Chimeric Antigen Receptor-T-Cell Therapy for B-Cell Hematological Malignancies: An Update of the Pivotal Clinical Trial Data

 , , and
, , and
Abstract
:1. Introduction
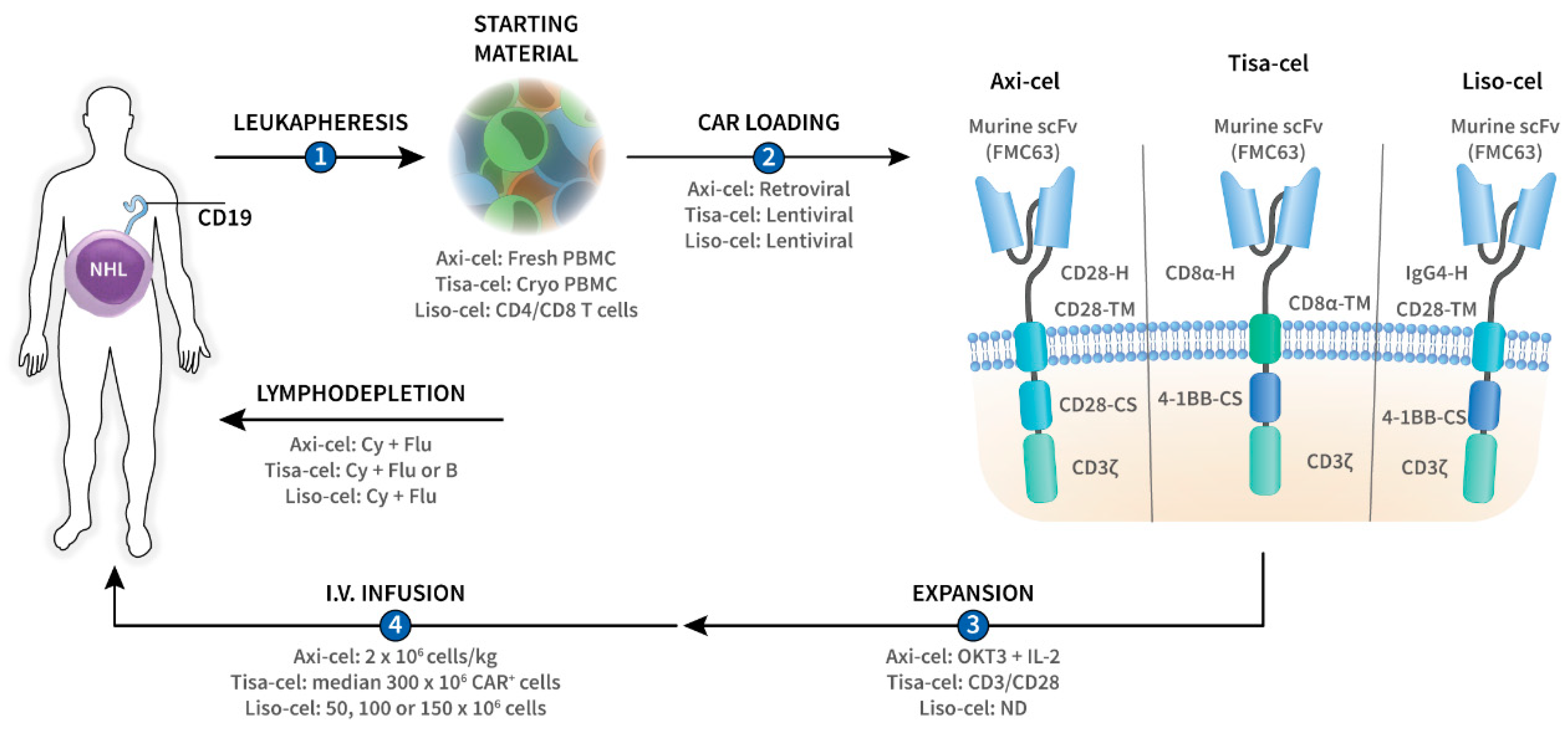
2. CAR-T-Cell Design
3. CAR-T-Cell Manufacturing and Administration
4. Efficacy and Toxicity of CAR-T-Cell Therapy in B-Cell Malignancies
4.1. Non-Hodgkin Lymphoma
4.2. B-Cell Acute Lymphoblastic Leukemia
4.3. Multiple Myeloma
4.4. Chronic Lymphocytic Leukemia
5. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- June, C.H.; Sadelain, M. Chimeric antigen receptor therapy. N. Engl. J. Med. 2018, 379, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Kolb, H.J. Donor leukocyte transfusions for treatment of leukemic relapse after bone marrow transplantation. EBMT Immunology and Chronic Leukemia Working Parties. Vox Sang. 1998, 74, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Liu, J.; Zhong, J.F.; Zhang, X. Engineering CAR-T cells. Biomark. Res. 2017, 5, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Stegen, S.J.; Hamieh, M.; Sadelain, M. The pharmacology of second-generation chimeric antigen receptors. Nat. Rev. Drug Discov. 2015, 14, 499–509. [Google Scholar] [CrossRef]
- Krause, A.; Guo, H.F.; Latouche, J.B.; Tan, C.; Cheung, N.K.; Sadelain, M. Antigen-dependent CD28 signaling selectively enhances survival and proliferation in genetically modified activated human primary T lymphocytes. J. Exp. Med. 1998, 188, 619–626. [Google Scholar] [CrossRef] [Green Version]
- Weinkove, R.; George, P.; Dasyam, N.; McLellan, A.D. Selecting costimulatory domains for chimeric antigen receptors: Functional and clinical considerations. Clin. Transl. Immunol. 2019, 8, e1049. [Google Scholar] [CrossRef] [Green Version]
- Long, A.H.; Haso, W.M.; Shern, J.F.; Wanhainen, K.M.; Murgai, M.; Ingaramo, M.; Smith, J.P.; Walker, A.J.; Kohler, M.E.; Venkateshwara, V.R.; et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat. Med. 2015, 21, 581–590. [Google Scholar] [CrossRef] [Green Version]
- Brentjens, R.J.; Curran, K.J. Novel cellular therapies for leukemia: CAR-modified T cells targeted to the CD19 antigen. Hematol. Am. Soc. Hematol. Educ. Program 2012, 2012, 143–151. [Google Scholar] [CrossRef] [Green Version]
- Ajina, A.; Maher, J. Strategies to address chimeric antigen receptor tonic signalling. Mol. Cancer Ther. 2018, 17, 1795–1815. [Google Scholar] [CrossRef] [Green Version]
- Maus, M.V.; June, C.H. Making better chimeric antigen receptors for adoptive T-cell therapy. Clin. Cancer Res. 2016, 22, 1875–1884. [Google Scholar] [CrossRef] [Green Version]
- Thieblemont, C.; Le Gouill, S.; Di Blasi, R.; Cartron, G.; Morschhauser, F.; Bachy, E.; Paillassa, J.; Bernard, S.; Tessoulin, B.; Gastine, T.; et al. Real-world results on CD19 CAR T-cell for 60 French patients with relapsed/refractory diffuse large B-cell lymphoma included in a temporary authorization for use program. Hematol. Oncol. 2019, 37, 301. [Google Scholar] [CrossRef]
- Klebanoff, C.A.; Khong, H.T.; Antony, P.A.; Palmer, D.C.; Restifo, N.P. Sinks, suppressors and antigen presenters: How lymphodepletion enhances T cell mediated tumor immunotherapy. Trends Immunol. 2005, 26, 111–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coiffier, B.; Lepage, E.; Briere, J.; Herbrecht, R.; Tilly, H.; Bouabdallah, R.; Morel, P.; Van Den Neste, E.; Salles, G.; Gaulard, P.; et al. CHOP chemotherapy plus rituximab compared with CHOP alone in elderly patients with diffuse large-B-cell lymphoma. N. Engl. J. Med. 2002, 346, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Crump, M.; Neelapu, S.S.; Farooq, U.; Van Den Neste, E.; Kuruvilla, J.; Westin, J.; Link, B.K.; Hay, A.; Cerhan, J.R.; Zhu, L.; et al. Outcomes in refractory diffuse large B-cell lymphoma: Results from the international SCHOLAR-1 study. Blood 2017, 130, 1800–1808. [Google Scholar] [CrossRef]
- Kochenderfer, J.N.; Dudley, M.E.; Kassim, S.H.; Somerville, R.P.; Carpenter, R.O.; Stetler-Stevenson, M.; Yang, J.C.; Phan, G.Q.; Hughes, M.S.; Sherry, R.M.; et al. Chemotherapy-refractory diffuse large B-cell lymphoma and indolent B-cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor. J. Clin. Oncol. 2015, 33, 540–549. [Google Scholar] [CrossRef] [Green Version]
- Kochenderfer, J.N.; Somerville, R.P.; Lu, T.; Shi, V.; Bot, A.; Rossi, J.; Xue, A.; Goff, S.L.; Yang, J.C.; Sherry, R.M.; et al. Lymphoma remissions caused by anti-CD19 chimeric antigen receptor T cells are associated with high serum interleukin-15 levels. J. Clin. Oncol. 2017, 35, 1803–1813. [Google Scholar] [CrossRef]
- Locke, F.L.; Ghobadi, A.; Jacobson, C.A.; Miklos, D.B.; Lekakis, L.J.; Oluwole, O.O.; Lin, Y.; Braunschweig, I.; Hill, B.T.; Timmerman, J.M.; et al. Long-term safety and activity of axicabtagene-ciloleucel in refractory large B-cell lymphoma (ZUMA-1): A single-arm, multicentre, phase 1–2 trial. Lancet Oncol. 2019, 20, 31–42. [Google Scholar] [CrossRef]
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544. [Google Scholar] [CrossRef]
- Schuster, S.J.; Bishop, M.R.; Tam, C.S.; Waller, E.K.; Borchmann, P.; McGuirk, J.P.; Jäger, U.; Jaglowski, S.; Andreadis, C.; Westin, J.R.; et al. Tisagenlecleucel in adult relapsed or refractory diffuse large B-cell lymphoma. N. Engl. J. Med. 2019, 380, 45–56. [Google Scholar] [CrossRef]
- Schuster, S.J.; Bishop, M.R.; Tam, C.; Waller, E.K.; Borchmann, P.; McGuirk, J.; Jäger, U.; Jaglowski, S.; Andreadis, C.; Westin, J.; et al. Global pivotal phase 2 trial of the CD19-targeted therapy CTL019 in adult patients with relapsed or refractory (R/R) diffuse large B-cell lymphoma (DLBCL)—An interim analysis. Hematol. Oncol. 2017, 35, 27. [Google Scholar] [CrossRef] [Green Version]
- Abramson, J.S.; Palomba, M.L.; Gordon, L.I.; Lunning, M.A.; Wang, M.L.; Arnason, J.E.; Mehta, A.; Purev, E.; Maloney, D.G.; Andreadis, C.; et al. Pivotal safety and efficacy results from Transcend NHL 001, a multicenter phase 1 study of lisocabtagene maraleucel (liso-cel) in relapsed/refractory (R/R) large B cell lymphomas. Blood 2019, 134, 241. [Google Scholar] [CrossRef]
- Abramson, J.S.; Palomba, M.L.; Gordon, L.I.; Lunning, M.A.; Arnason, J.E.; Forero-Torres, A.; Wang, M.; Albertson, T.M.; Allen, T.; Sutherland, C.; et al. CR rates in relapsed/refractory (R/R) aggressive B-NHL treated with the CD19-directed CAR T-cell product JCAR017 (TRANSCEND NHL001). J. Clin. Oncol. 2017, 35, 7513. [Google Scholar] [CrossRef]
- Abramson, J.; Palomba, M.L.; Gordon, L.; Lunning, M.; Arnason, J.; Wang, M.; Forero-Torres, A.; Albertson, T.; Allen, T.; Sutherland, C.; et al. High CR rates in relapsed/refractory (R/R) aggressive B-NHL treated with the CD19-directed CAR T cell product JCAR017 (TRANSCEND NHL 001). Hematol. Oncol. 2017, 35, 138. [Google Scholar] [CrossRef] [Green Version]
- Nastoupil, L.J.; Jain, M.D.; Spiegel, J.Y.; Ghobadi, A.; Lin, Y.; Dahiya, S.; Lunning, M.A.; Lekakis, L.J.; Reagan, P.M.; Oluwole, O.O.; et al. Axicabtagene ciloleucel (axi-cel) CD19 chimeric antigen receptor (CAR) T-cell therapy for relapsed/refractory large B-cell lymphoma: Real world experience. Blood 2018, 132, 91. [Google Scholar] [CrossRef]
- Jacobson, C.A.; Hunter, B.; Armand, P.; Kamihara, Y.; Ritz, J.; Rodig, S.J.; Wright, K.; Lipschitz, M.; Redd, R.A.; Maus, M.V.; et al. Axicabtagene ciloleucel in the real world: Outcomes and predictors of response, resistance and toxicity. Blood 2018, 132, 92. [Google Scholar] [CrossRef]
- Elsallab, M.; Levine, B.L.; Wayne, A.S.; Abou-El-Enein, M. CAR T-cell product performance in haematological malignancies before and after marketing authorisation. Lancet Oncol. 2020, 21, e104–e116. [Google Scholar] [CrossRef]
- Neelapu, S.S.; Tummala, S.; Kebriaei, P.; Wierda, W.; Gutierrez, C.; Locke, F.L.; Komanduri, K.V.; Lin, Y.; Jain, N.; Daver, N.; et al. Chimeric antigen receptor T-cell therapy - assessment and management of toxicities. Nat. Rev. Clin. Oncol. 2018, 15, 47–62. [Google Scholar] [CrossRef]
- Lee, D.; Santomasso, B.; Locke, F.; Ghobadi, A.; Turtle, C.J.; Brudno, J.N.; Maus, M.V.; Park, J.H.; Mead, E.; Pavletic, S.; et al. ASTCT consensus grading for cytokine release syndrome and neurologic toxicity associated with immune effector cells. Biol. Blood Marrow Transplant. 2019, 25, 625–638. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.W.; Gardner, R.; Porter, D.L.; Louis, C.U.; Ahmed, N.; Jensen, M.; Grupp, S.A.; Mackall, C.L. Current concepts in the diagnosis and management of cytokine release syndrome. Blood 2014, 124, 188–195. [Google Scholar] [CrossRef] [Green Version]
- Norelli, M.; Camisa, B.; Barbiera, G.; Falcone, L.; Purevdorj, A.; Genua, M.; Sanvito, F.; Ponzoni, M.; Doglioni, C.; Cristofori, P.; et al. Monocyte-derived IL-1 and IL-6 are differentially required for cytokine-release syndrome and neurotoxicity due to CAR T cells. Nat. Med. 2018, 24, 739–748. [Google Scholar] [CrossRef]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Danhof, S.; Hudecek, M.; Smith, E.L. CARs and other T cell therapies for MM: The clinical experience. Best Pract. Res. Clin. Haematol. 2018, 31, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.F.; Anderson, K.C.; Tai, Y.T. Targeting B cell maturation antigen (BCMA) in multiple myeloma: Potential uses of BCMA-based immunotherapy. Front. Immunol. 2018, 9, 1821. [Google Scholar] [CrossRef] [PubMed]
- Cohen, A.D. CAR T cells and other cellular therapies for multiple myeloma: 2018 update. Am. Soc. Clin. Oncol. Educ. Book 2018, 38, e6–e15. [Google Scholar] [CrossRef]
- Novak, A.J.; Darce, J.R.; Arendt, B.K.; Harder, B.; Henderson, K.; Kindsvogel, W.; Gross, J.A.; Greipp, P.R.; Jelinek, D.F. Expression of BCMA, TACI, and BAFF-R in multiple myeloma: A mechanism for growth and survival. Blood 2004, 103, 689–694. [Google Scholar] [CrossRef] [Green Version]
- Seckinger, A.; Delgado, J.A.; Moser, S.; Moreno, L.; Neuber, B.; Grab, A.; Lipp, S.; Merino, J.; Prosper, F.; Emde, M.; et al. Target expression, generation, preclinical activity, and pharmacokinetics of the BCMA-T cell bispecific antibody EM801 for multiple myeloma treatment. Cancer Cell 2017, 31, 396–410. [Google Scholar] [CrossRef] [Green Version]
- Tai, Y.T.; Acharya, C.; An, G.; Moschetta, M.; Zhong, M.Y.; Feng, X.; Cea, M.; Cagnetta, A.; Wen, K.; van Eenennaam, H.; et al. APRIL and BCMA promote human multiple myeloma growth and immunosuppression in the bone marrow microenvironment. Blood 2016, 127, 3225–3236. [Google Scholar] [CrossRef] [Green Version]
- Cohen, A.D.; Garfall, A.L.; Stadtmauer, E.A.; Melenhorst, J.J.; Lacey, S.F.; Lancaster, E.; Vogl, D.T.; Weiss, B.M.; Dengel, K.; Nelson, A.; et al. B cell maturation antigen-specific CAR T cells are clinically active in multiple myeloma. J. Clin. Investig. 2019, 130, 2210–2221. [Google Scholar] [CrossRef] [Green Version]
- Ali, S.A.; Shi, V.; Maric, I.; Wang, M.; Stroncek, D.F.; Rose, J.J.; Brudno, J.N.; Stetler-Stevenson, M.; Feldman, S.A.; Hansen, B.G.; et al. T cells expressing an anti-B-cell maturation antigen chimeric antigen receptor cause remissions of multiple myeloma. Blood 2016, 128, 1688–1700. [Google Scholar] [CrossRef]
- Brudno, J.N.; Maric, I.; Hartman, S.D.; Rose, J.J.; Wang, M.; Lam, N.; Stetler-Stevenson, M.; Salem, D.; Yuan, C.; Pavletic, S.; et al. T cells genetically modified to express an anti-B-cell maturation antigen chimeric antigen receptor cause remissions of poor-prognosis relapsed multiple myeloma. J. Clin. Oncol. 2018, 36, 2267–2280. [Google Scholar] [CrossRef]
- Raje, N.; Berdeja, J.; Lin, Y.; Siegel, D.; Jagannath, S.; Madduri, D.; Liedtke, M.; Rosenblatt, J.; Maus, M.V.; Turka, A.; et al. Anti-BCMA CAR T-cell therapy bb2121 in relapsed or refractory multiple myeloma. N. Engl. J. Med. 2019, 380, 1726–1737. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Chen, L.J.; Yang, S.S.; Sun, Y.; Wu, W.; Liu, Y.F.; Xu, J.; Zhuang, Y.; Zhang, W.; Weng, X.Q.; et al. Exploratory trial of a biepitopic CAR T-targeting B cell maturation antigen in relapsed/refractory multiple myeloma. Proc. Natl. Acad. Sci. USA 2019, 116, 9543–9551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, W.H.; Liu, J.; Wang, B.Y.; Chen, Y.X.; Cao, X.M.; Yang, Y.; Zhang, Y.L.; Wang, F.X.; Zhang, P.Y.; Lei, B.; et al. A phase 1, open-label study of LCAR-B38M, a chimeric antigen receptor T cell therapy directed against B cell maturation antigen, in patients with relapsed or refractory multiple myeloma. J. Hematol. Oncol. 2018, 11, 141. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; Cao, J.; Cheng, H.; Qiao, J.; Zhang, H.; Wang, Y.; Shi, M.; Lan, J.; Fei, X.; Jin, L.; et al. A combination of humanised anti-CD19 and anti-BCMA CAR T cells in patients with relapsed or refractory multiple myeloma: A single-arm, phase 2 trial. Lancet Haematol. 2019, 6, e521–e529. [Google Scholar] [CrossRef]
- Ma, T.; Shi, J.; Liu, H. Chimeric antigen receptor T cell targeting B cell maturation antigen immunotherapy is promising for multiple myeloma. Ann. Hematol. 2019, 98, 813–822. [Google Scholar] [CrossRef] [Green Version]
- Guo, B.; Chen, M.; Han, Q.; Hui, F.; Dai, H.; Zhang, W.; Zhang, Y.; Wang, Y.; Zhu, H.; Han, W.; et al. CD138-directed adoptive immunotherapy of chimeric antigen receptor (CAR)-modified T cells for multiple myeloma. J. Cell. Immunother. 2016, 2, 28–35. [Google Scholar] [CrossRef] [Green Version]
- Garfall, A.L.; Stadtmauer, E.A.; Hwang, W.T.; Lacey, S.F.; Melenhorst, J.J.; Krevvata, M.; Carroll, M.P.; Matsui, W.H.; Wang, Q.; Dhodapkar, M.V.; et al. Anti-CD19 CAR T cells with high-dose melphalan and autologous stem cell transplantation for refractory multiple myeloma. JCI Insight 2018, 3, e120505. [Google Scholar] [CrossRef]
- Nerreter, T.; Letschert, S.; Götz, R.; Doose, S.; Danhof, S.; Einsele, H.; Sauer, M.; Hudecek, M. Super-resolution microscopy reveals ultra-low CD19 expression on myeloma cells that triggers elimination by CD19 CAR-T. Nat. Commun. 2019, 10, 3137. [Google Scholar] [CrossRef]
- Boucher, K.; Parquet, N.; Widen, R.; Shain, K.; Baz, R.; Alsina, M.; Koomen, J.; Anasetti, C.; Dalton, W.; Perez, L.E. Stemness of B cell progenitors in multiple myeloma bone marrow. Clin. Cancer Res. 2012, 18, 6155–6168. [Google Scholar] [CrossRef] [Green Version]
- Pont, M.; Hill, T.; Cole, G.; Abbott, J.J.; Kelliher, J.; Salter, A.I.; Hudecek, M.; Comstock, M.L.; Rajan, A.; Patel, B.K.; et al. γ-secretase inhibition increases efficacy of BCMA-specific chimeric antigen receptor T cells in multiple myeloma. Blood 2019, 134, 1585–1597. [Google Scholar] [CrossRef]
- Timmers, M.; Roex, G.; Wang, Y.; Campillo-Davo, D.; Van Tendeloo, V.F.; Chu, Y.; Berneman, Z.N.; Luo, F.; Van Acker, H.H.; Anguille, S. Chimeric antigen receptor-modified T cell therapy in multiple myeloma: Beyond B cell maturation antigen. Front. Immunol. 2019, 10, 1613. [Google Scholar] [CrossRef] [PubMed]
- Porter, D.L.; Levine, B.L.; Kalos, M.; Bagg, A.; June, C.H. Chimeric antigen receptor modified T cells in chronic lymphoid leukemia. N. Engl. J. Med. 2011, 365, 725–733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemal, R.; Tournilhac, O. State-of-the-art for CAR T-cell therapy for chronic lymphocytic leukemia in 2019. J. Immunother. Cancer 2019, 7, 202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porter, D.L.; Hwang, W.T.; Frey, N.V.; Lacey, S.F.; Shaw, P.A.; Loren, A.W.; Bagg, A.; Marcucci, K.T.; Shen, A.; Gonzalez, V.; et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci. Transl. Med. 2015, 7, 303ra139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turtle, C.J.; Hay, K.A.; Hanafi, L.A.; Li, D.; Cherian, S.; Chen, X.; Wood, B.; Lozanski, A.; Byrd, J.C.; Heimfeld, S.; et al. Durable molecular remissions in chronic lymphocytic leukemia treated with CD19-specific chimeric antigen receptor-modified T cells after failure of ibrutinib. J. Clin. Oncol. 2017, 35, 3010–3020. [Google Scholar] [CrossRef] [PubMed]
- Fraietta, J.A.; Lacey, S.F.; Orlando, E.J.; Pruteanu-Malinici, I.; Gohil, M.; Lundh, S.; Boesteanu, A.C.; Wang, Y.; O’Connor, R.S.; Hwang, W.T.; et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat. Med. 2018, 24, 563–571. [Google Scholar] [CrossRef]
- Gauthier, J.; Hirayama, A.V.; Hay, K.A.; Li, D.; Lymp, J.; Sheih, A.; Purushe, J.; Pender, B.S.; Hawkins, R.M.; Vakil, A.; et al. Comparison of efficacy and toxicity of CD19-specific chimeric antigen receptor T-cells alone or in combination with ibrutinib for relapsed and/or refractory CLL. Blood 2018, 132, 299. [Google Scholar] [CrossRef]
- Gill, S.I.; Vides, V.; Frey, N.V.; Metzger, S.; O’Brien, M.; Hexner, E.; Mato, A.R.; Lacey, S.F.; Melenhorst, J.J.; Pequignot, E.; et al. Prospective clinical trial of anti-CD19 CAR T cells in combination with ibrutinib for the treatment of chronic lymphocytic leukemia shows a high response rate. Blood 2018, 132, 298. [Google Scholar] [CrossRef]
- Riches, J.C.; Davies, J.K.; McClanahan, F.; Fatah, R.; Iqbal, S.; Agrawal, S.; Ramsay, A.G.; Gribben, J.G. T cells from CLL patients exhibit features of T-cell exhaustion but retain capacity for cytokine production. Blood 2013, 121, 1612–1621. [Google Scholar] [CrossRef]
- Quintás-Cardama, A. What CAR will win the CD19 race? Mol. Cancer Ther. 2019, 18, 498–506. [Google Scholar] [CrossRef] [Green Version]
- Neelapu, S.S. Managing the toxicities of CAR T-cell therapy. Hematol. Oncol. 2019, 37 (Suppl. S1), 48–52. [Google Scholar] [CrossRef] [Green Version]
- Qin, J.S.; Johnstone, T.G.; Baturevych, A.; Hause, R.J.; Ragan, S.P.; Clouser, C.R.; Jones, J.C.; Ponce, R.; Krejsa, C.M.; Salmon, R.A.; et al. Antitumor potency of an anti-CD19 chimeric antigen receptor T-cell therapy, lisocabtagene maraleucel in combination with ibrutinib or acalabrutinib. J. Immunother. 2020. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Sun, Q.; Liang, X.; Chen, Z.; Zhang, X.; Zhou, X.; Li, M.; Tu, H.; Liu, Y.; Tu, S.; et al. Mechanisms of relapse after CD19 CAR T-cell therapy for acute lymphoblastic leukemia and its prevention and treatment strategies. Front. Immunol. 2019, 10, 2664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gardner, R.; Wu, D.; Cherian, S.; Fang, M.; Hanafi, L.-A.; Finney, O.; Smithers, H.; Jensen, M.C.; Riddell, S.R.; Maloney, D.G.; et al. Acquisition of a CD19-negative myeloid phenotype allows immune escape of MLL-rearranged B-ALL from CD19 CAR-T-cell therapy. Blood 2016, 127, 2406–2410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fry, T.J.; Shah, N.N.; Orentas, R.J.; Stetler-Stevenson, M.; Yuan, C.M.; Ramakrishna, S.; Wolters, P.; Martin, S.; Delbrook, C.; Yates, B.; et al. CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat. Med. 2018, 24, 20–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, N.; Maatman, T.; Hari, P.; Johnson, B. Multi targeted CAR-T cell therapies for B-cell malignancies. Front. Oncol. 2019, 9, 146. [Google Scholar] [CrossRef] [Green Version]
- Hill, L.; Lulla, P.; Heslop, H.E. CAR-T cell therapy for non-Hodgkin lymphomas: A new treatment paradigm. Adv. Cell Gene Ther. 2019, 2, e54. [Google Scholar] [CrossRef]
- Jacoby, E.; Shahani, S.A.; Shah, N.N. Updates on CAR T-cell therapy in B-cell malignancies. Immunol. Rev. 2019, 290, 39–59. [Google Scholar] [CrossRef]
- Smith, E.L.; Harrington, K.; Staehr, M.; Masakayan, R.; Jones, J.; Long, T.J.; Ng, K.Y.; Ghoddusi, M.; Purdon, T.J.; Wang, X.; et al. GPRC5D is a target for the immunotherapy of multiple myeloma with rationally designed CAR T cells. Sci. Transl. Med. 2019, 11, eaau7746. [Google Scholar] [CrossRef]
- Ghorashian, S.; Kramer, A.M.; Onuoha, S.; Wright, G.; Bartram, J.; Richardson, R.; Albon, S.J.; Casanovas-Company, J.; Castro, F.; Popova, B.; et al. Enhanced CAR T cell expansion and prolonged persistence in pediatric patients with ALL treated with a low-affinity CD19 CAR. Nat. Med. 2019, 25, 1408–1414. [Google Scholar] [CrossRef]
- Lynn, R.C.; Weber, E.W.; Sotillo, E.; Gennert, D.; Xu, P.; Good, Z.; Anbunathan, H.; Lattin, J.; Jones, R.; Tieu, V.; et al. c-Jun overexpression in CAR T cells induces exhaustion resistance. Nat. 2019, 576, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Ying, Z.; He, T.; Wang, X.; Zheng, W.; Lin, N.; Tu, M.; Xie, Y.; Ping, L.; Zhang, C.; Liu, W.; et al. Parallel comparison of 4-1BB or CD28 co-stimulated CD19-targeted CAR-T cells for B cell non-Hodgkin’s lymphoma. Mol. Ther. Oncolytics 2019, 15, 60–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majzner, R.G.; Rietberg, S.P.; Labanieh, L.; Sotillo, E.; Weber, E.W.; Lynn, R.C.; Theruvath, J.L.; Yuan, C.M.; Xu, P.; Nguyen, S.M.; et al. Low CD19 antigen density diminishes efficacy of CD19 CAR T cells and can be overcome by rational redesign of CAR signaling domains. Blood 2018, 132, 963. [Google Scholar] [CrossRef]
- Finney, O.C.; Brakke, H.M.; Rawlings-Rhea, S.; Hicks, R.; Doolittle, D.; Lopez, M.; Futrell, R.B.; Orentas, R.J.; Li, D.; Gardner, R.A.; et al. CD19 CAR T cell product and disease attributes predict leukemia remission durability. J. Clin. Investig. 2019, 129, 2123–2132. [Google Scholar] [CrossRef]
- Ardeshna, K.M.; Marzolini, M.A.V.; Norman, J.; Al-Hajj, M.; Thomas, S.; Faulkner, J.; Kotsopoulou, E.; Pule, M.; Peddareddigari, V.G.R.; Khokhar, N.Z.; et al. Phase 1/2 study of AUTO3 the first bicistronic chimeric antigen receptor (CAR) targeting CD19 and CD22 followed by an anti-PD1 in patients with relapsed/refractory (r/r) diffuse large B cell lymphoma (DLBCL): Results of cohort 1 and 2 of the Alexander study. Blood 2019, 134, 246. [Google Scholar] [CrossRef]
- Hill, B.T.; Roberts, Z.J.; Xue, A.; Rossi, J.M.; Smith, M.R. Rapid tumor regression from PD-1 inhibition after anti-CD19 chimeric antigen receptor T-cell therapy in refractory diffuse large B-cell lymphoma. Bone Marrow Transplant. 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, Y.; Lu, W.; Sun, R.; Jin, X.; Cheng, L.; He, X.; Wang, L.; Yuan, T.; Lyu, C.; Zhao, M. Anti-CD19 chimeric antigen receptor T cells in combination with nivolumab are safe and effective against relapsed/refractory B-cell non-hodgkin lymphoma. Front. Oncol. 2019, 9, 767. [Google Scholar] [CrossRef] [Green Version]
- Rafiq, S.; Yeku, O.O.; Jackson, H.J.; Purdon, T.J.; van Leeuwen, D.G.; Drakes, D.J.; Song, M.; Miele, M.M.; Li, Z.; Wang, P.; et al. Targeted delivery of a PD-1-blocking scFv by CAR-T cells enhances anti-tumor efficacy in vivo. Nat. Biotechnol. 2018, 36, 847–856. [Google Scholar] [CrossRef]
- Gardner, R.A.; Ceppi, F.; Rivers, J.; Annesley, C.; Summers, C.; Taraseviciute, A.; Gust, J.; Leger, K.J.; Tarlock, K.; Cooper, T.M.; et al. Preemptive mitigation of CD19 CAR T-cell cytokine release syndrome without attenuation of antileukemic efficacy. Blood 2019, 134, 2149–2158. [Google Scholar] [CrossRef]
- Bonifant, C.L.; Jackson, H.J.; Brentjens, R.J.; Curran, K.J. Toxicity and management in CAR T-cell therapy. Mol. Ther. Oncolytics 2016, 3, 16011. [Google Scholar] [CrossRef]
- Mestermann, K.; Giavridis, T.; Weber, J.; Rydzek, J.; Frenz, S.; Nerreter, T.; Mades, A.; Sadelain, M.; Einsele, H.; Hudecek, M. The tyrosine kinase inhibitor dasatinib acts as a pharmacologic on/off switch for CAR T cells. Sci. Transl. Med. 2019, 11, eaau5907. [Google Scholar] [CrossRef] [PubMed]
- Caruso, H.G.; Heimberger, A.B.; Cooper, L.J.N. Steering CAR T cells to distinguish friend from foe. Oncoimmunology 2019, 8, e1271857. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Cell Product | axi-cel | tisa-cel | liso-cel |
|---|---|---|---|
| Trial [ref.] | ZUMA-1 [17,18] | JULIET [19,20] | TRANSCEND [21,22,23] |
| N enrolled (infused) | 111 (101) | 165 (111) | 344 (269 + 25 #) |
| N response-evaluable | 101 | 93 | 256 |
| Best ORR (CR) | 83% (58%) | 52% (40%) | 73% (53%) |
| Median DoR | 11.1 mo (4.2 mo-n.e.) | Not reached (10.0 mo-n.e.) | Not reached (8.6 mo-n.e.) |
| Median PFS | 5.9 mo (3.3–15.0 mo) | NR | 6.8 mo (3.3–14.1 mo) |
| PFS rate | 24-mo PFS: 72% (for pts in CR at 3 mo) | 12-mo PFS: 83% (for pts in CR/PR at 3 mo) | 12-mo PFS: 65% (for pts in CR) |
| Median OS | Not reached (12.8 mo-n.e.) | 12.0 mo § (7.0 mo-n.e.) | 21.1 mo (13.3 mo-n.e.) |
| OS rate | Est. 24-mo OS: 50.5% | Est. 12-mo OS: 49% (90% for pts in CR) | Est. 12-mo OS: 58% (86% for pts in CR) |
| Cell Product | axi-cel | tisa-cel | liso-cel |
|---|---|---|---|
| Trial [ref.] | ZUMA-1 [17,18] | JULIET [19,20] | TRANSCEND [21,22,23] |
| CRS (gr. ≥ 3) | 92% (11%) | 58% (22%) | 42% (2%) |
| NT (gr. ≥ 3) | 67% (32%) | 21% (12%) | 30% (10%) |
| Tocilizumab use | 43% | 14% | 19% |
| Corticosteroid use | 27% | 10% | 21% |
| Trial Registry # [ref.] | N | ORR (CR) | Median PFS (95% CI) | CRS (gr. ≥ 3) | NT (gr. ≥ 3) |
|---|---|---|---|---|---|
| NCT02546167 [38] | 25 | 48% (8%) | NR | 88% (32%) | 32% (12%) |
| NCT02215967 [39,40] | 26 | 58% (8%) | NR | 69% (23%) | 4% (4%) |
| NCT02658929 [41] | 33 | 85% (45%) | 11.8 mo (6.2–17.8 mo) | 76% (6%) | 42% (3%) |
| NCT03090659 [42] | 17 | 88% (82%) | 12.2 mo (NR) | 100% (41%) | 0% (0%) |
| NCT03090659 [43] | 57 | 88% (74%) | 15.0 mo (11.0 mo-n.e.) | 89% (7%) | 2% (0%) |
| ChiCTR-17011272 [44] | 21 | 95% (57%) | 8.0 mo ¶ (NR) | 91% (5%) | 10% (NR) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roex, G.; Feys, T.; Beguin, Y.; Kerre, T.; Poiré, X.; Lewalle, P.; Vandenberghe, P.; Bron, D.; Anguille, S. Chimeric Antigen Receptor-T-Cell Therapy for B-Cell Hematological Malignancies: An Update of the Pivotal Clinical Trial Data. Pharmaceutics 2020, 12, 194. https://doi.org/10.3390/pharmaceutics12020194
Roex G, Feys T, Beguin Y, Kerre T, Poiré X, Lewalle P, Vandenberghe P, Bron D, Anguille S. Chimeric Antigen Receptor-T-Cell Therapy for B-Cell Hematological Malignancies: An Update of the Pivotal Clinical Trial Data. Pharmaceutics. 2020; 12(2):194. https://doi.org/10.3390/pharmaceutics12020194
Chicago/Turabian StyleRoex, Gils, Tom Feys, Yves Beguin, Tessa Kerre, Xavier Poiré, Philippe Lewalle, Peter Vandenberghe, Dominique Bron, and Sébastien Anguille. 2020. "Chimeric Antigen Receptor-T-Cell Therapy for B-Cell Hematological Malignancies: An Update of the Pivotal Clinical Trial Data" Pharmaceutics 12, no. 2: 194. https://doi.org/10.3390/pharmaceutics12020194