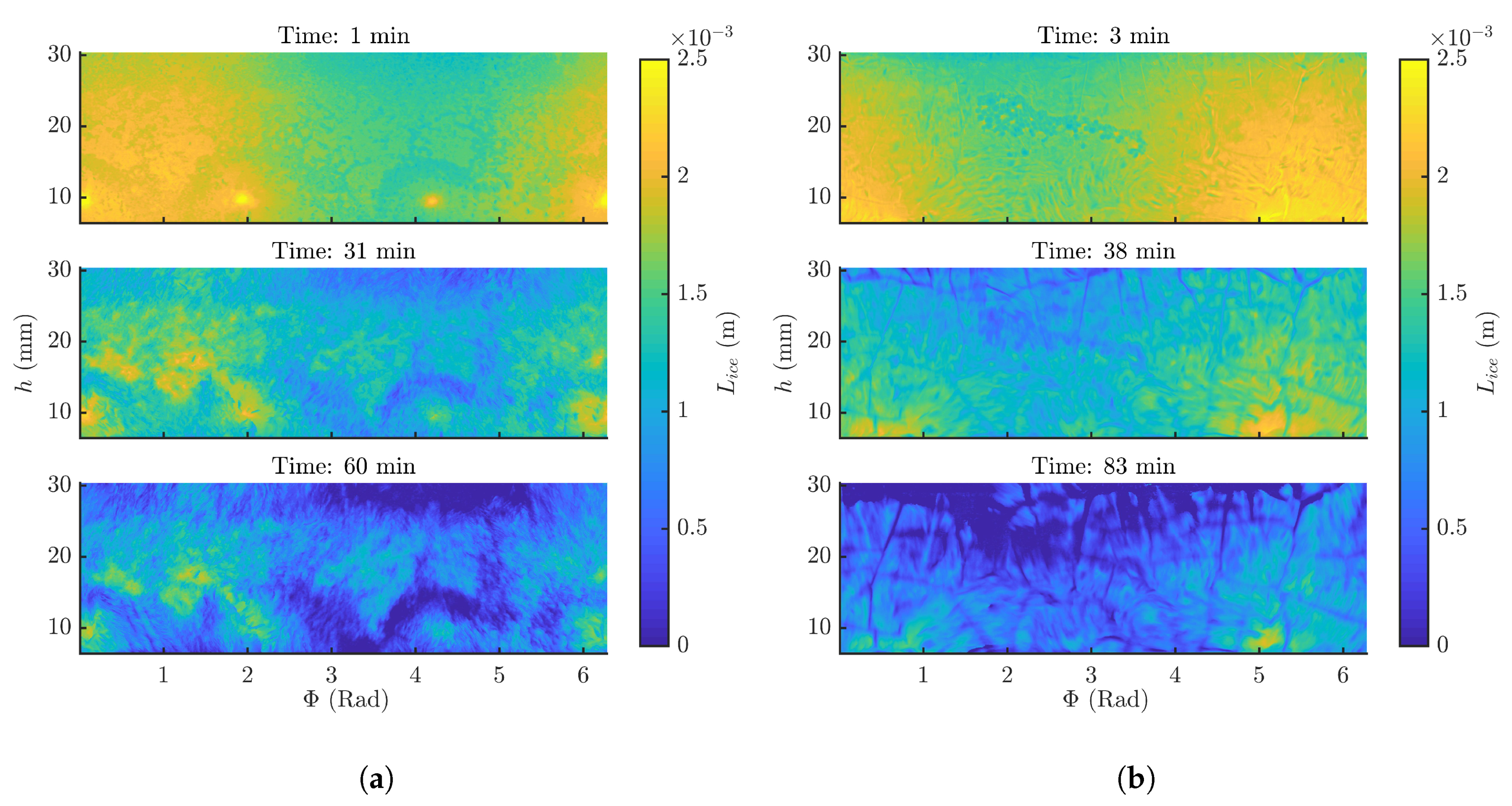
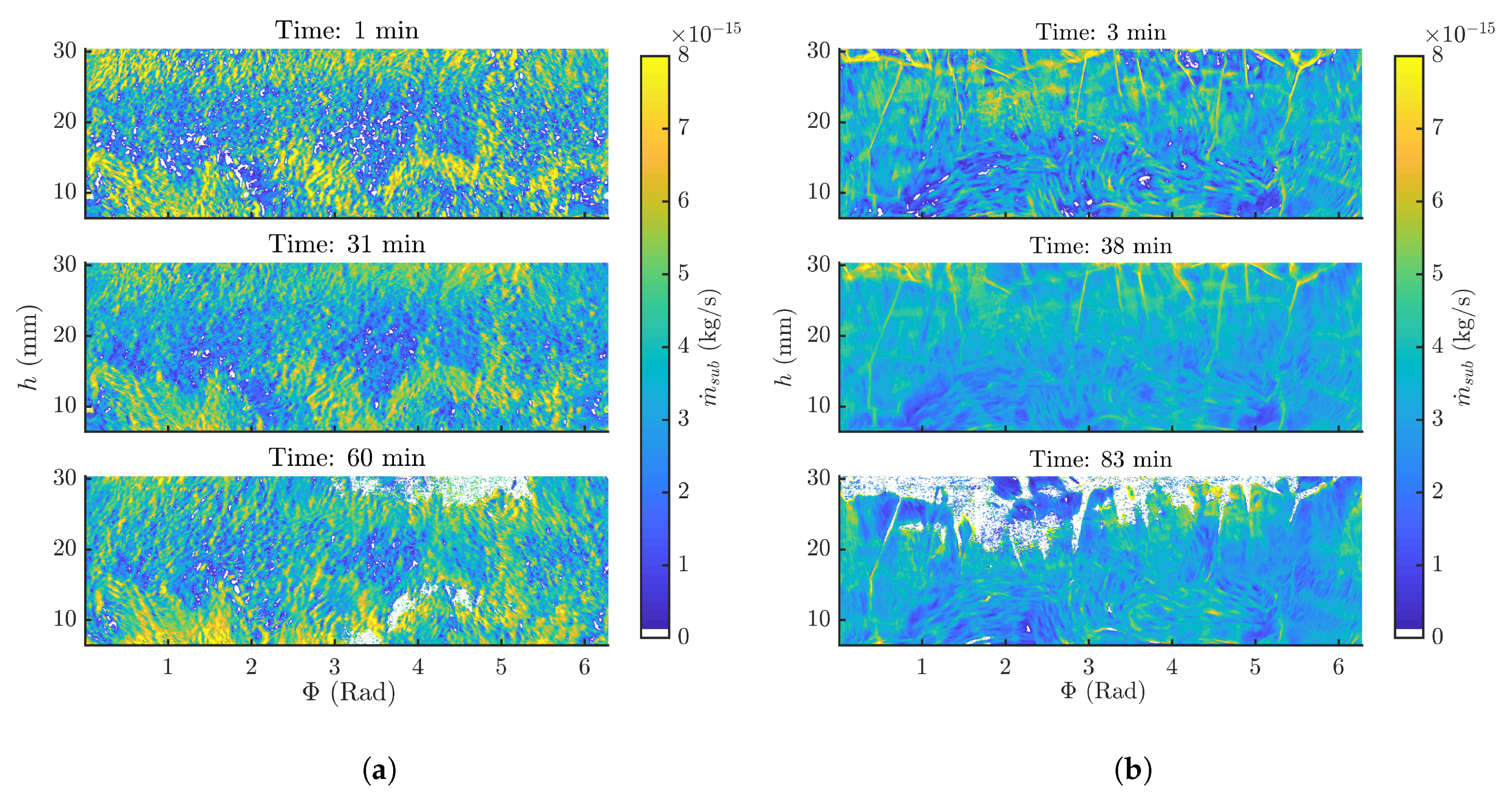
3.2. Frozen Product Thickness and Sublimation Rate
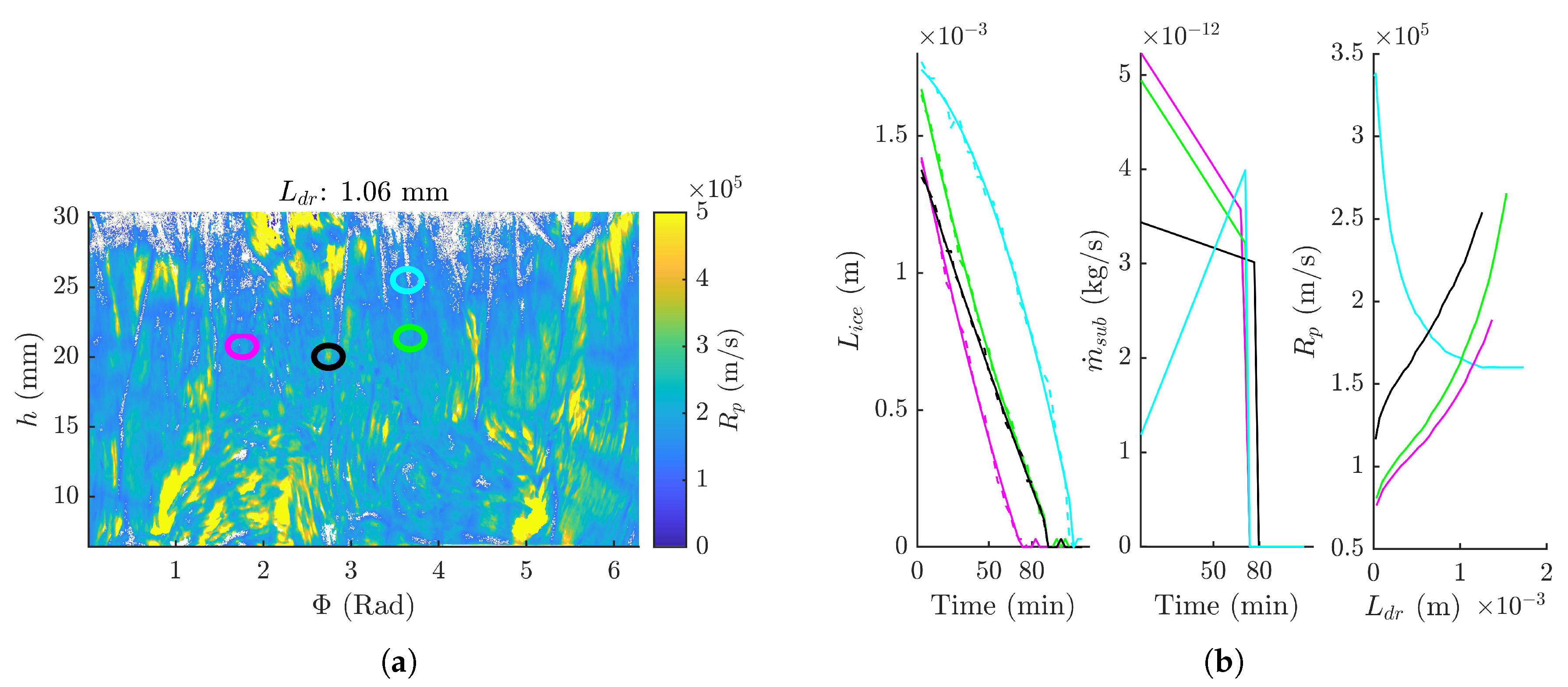
3.3. Dry Product Resistance Distribution
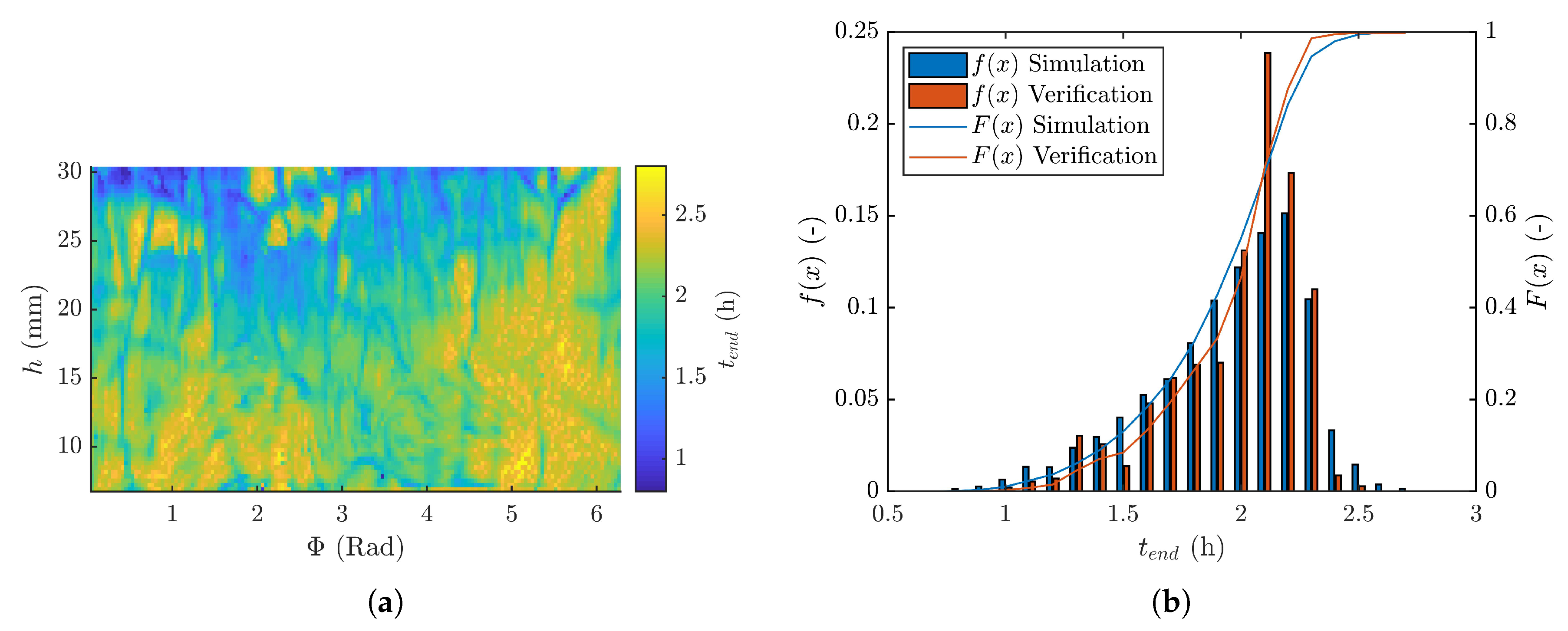
3.4. Primary Drying Time Distribution
3.5. Verification Experiment
4. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Nail, S.L.; Jiang, S.; Chongprasert, S.; Knopp, S.A. Fundamentals of freeze-drying. In Development and Manufacture of Protein Pharmaceuticals; Springer: New York, NY, USA, 2002; pp. 281–360. [Google Scholar]
- Tang, X.; Pikal, M. Design of freeze-drying processes for pharmaceuticals: Practical advice. Pharm. Res. 2004, 21, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Renteria Gamiz, A.G.; Van Bockstal, P.J.; De Meester, S.; De Beer, T.; Corver, J.; Dewulf,, J. Analysis of a pharmaceutical batch freeze dryer: Resource consumption, hotspots, and factors for potential improvement. Dry. Technol. 2019, 37, 1563–1582. [Google Scholar] [CrossRef]
- De Meyer, L.; Lammens, J.; Mortier, S.T.F.C.; Vanbillemont, B.; Van Bockstal, P.J.; Corver, J.; Nopens, I.; Vervaet, C.; De Beer, T. Modelling the primary drying step for the determination of the optimal dynamic heating pad temperature in a continuous pharmaceutical freeze-drying process for unit doses. Int. J. Pharm. 2018, 90, 13591–13599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Bockstal, P.J.; Corver, J.; De Meyer, L.; Vervaet, C.; De Beer, T. Thermal Imaging as a Noncontact Inline Process Analytical Tool for Product Temperature Monitoring during Continuous Freeze-Drying of Unit Doses. Anal. Chem. 2019, 37, 1563–1582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lammens, J.; Mortier, S.T.F.C.; De Meyer, L.; Vanbillemont, B.; Van Bockstal, P.J.; Van Herck, S.; Corver, J.; Nopens, I.; Vanhoorne, V.; De Geest, B.G.; et al. The relevance of shear, sedimentation and diffusion during spin freezing, as potential first step of a continuous freeze-drying process for unit doses. Int. J. Pharm. 2018, 539, 1–10. [Google Scholar] [CrossRef] [PubMed]
- De Meyer, L.; Lammens, J.; Vanbillemont, B.; Van Bockstal, P.J.; Corver, J.; Vervaet, C.; Friess, W.; De Beer, T. Dual chamber cartridges in a continuous pharmaceutical freeze-drying concept: Determination of the optimal dynamic infrared heater temperature during primary drying. Int. J. Pharm. 2019, 570, 118631. [Google Scholar] [CrossRef]
- Brouckaert, D.; De Meyer, L.; Vanbillemont, B.; Van Bockstal, P.J.; Lammens, J.; Mortier, S.T.F.C.; Corver, J.; Vervaet, C.; Nopens, I.; De Beer, T. Potential of near-infrared chemical imaging as process analytical technology tool for continuous freeze-drying. Anal. Chem. 2018, 90, 4354–4362. [Google Scholar] [CrossRef]
- Velardi, S.A.; Barresi, A.A. Development of simplified models for the freeze-drying process and investigation of the optimal operating conditions. Chem. Eng. Res. Des. 2008, 86, 9–22. [Google Scholar] [CrossRef]
- Kuu, W.Y.; McShane, J.; Wong, J. Determination of mass transfer coefficients during freeze drying using modeling and parameter estimation techniques. Int. J. Pharm. 1995, 124, 241–252. [Google Scholar] [CrossRef]
- Pikal, M.J. Use of laboratory data in freeze drying process design: Heat and mass transfer coefficients and the computer simulation of freeze drying. PDA J. Pharm. Sci. Technol. 1985, 39, 115–139. [Google Scholar]
- Mortier, S.T.F.C.; Van Bockstal, P.J.; Corver, J.; Nopens, I.; Gernaey, K.V.; De Beer, T. Uncertainty analysis as essential step in the establishment of the dynamic Design Space of primary drying during freeze-drying. Eur. J. Pharm. Biopharm. 2016, 103, 71–83. [Google Scholar] [CrossRef] [PubMed]
- Vanbillemont, B.; Nicolaï, N.; Leys, L.; De Beer, T. Model-Based Optimisation and Control Strategy for the Primary Drying Phase of a Lyophilisation Process. Pharmaceutics 2020, 12, 181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, S.M.; Nail, S.L.; Pikal, M.J.; Geidobler, R.; Winter, G.; Hawe, A.; Davagnino, J.; Gupta, S.R. Lyophilized drug product cake appearance: What is acceptable? J. Pharm. Sci. 2017, 106, 1706–1721. [Google Scholar] [CrossRef] [PubMed]
- Goshima, H.; Do, G.; Nakagawa, K. Impact of ice morphology on design space of pharmaceutical freeze-drying. J. Pharm. Sci. 2016, 105, 1920–1933. [Google Scholar] [CrossRef] [PubMed]
- Pisano, R.; Barresi, A.A.; Capozzi, L.C.; Novajra, G.; Oddone, I.; Vitale-Brovarone, C. Characterization of the mass transfer of lyophilized products based on X-ray micro-computed tomography images. Dry. Technol. 2017, 35, 933–938. [Google Scholar] [CrossRef]
- Pisano, R.; Fissore, D.; Barresi, A.A.; Brayard, P.; Chouvenc, P.; Woinet, B. Quality by design: Optimization of a freeze-drying cycle via design space in case of heterogeneous drying behavior and influence of the freezing protocol. Pharm. Dev. Technol. 2013, 18, 280–295. [Google Scholar] [CrossRef]
- Kasper, J.C.; Friess, W. The freezing step in lyophilization: Physico-chemical fundamentals, freezing methods and consequences on process performance and quality attributes of biopharmaceuticals. Eur. J. Pharm. Biopharm. 2011, 78, 248–263. [Google Scholar] [CrossRef]
- Capozzi, L.C.; Trout, B.L.; Pisano, R. From Batch to Continuous: Freeze-Drying of Suspended Vials for Pharmaceuticals in Unit-Doses. Ind. Eng. Chem. 2019, 58, 1635–1649. [Google Scholar] [CrossRef]
- Oddone, I.; Van Bockstal, P.J.; De Beer, T.; Pisano, R. Impact of vacuum-induced surface freezing on inter- and intra-vial heterogeneity. Eur. J. Pharm. Biopharm. 2016, 103, 167–178. [Google Scholar] [CrossRef] [Green Version]
- Kuu, W.Y.; O’Bryan, K.R.; Hardwick, L.M.; Paul, T.W. Product mass transfer resistance directly determined during freeze-drying cycle runs using tunable diode laser absorption spectroscopy (TDLAS) and pore diffusion model. Pharm. Dev. Technol. 2011, 16, 343–357. [Google Scholar] [CrossRef]
- Fissore, D.; Pisano, R.; Velardi, S.; Barresi, A.A.; Galan, M. PAT tools for the optimization of the freeze-drying process. Pharm. Eng. 2009, 29, 58–70. [Google Scholar]
- Hottot, A.; Vessot, S.; Andrieu, J. Determination of mass and heat transfer parameters during freeze-drying cycles of pharmaceutical products. PDA J. Pharm. Sci. Technol. 2005, 59, 138–153. [Google Scholar] [PubMed]
- Kuu, W.Y.; Hardwick, L.M.; Akers, M.J. Rapid determination of dry layer mass transfer resistance for various pharmaceutical formulations during primary drying using product temperature profiles. Int. J. Pharm. 2006, 313, 99–113. [Google Scholar] [CrossRef] [PubMed]
- Maire, E.; Withers, P.J. Quantitative X-ray tomography. Int. Mater. Rev. 2014, 313, 1–43. [Google Scholar] [CrossRef] [Green Version]
- Haeuser, C.; Goldbach, P.; Huwyler, J.; Friess, W.; Allmendinger, A. Imaging techniques to characterize cake appearance of freeze-dried products. J. Pharm. Sci. 2018, 107, 2810–2822. [Google Scholar] [CrossRef]
- Bultreys, T.; Boone, M.A.; Boone, M.N.; De Schryver, T.; Masschaele, B.; Van Hoorebeke, L.; Cnudde, V. Fast laboratory-based micro-computed tomography for pore-scale research: Illustrative experiments and perspectives on the future. Adv. Water Resour. 2016, 95, 341–351. [Google Scholar] [CrossRef]
- Mokso, R.; Marone, F.; Haberthür, D.; Schittny, J.C.; Mikuljan, G.; Isenegger, A.; Stampanoni, M. Following dynamic processes by X-ray tomographic microscopy with sub-second temporal resolution. AIP Conf. Proc. 2010, 1365, 38–41. [Google Scholar]
- García-Moreno, F.; Kamm, P.H.; Neu, T.R.; Bülk, F.; Mokso, R.; Schlepütz, C.M.; Stampanoni, M.; Banhart, J. Using X-ray tomoscopy to explore the dynamics of foaming metal. Nat. Commun. 2019, 10, 1–9. [Google Scholar] [CrossRef]
- Corver, J.; Van Bockstal, P.J.; De Beer, T. A continuous and controlled pharmaceutical freeze-drying technology for unit doses. Eur. Pharm. Rev. 2017, 22, 51–53. [Google Scholar]
- Dierick, M.; Van Loo, D.; Masschaele, B.; Van den Bulcke, J.; Van Acker, J.; Cnudde, V.; Van Hoorebeke, L. Recent micro-CT scanner developments at UGCT. Nucl. Instrum. Meth. B 2014, 324, 35–40. [Google Scholar] [CrossRef] [Green Version]
- Murphy, D.M.; Koop, T. Review of the vapour pressures of ice and supercooled water for atmospheric applications. Q. J. R. Meteorol. Soc. 2005, 131, 1539–1565. [Google Scholar] [CrossRef]
- Ullrich, S.; Seyferth, S.; Lee, G. Measurement of shrinkage and cracking in lyophilized amorphous cakes. Part II: Kinetics. Pharm. Res. 2015, 32, 2503–2515. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, K.; Hottot, A.; Vessot, S.; Andrieu, J. Modeling of freezing step during freeze-drying of drugs in vials. AIChE J. 2007, 53, 1362–1372. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Description | Symbol | Value | Unit |
|---|---|---|---|
| Field of view angle | 45.1 | ||
| Latent heat of sublimation | - | J/mol | |
| Time resolution prediction | 60 | s | |
| Dried layer porosity | 0.97 | (-) | |
| Mass density ice | 918 | kg/m | |
| Azimuth (cylindrical coordinate) | - | Rad | |
| coefficient | 9.550426 | Pa | |
| coefficient | 5723.2658 | K | |
| coefficient | 3.53068 | 1/K | |
| coefficient | 0.00728332 | Pa | |
| coefficient | 4.68 × 10 | J/mol | |
| coefficient | 35.9 | J/molK | |
| coefficient | 0.0741 | J/molK | |
| coefficient | 542 | J/mol | |
| coefficient | 124 | K | |
| Product surface | - | m | |
| Product surface entire vial | - | m | |
| Distance camera - vial | m | ||
| Height (cylindrical coordinate) | h | - | m |
| Height of vial | m | ||
| Horizontal field of view | m | ||
| Dried product thickness | - | m | |
| Frozen product thickness | - | m | |
| Initial frozen product thickness | - | m | |
| Integrated frozen product length | - | m | |
| Sublimation rate | - | kg/s | |
| Molecular weight water | M | kg/mol | |
| Height segmentations (Section 2.6/Section 2.7) | m | 1000/100 | (-) |
| Azimuthal segmentations (Section 2.6/Section 2.7) | n | 1600/160 | (-) |
| Chamber Pressure (Section 2.6/Section 2.7 and Section 2.8) | 5/10 | Pa | |
| Partial water vapour Pressure | - | Pa | |
| Total power towards vial | 1.2 | W | |
| Radius (cylindrical coordinate) | r | - | m |
| Radius frozen product | - | m | |
| Radius inner vial wall | m | ||
| Dried layer resistance | - | m/s | |
| Time point | - | min | |
| Primary drying time | - | h | |
| Collapse temperature (BSA/mannitol) | −9/−2 | C | |
| Sublimation interface temperature | - | K | |
| Product temperature | - | K | |
| Volume cylindrical segment | V | - | m |
| Length (cartesian coordinate) | x | - | m |
| Width (cartesian coordinate) | y | - | m |
| Depth (cartesian coordinate) | z | - | m |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vanbillemont, B.; Lammens, J.; Goethals, W.; Vervaet, C.; Boone, M.N.; De Beer, T. 4D Micro-Computed X-ray Tomography as a Tool to Determine Critical Process and Product Information of Spin Freeze-Dried Unit Doses. Pharmaceutics 2020, 12, 430. https://doi.org/10.3390/pharmaceutics12050430
Vanbillemont B, Lammens J, Goethals W, Vervaet C, Boone MN, De Beer T. 4D Micro-Computed X-ray Tomography as a Tool to Determine Critical Process and Product Information of Spin Freeze-Dried Unit Doses. Pharmaceutics. 2020; 12(5):430. https://doi.org/10.3390/pharmaceutics12050430
Chicago/Turabian StyleVanbillemont, Brecht, Joris Lammens, Wannes Goethals, Chris Vervaet, Matthieu N. Boone, and Thomas De Beer. 2020. "4D Micro-Computed X-ray Tomography as a Tool to Determine Critical Process and Product Information of Spin Freeze-Dried Unit Doses" Pharmaceutics 12, no. 5: 430. https://doi.org/10.3390/pharmaceutics12050430