The Role of Functional Excipients in Solid Oral Dosage Forms to Overcome Poor Drug Dissolution and Bioavailability


Abstract
:
1. Introduction
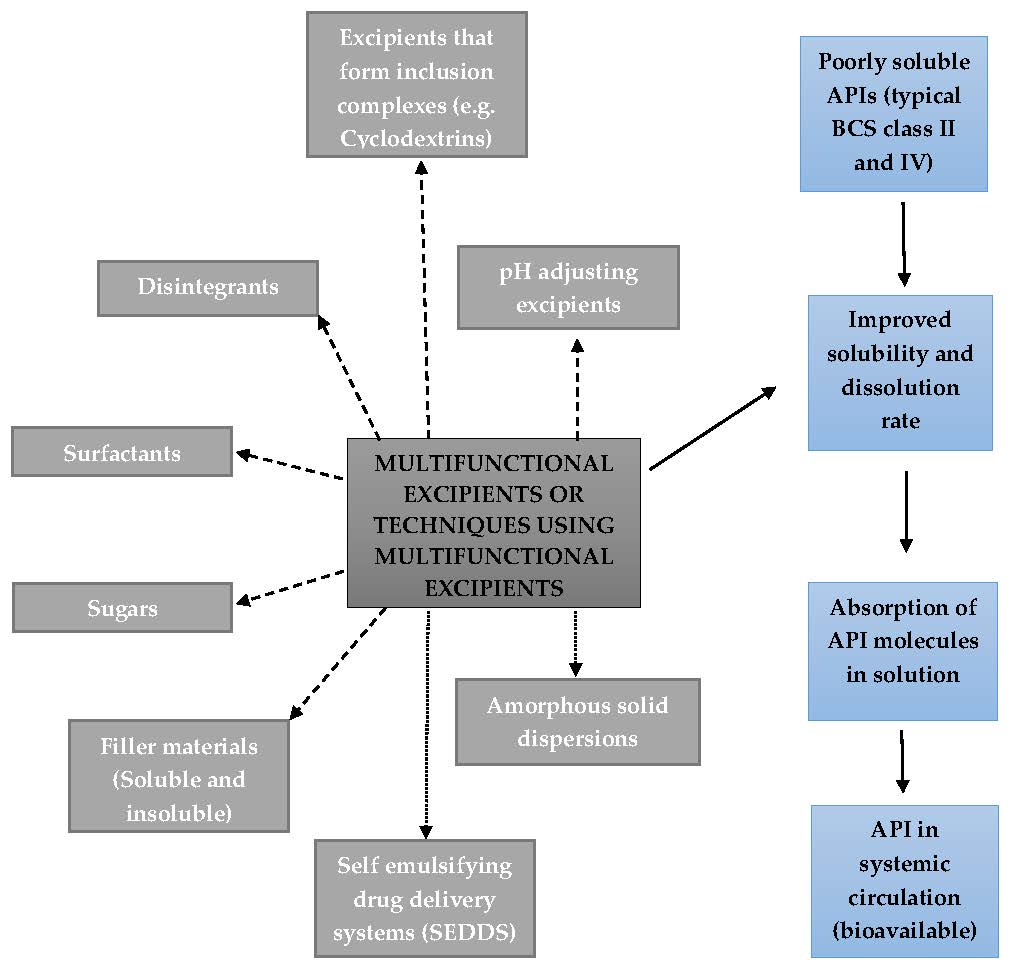
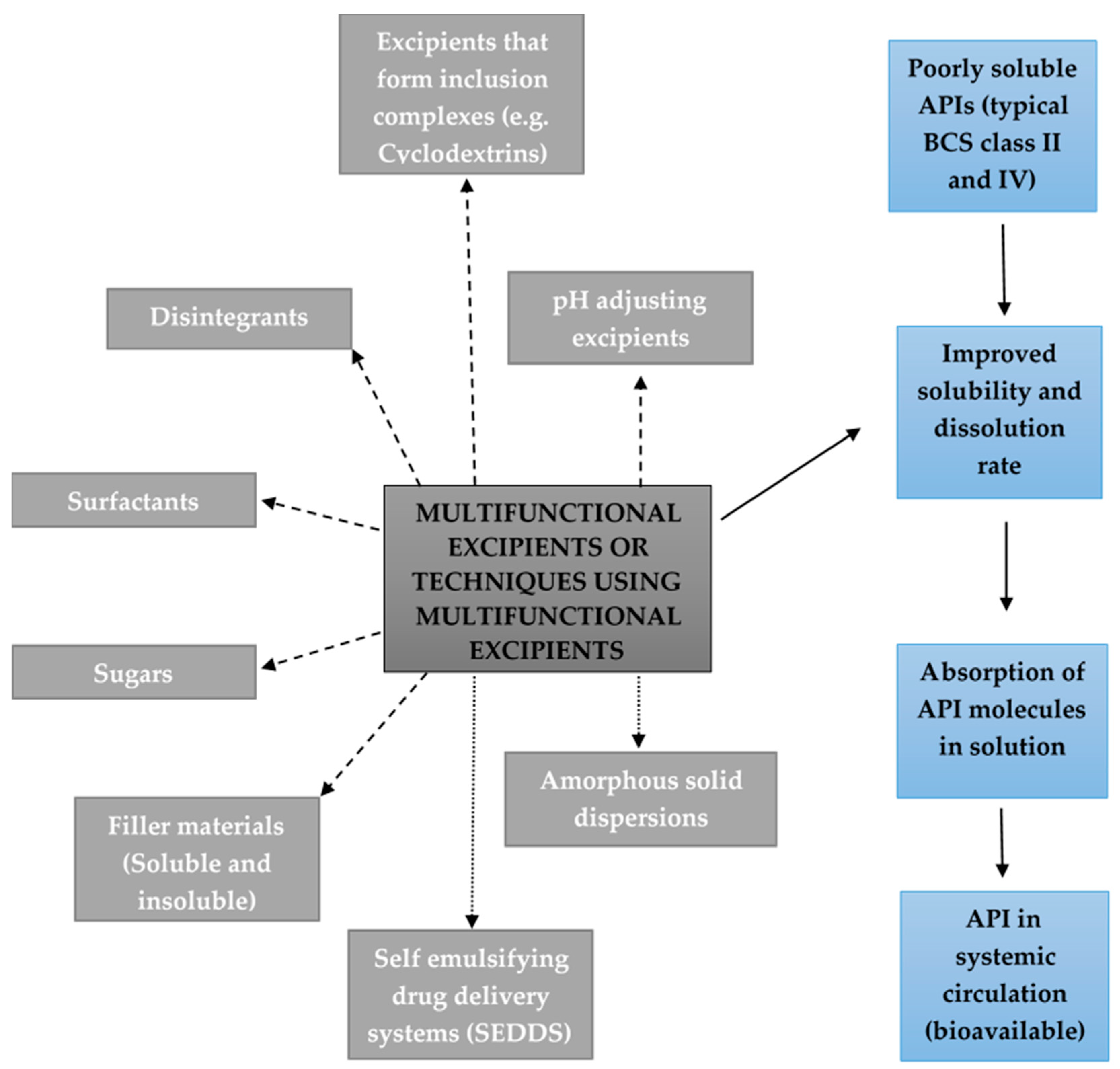
2. Excipients Added to Solid Oral Dosage Forms to Improve Drug Solubility and/or Dissolution
2.1. Excipients that Form Inclusion Complexes with Drug Molecules
2.2. Disintegrants
2.3. pH Adjusting Excipients
2.4. Amorphous Solid Dispersions
2.5. Surfactants
2.6. Self-Emulsifying Drug Delivery Systems (SEDDS)
2.7. Sugars
2.8. Soluble and Insoluble Filler Materials
3. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Elder, D.P.; Kuentz, M.; Holm, R. Pharmaceutical excipients—Quality, regulatory and biopharmaceutical considerations. Eur. J. Pharm. Sci. 2016, 87, 88–99. [Google Scholar] [CrossRef] [PubMed]
- Abrantes, C.G.; Duarte, D.; Reis, C.P. An Overview of Pharmaceutical Excipients: Safe or Not Safe. J. Pharm. Sci. 2016, 105, 2019–2026. [Google Scholar] [CrossRef] [PubMed]
- Haywood, A.; Glass, B.D. Pharmaceutical excipients—Where do we begin? Aust. Prescr. 2011, 34, 112–114. [Google Scholar] [CrossRef]
- Monsuur, F.; Poncher, J. Raising expectations of excipients. Chim. Oggi Chem. Today 2010, 28, 1–8. [Google Scholar]
- Taylor, P. Focus on excipient quality: Evolving regulations require a fresh approach. Chim. Oggi Chem. Today 2014, 32, 49–52. [Google Scholar]
- Guth, F.; Schiffter, H.A.; Kolter, K. Novel excipients—From concept to launch. Chim. Oggi Chem. Today 2013, 31, 78–81. [Google Scholar]
- Kanojia, N.; Kaur, L.; Nagpal, M.; Bala, R. Modified Excipients in Novel Drug Delivery: Need of the Day. J. Pharm. Technol. Res. Manag. 2013, 1, 81–107. [Google Scholar] [CrossRef]
- Crowley, P.J.; Martini, L.G. Formulation design: New drugs from old. Drug Discov, Today 2004, 1, 537–542. [Google Scholar] [CrossRef]
- Junyaprasert, V.B.; Morakul, B. Nanocrystals for enhancement of oral bioavailability of poorly water-soluble drugs. Asian J. Pharm. Sci. 2015, 10, 13–23. [Google Scholar] [CrossRef] [Green Version]
- Garcίa-Arieta, A. Interactions between active pharmaceutical ingredients and excipients affecting bioavailability: Impact on bioequivalence. Eur. J. Pharm. Sci. 2014, 65, 89–97. [Google Scholar] [CrossRef]
- Loftsson, T.; Brewster, M.E. Pharmaceutical apllications of cyclodextrins. 1. Drug solubilisation and stabilization. J. Pharm. Sci. 1996, 85, 1017–1025. [Google Scholar] [CrossRef] [PubMed]
- Saharan, V.A.; Kukkar, V.; Kataria, M.; Gera, M.; Choudhury, P.K. Dissolution Enhancement of Drugs. Part I: Technologies and Effect of Carriers. Int. J. Health Res. 2009, 2, 1–20. [Google Scholar] [CrossRef]
- Arias, K.; Robinson, S.G.; Lyngaas, S.S.; Cherala, S.S.; Hartzell, M.; Mei, S.; Vilic, A.; Girel, J.K.; Kuemmell, A.; Vrettas, J.S.; et al. Minocycline and Tigecycline form higher-order Ca2+ complexes of stronger affinity than Tetracycline. Inorg. Chem. Acta 2016, 441, 181–191. [Google Scholar] [CrossRef]
- Conceição, J.; Adeoye, O.; Cabral-Marques, H.M.; Lobo, J.M.S. Cyclodextrins as excipients in tablet formulations. Drug Discov. Today 2018, 23, 1274–1284. [Google Scholar] [CrossRef] [PubMed]
- Loftsson, T. Excipient Pharmacokinetics and Profiling. Int. J. Pharm. 2015, 480, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Brewster, M.E.; Loftsson, T. Cyclodextrins as pharmaceutical solubilizers. Adv. Drug Deliv. Rev. 2007, 59, 645–666. [Google Scholar] [CrossRef] [PubMed]
- Kou, W.; Cai, C.; Xu, S.; Wang, H.; Liu, J.; Yang, D.; Zhang, T. In vitro and in vivo evaluation of novel immediate release carbamazepine tablets: Complexation with hydroxypropyl-β-cyclodextrin in the presence of HPMC. Int. J. Pharm. 2011, 409, 75–80. [Google Scholar] [CrossRef]
- Anraku, M.; Hiraga, A.; Iohara, D.; Pipkin, J.D.; Uekama, K.; Hirayama, F. Slow-release of famotidinefrom tablets consisting of chitosan/sulfobutyl ether β-cyclodextrin composites. Int. J. Pharm. 2015, 487, 142–147. [Google Scholar] [CrossRef]
- Palem, C.R.; Battu, S.K.; Gannu, R.; Yamsani, V.V.; Repka, M.A.; Yamsani, M.R. Role of cyclodextrin complexation in felodipine-sustained release matrix tablets intended for oral transmucosal deliver: In vitro and ex vivo characterization. Pharm. Dev. Technol. 2012, 17, 321–332. [Google Scholar] [CrossRef]
- Songsurang, K.; Pakdeebumrung, J.; Praphairaksit, N.; Muangsin, N. Sustained release of amoxicillin from ethyl cellulose-coated amoxicillin/chitosan-cyclodextrin-based tablets. AAPS PharmSciTech 2010, 12, 35–45. [Google Scholar] [CrossRef] [Green Version]
- Desai, S.; Poddar, A.; Sawant, K. Formulation of cyclodextrin inclusion complex-based orally disintegrating tablet of esclicarbazeine acetate for improved oral bioavailability. Mater. Sci. Eng. C 2016, 58, 826–834. [Google Scholar] [CrossRef] [PubMed]
- Gerber, W.; Hamman, J.H.; Steyn, J.D. Excipient-drug pharmacokinetic interactions: Effect of disintegrants on efflux across excised pig intestinal tissues. J. Food Drug Anal. 2018, 26, S115–S124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Onuki, Y.; Kosugi, A.; Hamaguchi, M.; Marumo, Y.; Kumada, S.; Hirai, D.; Ikeda, J.; Hayashi, Y. A comparative study of disintegration actions of various disintegrants using Kohonen’s self-organizing maps. J. Drug Deliv. Sci. Technol. 2018, 43, 141–148. [Google Scholar] [CrossRef]
- Suryadevara, V.; Lankapalli, S.R.; Danda, L.H.; Pendyala, V.; Katta, V. Studies on jackfruit seed starch as a novel natural superdisintegrant for the design and evaluation of irbesartan fast dissolving tablets. Integr. Med. Res. 2017, 6, 280–291. [Google Scholar] [CrossRef] [PubMed]
- Husseiny, R.A.; Abu Lila, A.S.; Abdallah, M.H.; El-ghamry, H.A. Fast disintegrating tablet of Valsartan for the treatment of pediatric hypertension: In vitro and in vivo evaluation. J. Drug Deliv. Sci. Technol. 2018, 43, 194–200. [Google Scholar] [CrossRef]
- Meyers, R.S.; Siu, A. Pharmacotherapy Review of Chronic Pediatric Hypertension. Clin. Ther. 2011, 33, 1331–1356. [Google Scholar] [CrossRef]
- Jambhekar, S.S.; Breen, P.J. Drug dissolution: Significance of physicochemical properties and physiological conditions. Drug Discov. Today 2013, 18, 1173–1184. [Google Scholar] [CrossRef]
- Martins, G.B.; Tarouco, F.; Rosa, C.E.; Robaldo, R.B. The utilization of sodium bicarbonate, calcium carbonate or hydroxide in biofloc system: Water quality, growth performance and oxidative stress of nile tilapia (Oreochromis niloticus). Aquaculture 2017, 468, 10–17. [Google Scholar] [CrossRef]
- Kataoka, M.; Fukahari, M.; Ikemura, A.; Kubota, A.; Hipashino, H.; Sakuma, S.; yamashira, S. Effects of gastric pH on oral drug absorption: In vitro assessment using a dissolution/permeation system reflecting the gastric dissolution process. Eur. J. Pharm. Biopharm. 2016, 101, 103–111. [Google Scholar] [CrossRef]
- Adachi, M.; Hinatsu, Y.; Katsumi, H.; Sakane, T.; Nakatani, M.; Wada, K.; Yamamoto, A. Improved dissolution and absorption of Ketoconazole in the presence of organic acids as pH-modifiers. Eur. J. Pharm. Sci. 2015, 76, 225–230. [Google Scholar] [CrossRef]
- Grattan, T.; Hickman, R.; Darby-Dowman, A.; Hayward, M.; Boyce, M.; Warrington, S. A five way crossover human volunteer study to compare the pharmacokinetics of paracetamol following oral administration of two commercially available paracetamol tablets and three development tablets containing paracetamol in combination with sodium bicarbonate or calcium carbonate. Eur. J. Pharm. Biopharm. 2000, 49, 225–229. [Google Scholar] [PubMed]
- Tran, T.T.; Tran, P.H.; Lee, B. Dissolution-modulation mechanism of alkalizers and polymers in a nanoemulsifying solid dispersion containing ionizable and poorly water-soluble drug. Eur. J. Pharm. Biopharm. 2009, 72, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Kang, W.; Van Nguyen, H.; Park, C.; Choi, Y.; Lee, B. Modulation of microenvironmental pH for dual release and reduced in vivo gastrointestinal bleeding of aceclofenac using hydroxypropyl methylcellulose-based bilayered matrix tablet. Eur. J. Pharm. Sci. 2017, 102, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Jermain, S.V.; Brough, C.; Williams, R.O., III. Amorphous solid dispersions and nanocrystal technologies for poorly water-soluble drug delivery—An update. Int. J. Pharm. 2018, 535, 379–392. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.; Chung, Y.; Cheah, X.; Tan, E.Y.; Quah, J. The characterization and dissolution performances of spray dried solid dispersion of ketoprofen in hydrophilic carriers. Asian J. Pharm. Sci. 2015, 10, 372–385. [Google Scholar] [CrossRef] [Green Version]
- Herbrink, M.; Schellens, J.H.M.; Beijnen, J.H.; Nuijen, B. Improving the solubility of nilotinib through novel spray-dried solid dispersions. Int. J. Pharm. 2017, 529, 294–302. [Google Scholar] [CrossRef]
- Hens, B.; Corsetti, M.; Brouwers, J.; Augustijns, P. Gastrointestinal and Systemic Monitoring of Posaconazole in Humans after Fasted and Fed State Administration of a Solid Dispersion. J. Pharm. Sci. 2016, 105, 2904–2912. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.; Lee, S.; Jang, W.S.; Byeon, J.C.; Park, J. Solid dispersion of dutasteride using the solvent evaporation method: Approaches to improve dissolution rate and oral bioavailability in rats. Mater. Sci. Eng. C 2018, 90, 387–396. [Google Scholar] [CrossRef]
- Neophytou, C.M.; Constantinou, C.; Papageorgis, P.; Constantinou, A.I. D-alpha-tocopheryl polyethylene glycol succinate (TPGS) induces cell cycle arrest and apoptosis selectively in Survivin-overexpressing breast cancer cells. Biochem. Pharmacol. 2014, 89, 31–42. [Google Scholar] [CrossRef]
- Mirzaei, H.; Shakeri, A.; Rashidi, B.; Jalili, A.; Banikazemi, Z.; Sahebkar, A. Photosomal curcumin: A review of pharmacokinetic, experimental and clinical studies. Biomed. Pharmacother. 2017, 85, 102–112. [Google Scholar] [CrossRef]
- Huang, R.; Han, J.; Wang, R.; Zhao, X.; Qiao, H.; Chen, L.; Li, W.; Di, L.; Zhang, W.; Li, J. Surfactant-free solid dispersion of BCS class IV drug in an amorphous chitosan oligosaccharide matrix for concomitant dissolution in vitro—Permeability increase. Eur. J. Pharm. Sci. 2019, 130, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Stoyanova, K.; Vinarov, Z.; Tcholakova, S. Improving Ibuprofen solubility by surfactant-facilitated self-assembly into mixed micelles. J. Drug Deliv. Sci. Technol. 2016, 36, 208–215. [Google Scholar] [CrossRef]
- Dun, J.; Osei-Yeboah, F.; Boulas, P.; Lin, Y.; Sun, C.C. A systemic evaluation of dual functionality of sodium lauryl sulfate as a tablet lubricant and wetting enhancer. Int. J. Pharm. 2018, 552, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Alizadeh, M.N.; Shayanfar, A.; Jouyban, A. Solubilization of drugs using sodium lauryl sulfate: Experimental data and modeling. J. Mol. Liq. 2018, 268, 410–414. [Google Scholar] [CrossRef]
- Rege, B.D.; Yu, L.X.; Hussain, A.S.; Polli, J.E. Effect of Common Excipients on Caco-2 Transport of Low-Permeability Drugs. J. Pharm. Sci. 2001, 90, 1776–1786. [Google Scholar] [CrossRef] [PubMed]
- Varma, M.V.S.; Panchagnula, R. Enhanced oral paclitaxel absorption with vitamin E-TPGS: Effect on solubility and permeability in vitro, in situ and in vivo. Eur. J. Pharm. Sci. 2005, 25, 445–453. [Google Scholar] [CrossRef] [PubMed]
- Dokania, S.; Joshi, A.K. Self-microemulsifying drug delivery system (SMEDDS)—Challenges and road ahead. Drug Deliv. 2015, 22, 675–690. [Google Scholar] [CrossRef] [Green Version]
- Joyce, P.; Dening, T.J.; Meola, T.R.; Schultz, H.B.; Holm, R.; Thomas, N.; Prestidge, C.A. Solidification to improve the biopharmaceutical performance of SEDDS: Opportunities and challenges. Adv. Drug Deliv. Rev. 2019, 142, 102–117. [Google Scholar] [CrossRef]
- Mahmood, A.; Bernkop-Schnürch, A. SEDDS: A game changing approach for the oral administration of hydrophilic macromolecular drugs. Adv. Drug Deliv. Rev. 2019, 142, 91–101. [Google Scholar] [CrossRef]
- Chaudhary, S.; Aqil, M.; Sultana, Y.; Kalam, M.A. Self-nanoemulsifying drug delivery system of nabumetone improved its oral bioavailability and anti-inflammatory effects in rat model. J. Drug Deliv. Sci. Technol. 2019, 51, 736–745. [Google Scholar] [CrossRef]
- Kang, B.K.; Lee, J.S.; Chon, S.K.; Jeong, S.Y.; Yuk, S.H.; Khang, G.; Lee, H.B.; Cho, S.H. Development of self-microemulsifying drug delivery systems (SMEDDS) for oral bioavailability enhancement of simvastatin in beagle dogs. Int. J. Pharm. 2004, 274, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Bowe, K.E. Recent advances in sugar-based excipients. Pharm. Sci. Technol. Today 1998, 1, 166–173. [Google Scholar] [CrossRef]
- Szűts, A.; Szabό-Révész, P. Sucrose esters as natural surfactants in drug delivery systems—A mini-review. Int. J. Pharm. 2012, 433, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Szűts, A.; Láng, P.; Ambrus, R.; Kiss, L.; Deli, M.A.; Szabό-Révész, P. Applicability of sucrose laurate as surfactant in solid dispersions prepared by melt technology. Int. J. Pharm. 2011, 410, 107–110. [Google Scholar] [CrossRef]
- Jaipal, A.; Pandey, M.M.; Charde, S.Y.; Raut, P.P.; Prasanth, K.V.; Prasad, R.G. Effect of HPMC and mannitol on drug release and bioadhesion behavior of buccal discs of buspirone hydrochloride: In-vitro and in-vivo pharmacokinetic studies. Saudi Pharm. J. 2015, 23, 315–326. [Google Scholar] [CrossRef] [Green Version]
- Nassab, P.R.; Rajkό, R.; Szabό-Révész, P. Physicochemical characterization of meloxicam-mannitol binary system. J. Pharm. Biomed. Anal. 2006, 41, 1191–1197. [Google Scholar] [CrossRef]
- Yadav, P.S.; Kumar, V.; Singh, U.P.; Bhat, H.R.; Mazumber, B. Physicochemical characterization and in vitro dissolution studies of solid dispersions of ketoprofen with PVP K30 and D-mannitol. Saudi Pharm. J. 2011, 21, 77–84. [Google Scholar] [CrossRef] [Green Version]
- Medina, M.R.; Kumar, V. Evaluation of cellulose II powders as a potential multifunctional excipient in tablet formulation. Int. J. Pharm. 2006, 322, 31–35. [Google Scholar] [CrossRef]
- Kundu, D.; Banerjee, T. Development of microcrystalline cellulose based hydrogels for the in vitro delivery of Cephalexin. Heliyon 2020, 6. [Google Scholar] [CrossRef] [Green Version]
- Sheng, F.; Chow, P.S.; Hu, J.; Cheng, S.; Guo, L.; Dong, Y. Preparation of quercetin nanorod/microcrystalline cellulose formulation via fluid bed coating crystallization for dissolution enhancement. Int. J. Pharm. 2020, 576, 118983. [Google Scholar] [CrossRef]
- Hebbink, G.A.; Dickhoff, B.H.J. Application of Lactose in the Pharmaceutical Industry; Chapter 5; DFE Pharma: Wageningen, The Netherlands, 2019; pp. 175–229. [Google Scholar]
- Li, C.; Li, C.X.; Le, Y.; Chen, J. Formation of bicalutamide nanodispersion for dissolution rate enhancement. Int. J. Pharm. 2011, 404, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Ganesh, M.; Ubaidulla, U.; Rathnam, G.; Jang, H.T. Chitosan-telmisartan polymeric cocrystals for improving oral absorption: In vitro and in vivo evaluation. Int. J. Biol. 2019, 131, 879–885. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; Sharma, R.; Jain, D.K.; Saraf, A. Enhancement of oral bioavailability of poorly water soluble carvedilol by chitosan nanoparticles: Optimization and pharmacokinetic study. Int. J. Biol. Macromol. 2019, 135, 246–260. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Excipient | Excipient Subdivision | Drug Example | In Vitro/In Vivo | Dosage Form Type | Reference |
|---|---|---|---|---|---|
| Cyclodextrin | β-Cyclodextrin | Eslicarbazepine | in vitro + in vivo | Orodispersable tablet (solid dispersion) | [21] |
| HP(2-Hydroxypropyl)-β-Cyclodextrin | Carbamazepine, Naproxen | in vitro + in vivo | Immediate release tablet/ Enteric coated tablet | [14,17] | |
| Disintegrants | Croscarmellose Sodium and Jackfruit starch | Irbesartan | in vitro + in vivo | Direct compressed fast disintegrating tablet | [24] |
| Sodium starch glycolate and crospovidone | Valsartan | in vitro + in vivo | Direct compressed fast disintegrating tablets | [25] | |
| pH adjusting excipients | Citric acid | Ketoconazole | in vitro + in vivo | Physical mixture (granules) | [30] |
| Tartaric acid | Ketoconazole | in vitro + in vivo | Physical mixture (granules) | [30] | |
| Sodium Hydrogen Carbonate | Paracetamol | in vitro + in vivo | Controlled release matrix tablet | [33] | |
| Calcium Carbonate | Paracetamol | in vitro + in vivo | Controlled release matrix tablet | [33] | |
| di-Sodium Carbonate | Aceclofenac | in vitro + in vivo | Controlled release matrix tablet | [33] | |
| di-Sodium Carbonate | Aceclofenac | in vitro + in vivo | Nanoemulsifying GUC (Gelucire 44/14)-based solid dispersions | [32] | |
| Solid dispersions | Tocopherol polyethyleneglycol-1000-succinate | Dutasteride | in vitro + in vivo | Physical mixture (solid dispersion) | [38] |
| Polyethylene glycol, polyvinyl acetate and polyvinylcaprolactame-based graft co-polymer | Nilotinib | in vitro | Encapsulated physical mixture (spray-dried mixture) | [36] | |
| Hydroxypropyl methylcellulose acetate succinate | Posaconazole | in vitro + in vivo | Delayed release tablet | [37] | |
| Chitosan | Curcumin | in vitro + in vivo | Amorphous solid dispersion | [41] | |
| Surfactant | Sodium lauryl sulphate | Celecoxib, Tramadol, Methocarbamol Diazepam, Alprazolam, Buspirone, Gabapentin and Acetaminophen | in vitro | Direct compressed tablet | [43] |
| Tween 20, Tween 40, Tween 60, Tween 80, sodium dodecyl sulphate and sodium lauryl ethoxy (3) sulphate | Ibuprofen | in vitro | Oral solution | [42] | |
| D-α-tocopherol polyethylene glycol 1000 succinate | Paclitaxel | in vitro + in vivo | Oral mixture | [46] | |
| D-α-tocopherol polyethylene glycol 1000 succinate | Dutasteride | in vitro + in vivo | Physical mixture (solid dispersion) | [38] | |
| SNEDDS | Capryol-90, Tween 80 and PEG-400 | Nabumetone | in vitro + in vivo | Oral SNEDDS | [50] |
| SMEDDS | capryol 90, lauroglycol 90, carbitol, PEG 400, polypropylene glycol and cremophor EL | Simvastatin | in vitro + in vivo | Oral SMEDDS | [51] |
| Mucoadhesive/ Mucopenetrating polymer | Chitosan | Telmisartan | in vitro + in vivo | Oral co-crystals | [63] |
| Chitosan | Carvedilol | in vitro + in vivo | Oral nanoparticles | [64] | |
| Sugars | Sucrose laurate | Gemfibrozil | in vitro | Oral solid dispersion | [54] |
| Mannitol | Ketoprofen | in vitro + in vivo | Oral co-crystals | [57] | |
| Mannitol | Meloxicam | in vitro + in vivo | Oral co-crystals | [56] | |
| Soluble and insoluble fillers | MCC | Quercetin | in vitro + in vivo | Oral co-crystals | [60] |
| Lactose | Bicalutamide | in vitro | Oral nanodispersion | [62] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
van der Merwe, J.; Steenekamp, J.; Steyn, D.; Hamman, J. The Role of Functional Excipients in Solid Oral Dosage Forms to Overcome Poor Drug Dissolution and Bioavailability. Pharmaceutics 2020, 12, 393. https://doi.org/10.3390/pharmaceutics12050393
van der Merwe J, Steenekamp J, Steyn D, Hamman J. The Role of Functional Excipients in Solid Oral Dosage Forms to Overcome Poor Drug Dissolution and Bioavailability. Pharmaceutics. 2020; 12(5):393. https://doi.org/10.3390/pharmaceutics12050393
Chicago/Turabian Stylevan der Merwe, Jannes, Jan Steenekamp, Dewald Steyn, and Josias Hamman. 2020. "The Role of Functional Excipients in Solid Oral Dosage Forms to Overcome Poor Drug Dissolution and Bioavailability" Pharmaceutics 12, no. 5: 393. https://doi.org/10.3390/pharmaceutics12050393