Development of a Biodegradable Subcutaneous Implant for Prolonged Drug Delivery Using 3D Printing

 ,
,  ,
,  ,
,  and
and 
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
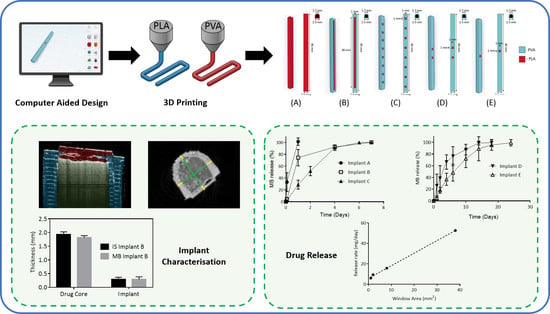
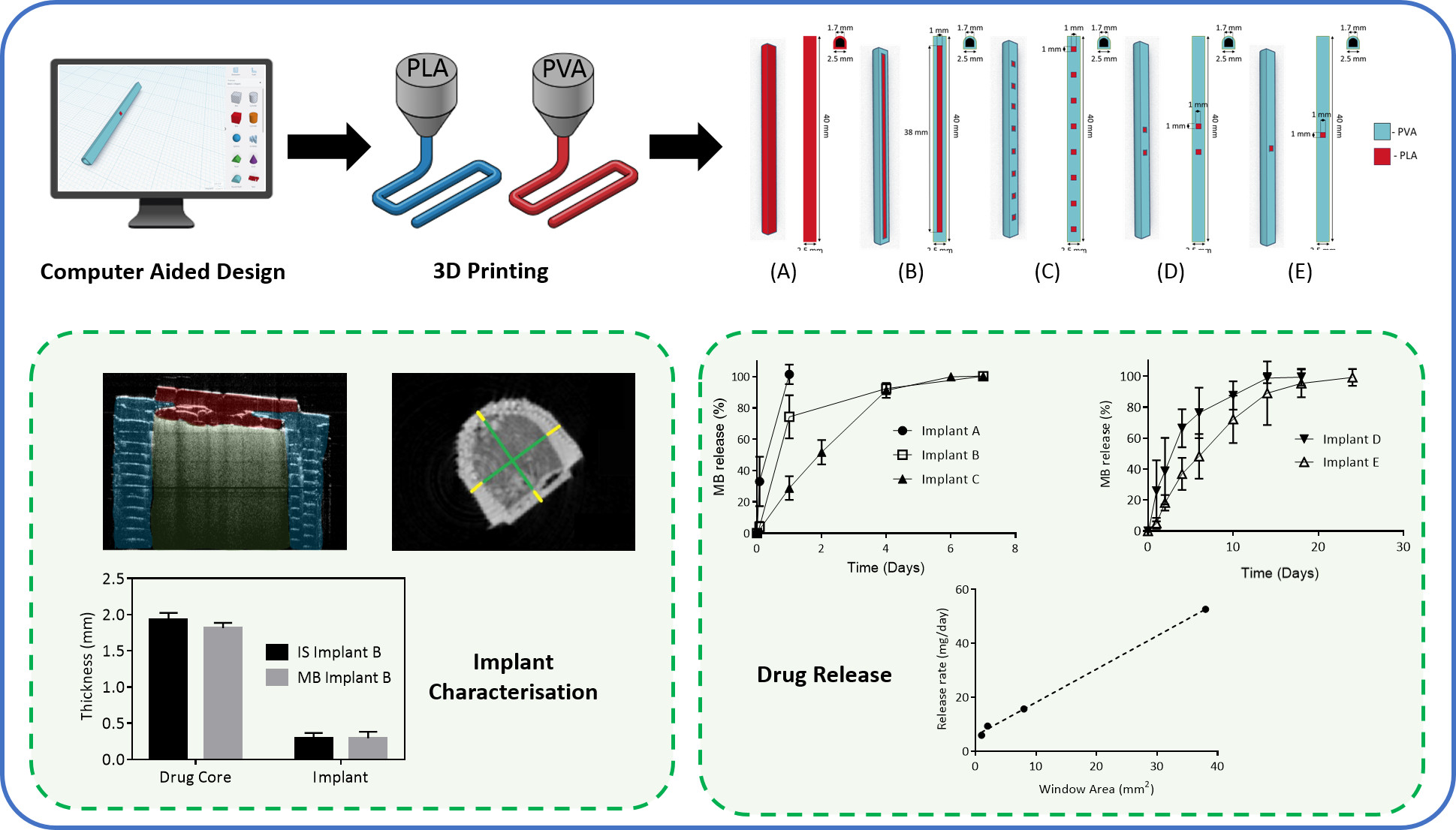
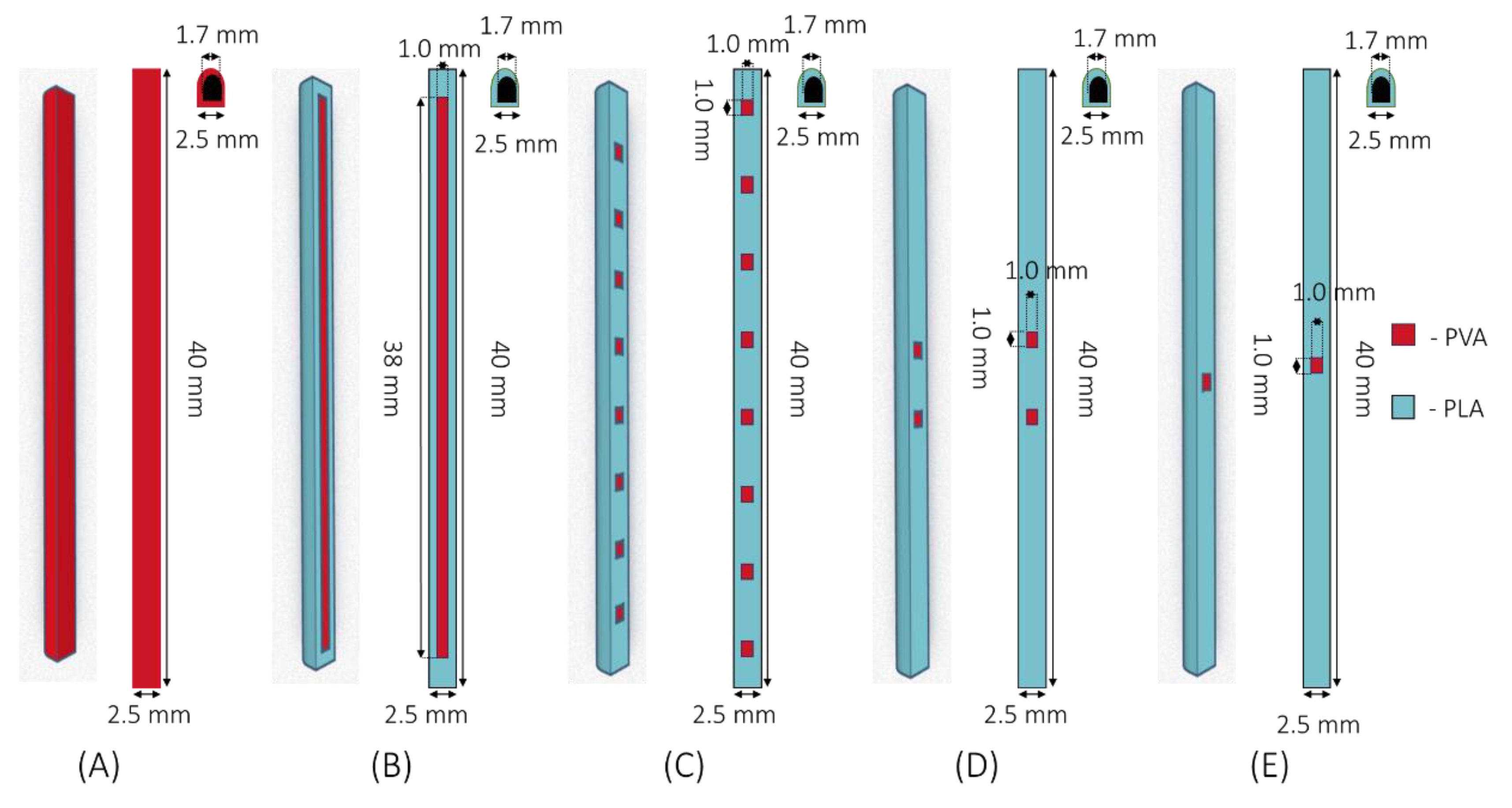
2.2.1. Implant Designs
2.2.2. Implant Characterisation
2.2.3. Analytical Methods
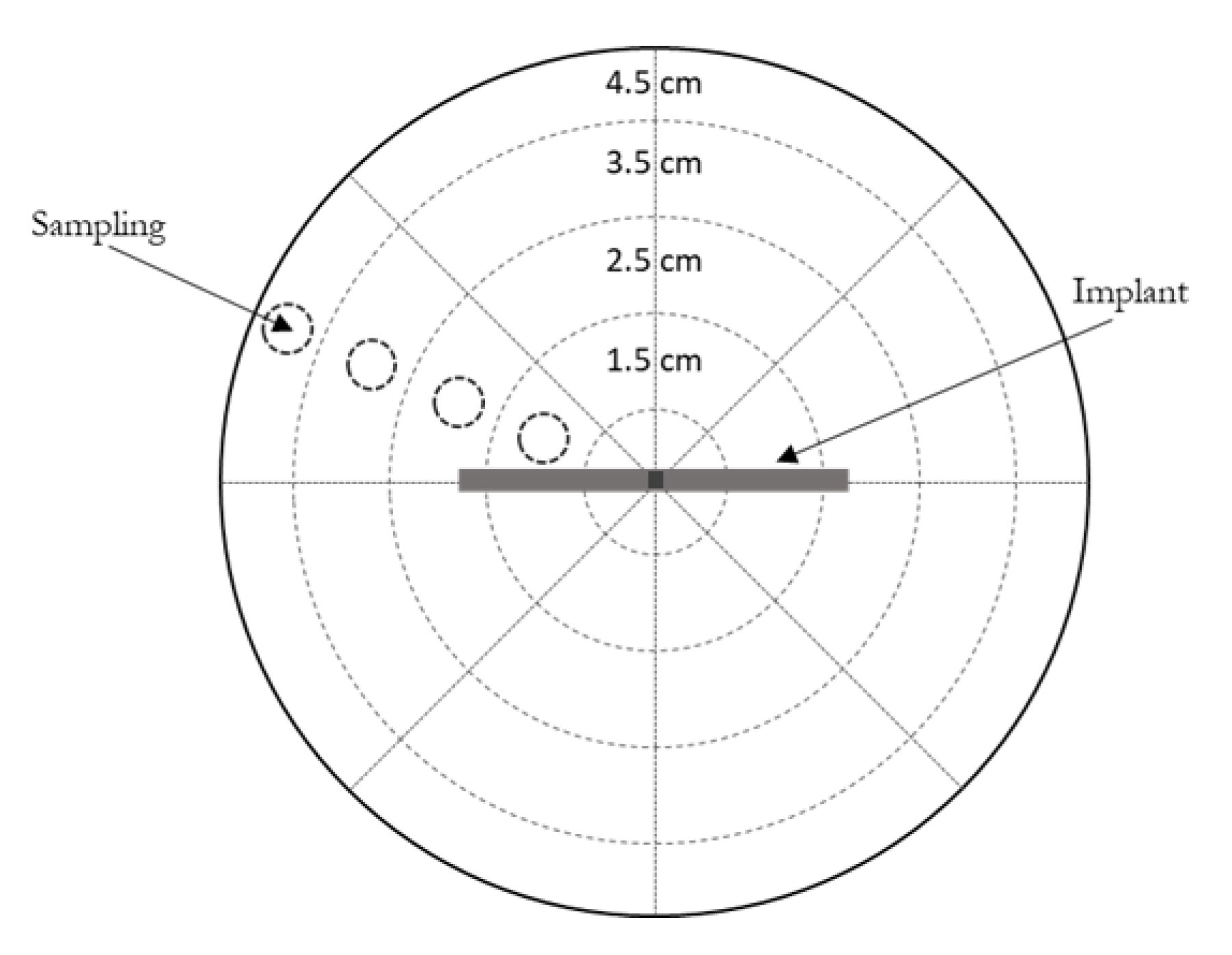
2.2.4. In Vitro Drug Release Experiments
2.3. Data Analysis
3. Results and Discussion
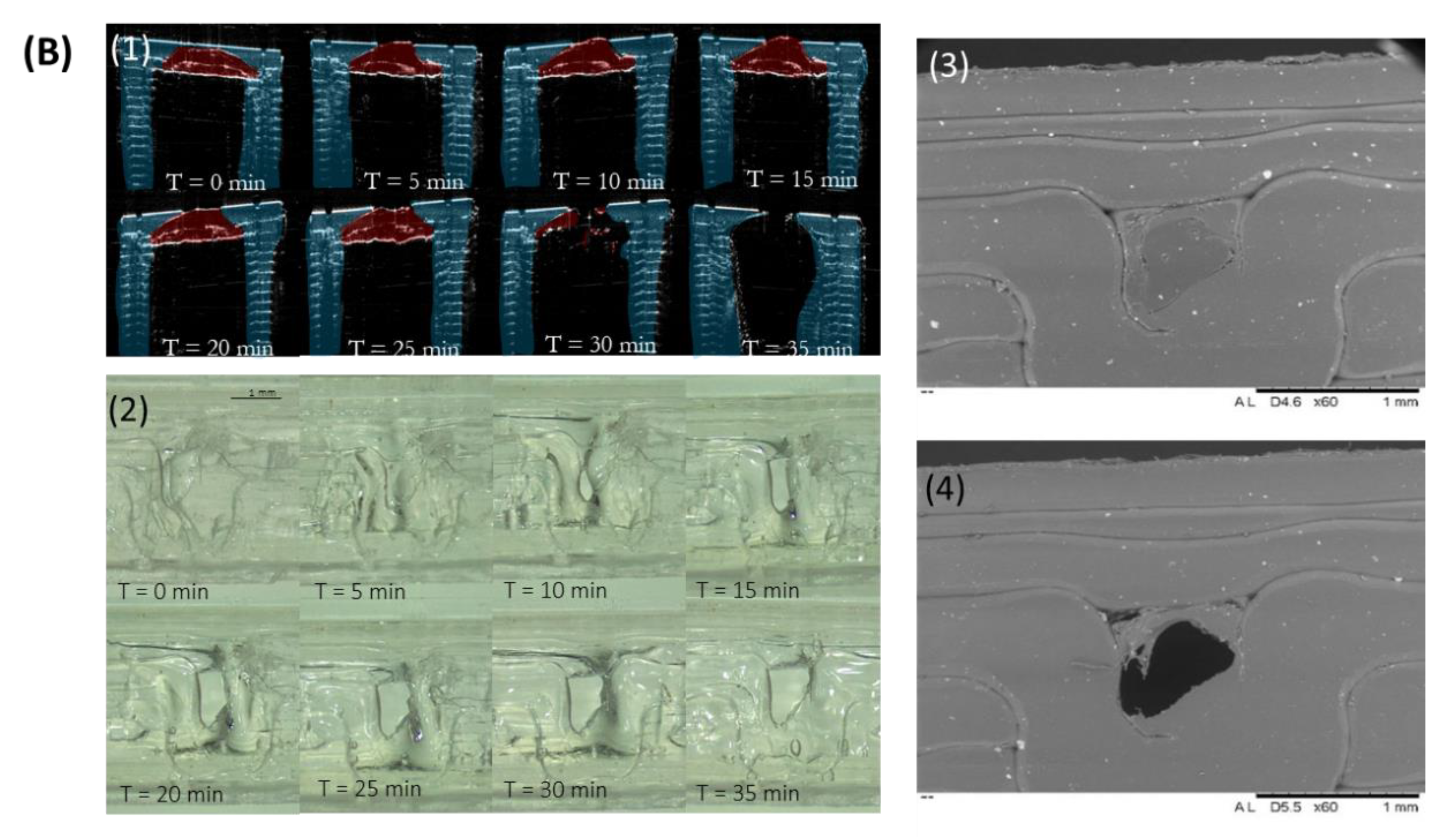
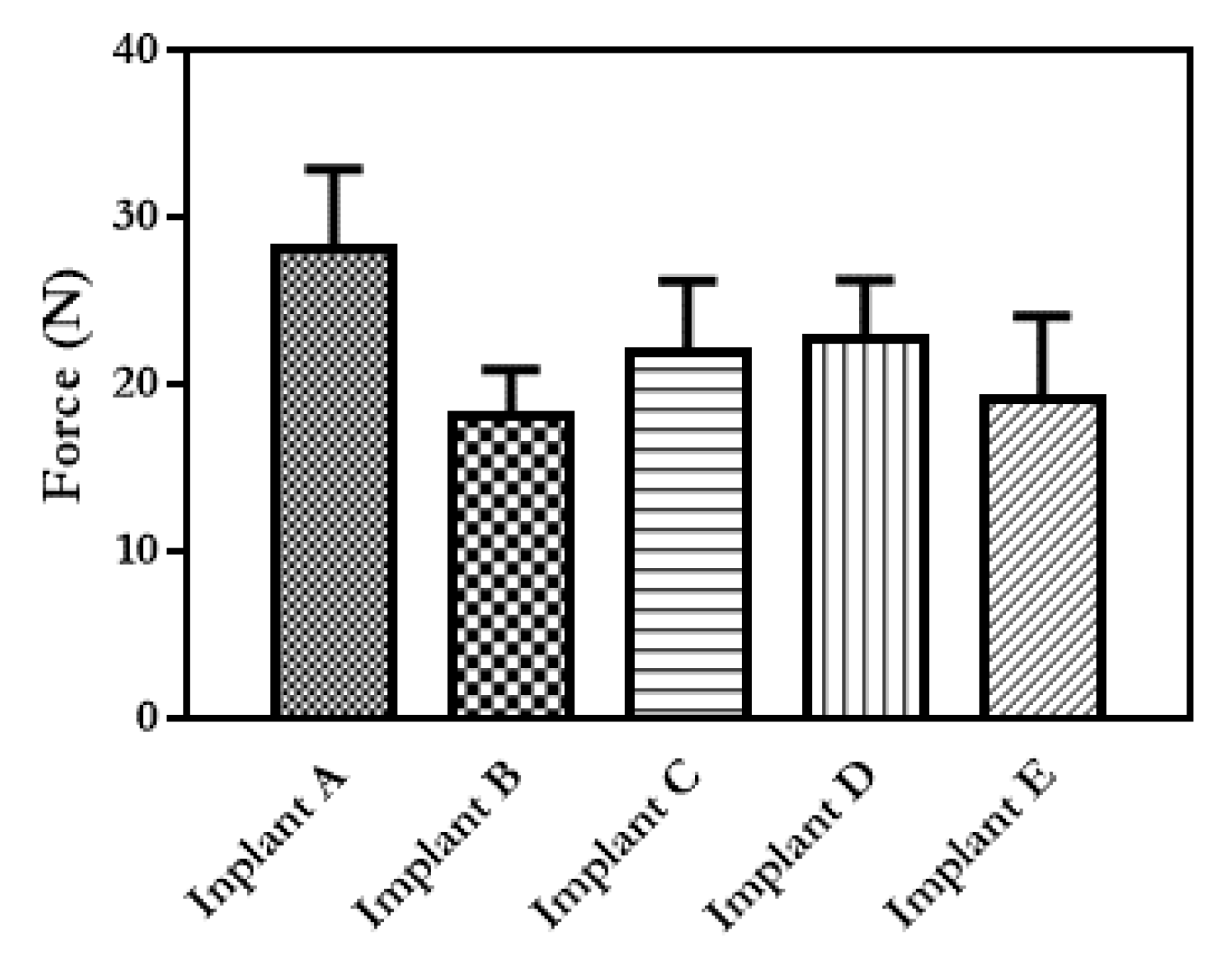
3.1. Implant Design and Characterisation
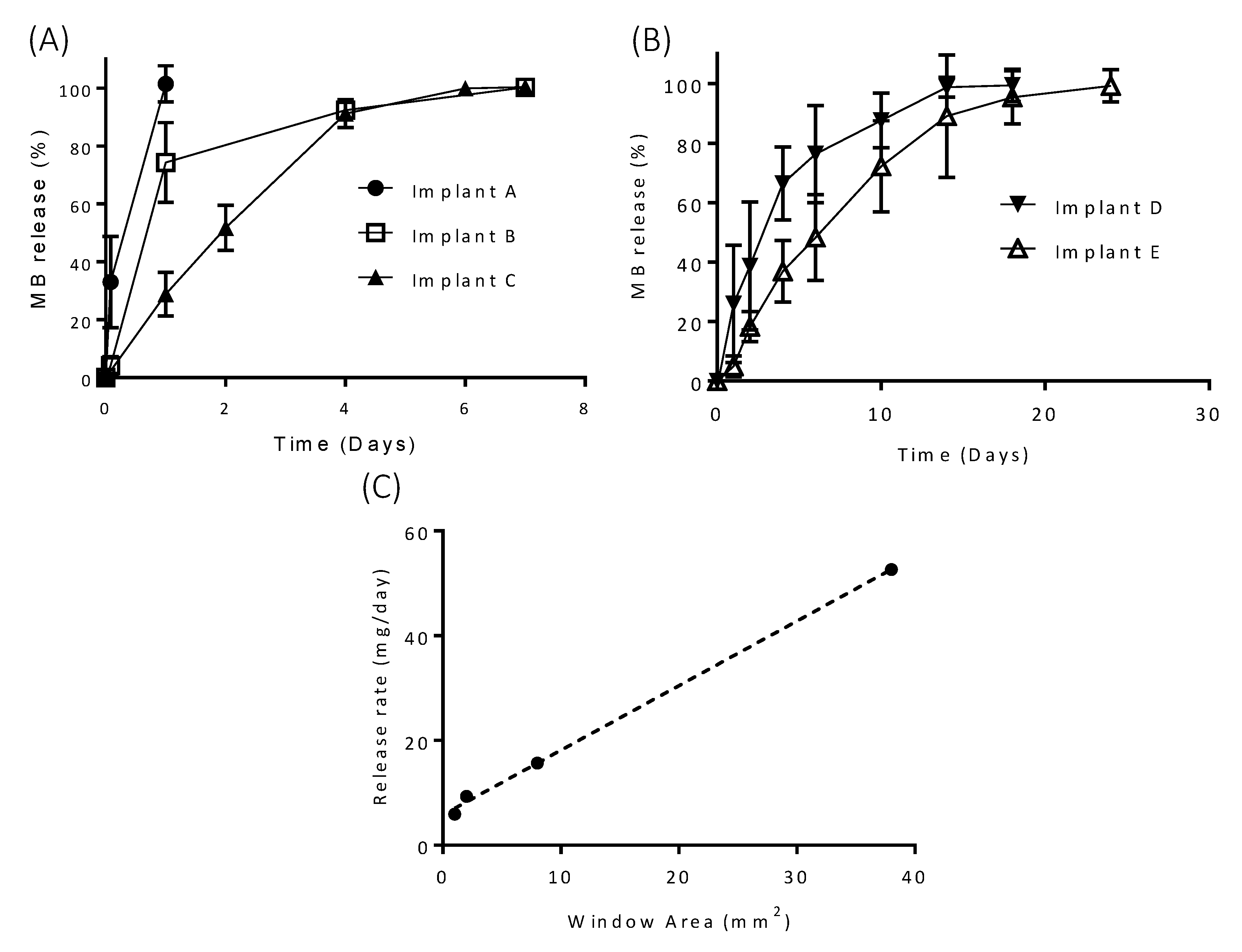
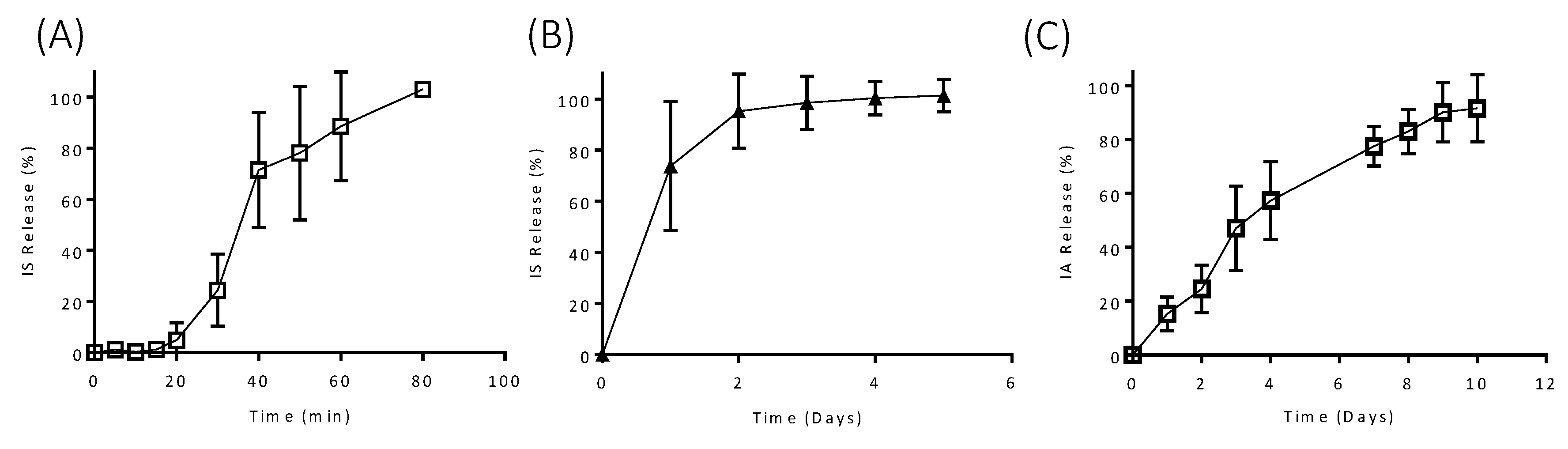
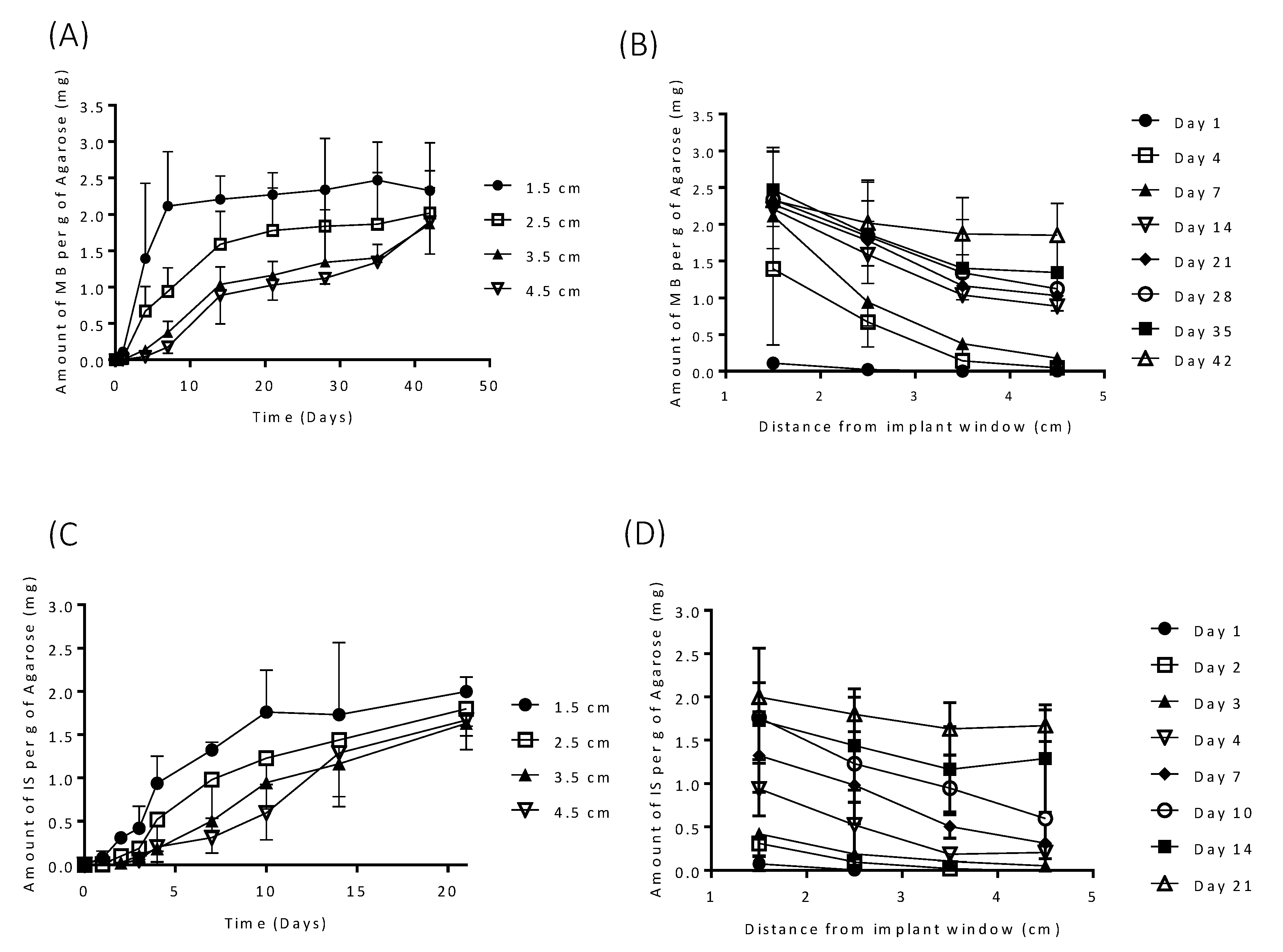
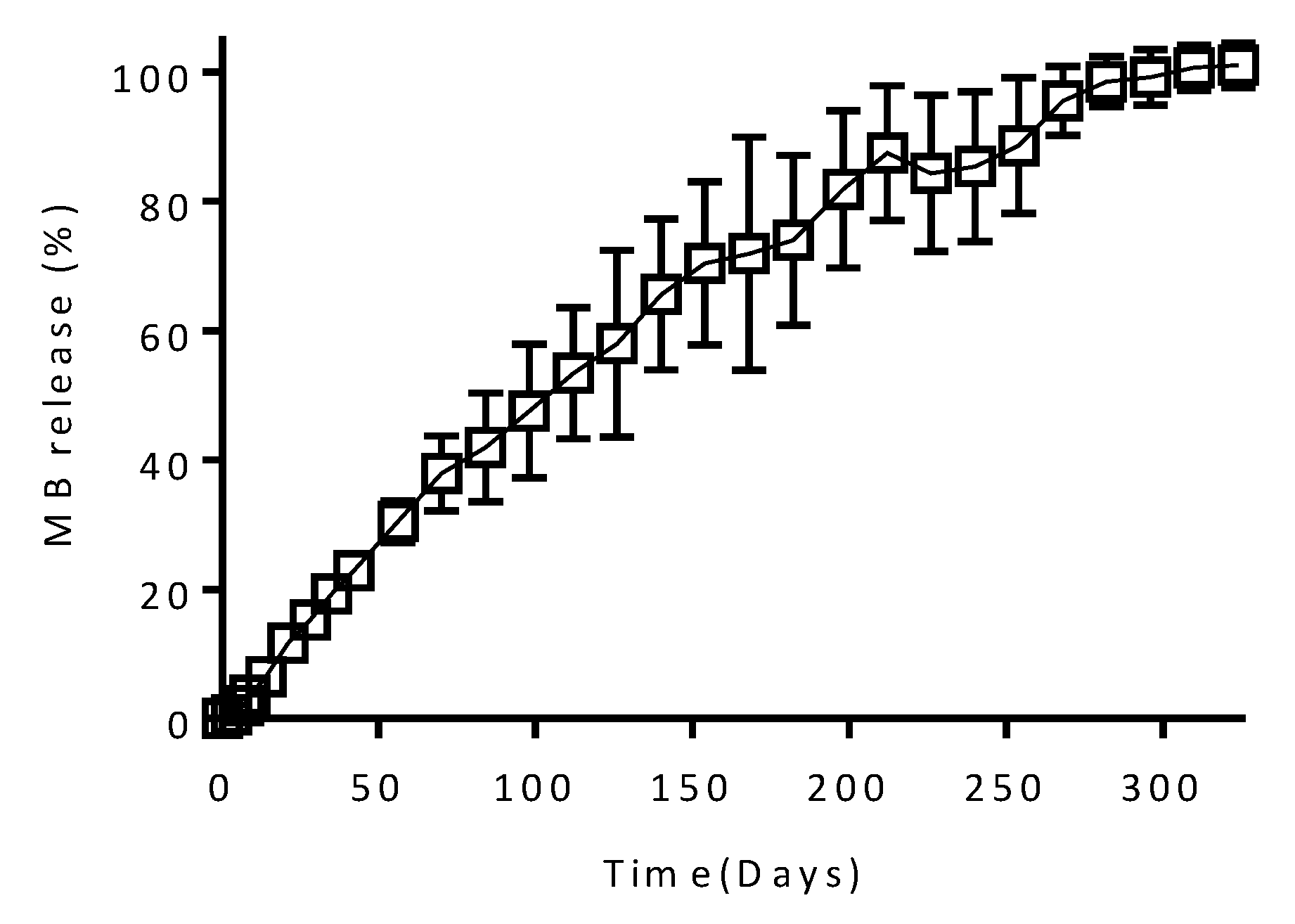
3.2. In Vitro Drug Release
3.3. In Vitro Drug Release from Coated Implants
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Rajgor, N.; Bhaskar, V.; Patel, M. Implantable drug delivery systems: An overview. Syst. Rev. Pharm. 2011, 2, 91. [Google Scholar] [CrossRef]
- Langer, R. New methods of drug delivery. Science 1990, 249, 1527–1533. [Google Scholar] [CrossRef]
- Dash, A.K.; Cudworth, G.C. Therapeutic applications of implantable drug delivery systems. J. Pharmacol. Toxicol. Methods 1998, 40, 1–12. [Google Scholar] [CrossRef]
- Wang, Y.; Sun, L.; Mei, Z.; Zhang, F.; He, M.; Fletcher, C.; Wang, F.; Yang, J.; Bi, D.; Jiang, Y.; et al. 3D printed biodegradable implants as an individualized drug delivery system for local chemotherapy of osteosarcoma. Mater. Des. 2020, 186, 108336. [Google Scholar] [CrossRef]
- Zhou, H.; Hernandez, C.; Goss, M.; Gawlik, A.; Exner, A. Biomedical imaging in implantable drug delivery systems. Curr. Drug Targets 2015, 16, 672–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fialho, S.L.; da Silva Cunha, A. Manufacturing techniques of biodegradable implants intended for intraocular application. Drug Deliv. 2005, 12, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Meng, E.; Hoang, T. Micro- and nano-fabricated implantable drug-delivery systems. Ther. Deliv. 2012, 3, 1457–1467. [Google Scholar] [CrossRef] [Green Version]
- Rabin, C.; Liang, Y.; Ehrlichman, R.S.; Budhian, A.; Metzger, K.L.; Majewski-Tiedeken, C.; Winey, K.I.; Siegel, S.J. In vitro and in vivo demonstration of risperidone implants in mice. Schizophr. Res. 2008, 98, 66–78. [Google Scholar] [CrossRef] [Green Version]
- Schlesinger, E.; Johengen, D.; Luecke, E.; Rothrock, G.; McGowan, I.; van der Straten, A.; Desai, T. A tunable, biodegradable, thin-film polymer device as a long-acting implant delivering tenofovir alafenamide fumarate for HIV pre-exposure prophylaxis. Pharm. Res. 2016, 33, 1649–1656. [Google Scholar] [CrossRef]
- Stewart, S.; Domínguez-Robles, J.; Donnelly, R.; Larrañeta, E. Implantable polymeric drug delivery devices: Classification, manufacture, materials, and clinical Applications. Polymers (Basel) 2018, 10, 1379. [Google Scholar] [CrossRef] [Green Version]
- Mansour, D. Nexplanon®: What Implanon® did next. J. Fam. Plan. Reprod. Heal. Care 2010, 36, 187–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palomba, S.; Falbo, A.; Di Cello, A.; Materazzo, C.; Zullo, F. Nexplanon: The new implant for long-term contraception: A comprehensive descriptive review. Gynecol. Endocrinol. 2012, 28, 710–721. [Google Scholar] [CrossRef] [PubMed]
- Schlegel, P. A review of the pharmacokinetic and pharmacological properties of a once-yearly administered histrelin acetate implant in the treatment of prostate cancer. BJU Int. 2009, 103, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Bagshaw, K.R.; Hanenbaum, C.L.; Carbone, E.J.; Lo, K.W.; Laurencin, C.T.; Walker, J.; Nair, L.S. Pain management via local anesthetics and responsive hydrogels. Ther. Deliv. 2015, 6, 165–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gimeno, M.; Pinczowski, P.; Pérez, M.; Giorello, A.; Martínez, M.Á.; Santamaría, J.; Arruebo, M.; Luján, L. A controlled antibiotic release system to prevent orthopedic-implant associated infections: An in vitro Study. Eur. J. Pharm. Biopharm. 2015, 96, 264–271. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.; Mei, L.; Song, C.; Cui, X.; Wang, P. The in vivo degradation, absorption and excretion of PCL-based implant. Biomaterials 2006, 27, 1735–1740. [Google Scholar] [CrossRef] [PubMed]
- Ulery, B.D.; Nair, L.S.; Laurencin, C.T. Biomedical applications of biodegradable polymers. J. Polym. Sci. Part B Polym. Phys. 2011, 49, 832–864. [Google Scholar] [CrossRef] [Green Version]
- Kumari, A.; Yadav, S.K.; Yadav, S.C. Biodegradable polymeric nanoparticles based drug delivery systems. Colloids Surfaces B Biointerfaces 2010, 75, 1–18. [Google Scholar] [CrossRef]
- Cockshott, I.D. Clinical pharmacokinetics of goserelin. Clin. Pharmacokinet. 2000, 39, 27–48. [Google Scholar] [CrossRef]
- Park, E.J.; Amatya, S.; Kim, M.S.; Park, J.H.; Seol, E.; Lee, H.; Shin, Y.-H.; Na, D.H. Long-acting injectable formulations of antipsychotic drugs for the treatment of schizophrenia. Arch. Pharm. Res. 2013, 36, 651–659. [Google Scholar] [CrossRef]
- Khaled, S.A.; Burley, J.C.; Alexander, M.R.; Roberts, C.J. Desktop 3D printing of controlled release pharmaceutical bilayer tablets. Int. J. Pharm. 2014, 461, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Barrett, S.E.; Teller, R.S.; Forster, S.P.; Li, L.; Mackey, M.A.; Skomski, D.; Yang, Z.; Fillgrove, K.L.; Doto, G.J.; Wood, S.L.; et al. Extended-duration MK-8591-eluting implant as a candidate for HIV treatment and prevention. Antimicrob. Agents Chemother. 2018, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gunawardana, M.; Remedios-Chan, M.; Miller, C.S.; Fanter, R.; Yang, F.; Marzinke, M.A.; Hendrix, C.W.; Beliveau, M.; Moss, J.A.; Smith, T.J.; et al. Pharmacokinetics of long-acting tenofovir alafenamide (GS-7340) subdermal implant for HIV prophylaxis. Antimicrob. Agents Chemother. 2015, 59, 3913–3919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domínguez-Robles, J.; Martin, N.; Fong, M.; Stewart, S.; Irwin, N.; Rial-Hermida, M.; Donnelly, R.; Larrañeta, E. Antioxidant PLA composites containing lignin for 3D printing applications: A potential material for healthcare applications. Pharmaceutics 2019, 11, 165. [Google Scholar] [CrossRef] [Green Version]
- Mathew, E.; Domínguez-Robles, J.; Stewart, S.A.; Mancuso, E.; O’Donnell, K.; Larrañeta, E.; Lamprou, D.A. Fused deposition modeling as an effective tool for anti-infective dialysis catheter fabrication. ACS Bbomaterials Sci. Eng. 2019, 5, 6300–6310. [Google Scholar] [CrossRef]
- Domínguez-Robles, J.; Mancinelli, C.; Mancuso, E.; García-Romero, I.; Gilmore, B.F.; Casettari, L.; Larrañeta, E.; Lamprou, D.A. 3D printing of drug-loaded thermoplastic polyurethane meshes: A potential material for soft tissue reinforcement in vaginal surgery. Pharmaceutics 2020, 12, 63. [Google Scholar] [CrossRef] [Green Version]
- Donnelly, R.F.; Majithiya, R.; Singh, T.R.R.; Morrow, D.I.J.J.; Garland, M.J.; Demir, Y.K.; Migalska, K.; Ryan, E.; Gillen, D.; Scott, C.J.; et al. Design, optimization and characterisation of polymeric microneedle arrays prepared by a novel laser-based micromoulding technique. Pharm. Res. 2011, 28, 41–57. [Google Scholar] [CrossRef] [Green Version]
- The British Pharmacopeia Commission British Pharmacopoeia. Available online: https://www.pharmacopoeia.com/bp-2020?date=2020-01-01 (accessed on 30 August 2019).
- Hoang Thi, T.H.; Chai, F.; Leprêtre, S.; Blanchemain, N.; Martel, B.; Siepmann, F.; Hildebrand, H.F.; Siepmann, J.; Flament, M.P. Bone implants modified with cyclodextrin: Study of drug release in bulk fluid and into agarose gel. Int. J. Pharm. 2010, 15, 74–85. [Google Scholar] [CrossRef]
- Larrañeta, E.; Martínez-Ohárriz, C.; Vélaz, I.; Zornoza, A.; Machín, R.; Isasi, J.R. In vitro release from reverse poloxamine/α-cyclodextrin matrices: Modelling and comparison of dissolution profiles. J. Pharm. Sci. 2014, 103, 197–206. [Google Scholar] [CrossRef] [Green Version]
- Costa, P.; Sousa Lobo, J.M. Modeling and comparison of dissolution profiles. Eur. J. Pharm. Sci. 2001, 13, 123–133. [Google Scholar] [CrossRef]
- Funk, S.; Miller, M.M.; Mishell, D.R.; Archer, D.F.; Poindexter, A.; Schmidt, J.; Zampaglione, E. Safety and efficacy of Implanon™, a single-rod implantable contraceptive containing etonogestrel. Contraception 2005, 71, 319–326. [Google Scholar] [CrossRef] [PubMed]
- George, E.; Liacouras, P.; Rybicki, F.J.; Mitsouras, D. Measuring and establishing the accuracy and reproducibility of 3D printed medical models. Radiographics 2017, 37, 1424–1450. [Google Scholar] [CrossRef] [PubMed]
- Drugbank Methylene Blue. Available online: https://www.drugbank.ca/drugs/DB09241 (accessed on 30 October 2019).
- Sigma-Aldrich Ibuprofen Sodium Salt. Available online: https://www.sigmaaldrich.com/catalog/product/sial/i1892?lang=en®ion=GB (accessed on 8 March 2019).
- Drugbank Ibuprofen. Available online: https://www.drugbank.ca/drugs/DB01050 (accessed on 30 October 2019).
- Goyanes, A.; Robles Martinez, P.; Buanz, A.; Basit, A.W.; Gaisford, S. Effect of geometry on drug release from 3D printed tablets. Int. J. Pharm. 2015, 494, 657–663. [Google Scholar] [CrossRef] [PubMed]
- Chong, S.-F.; Smith, A.A.A.; Zelikin, A.N. Microstructured, functional PVA hydrogels through bioconjugation with oligopeptides under physiological conditions. Small 2013, 9, 942–950. [Google Scholar] [CrossRef] [PubMed]
- Khizer, Z.; Akram, M.R.; Sarfraz, R.M.; Nirwan, J.S.; Farhaj, S.; Yousaf, M.; Hussain, T.; Lou, S.; Timmins, P.; Conway, B.R.; et al. Plasticiser-free 3D printed hydrophilic matrices: Quantitative 3D surface texture, mechanical, swelling, erosion, drug release and pharmacokinetic studies. Polymers (Basel) 2019, 11, 1095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chai, X.; Chai, H.; Wang, X.; Yang, J.; Li, J.; Zhao, Y.; Cai, W.; Tao, T.; Xiang, X. Fused deposition modeling (FDM) 3D printed tablets for intragastric floating delivery of domperidone. Sci. Rep. 2017, 7, 2829. [Google Scholar] [CrossRef] [Green Version]
- Horal, M. 3D printing implants for fracture healing studies in rat; Lund University: Lund, Sweden, 2015. [Google Scholar]
- Auras, R.; Lim, L.-T.; Selke, S.E.M.; Tsuji, H. Poly(lactic acid): Synthesis, structures, properties, processing and applications; Auras, R., Lim, L.-T., Selke, S.E.M., Tsuji, H., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2010. [Google Scholar]
- Fung, D.Y.C.; Miller, R.D. Effect of dyes on bacterial growth. Appl. Microbiol. 1973, 25, 793–799. [Google Scholar] [CrossRef] [Green Version]
- Lichstein, H.C. Studies of the effect of sodium azide on microbic growth and respiration: III. The effect of sodium azide on the gas metabolism of B. subtilis and P. aeruginosa and the influence of pyocyanine on the gas exchange of a pyocyanine-free Strain of P. aerugino. J. Bacteriol. 1944, 47, 239–251. [Google Scholar] [CrossRef] [Green Version]
- Herrera, L.C.; Tesoriero, M.V.; Hermida, L.G. In vitro release testing of PLGA microspheres with franz diffusion cells. Dissolution Technol. 2012, 19, 6–11. [Google Scholar] [CrossRef]
- Kelm, J.; Regitz, T.; Schmitt, E.; Jung, W.; Anagnostakos, K. In vivo and in vitro studies of antibiotic release from and bacterial growth inhibition by antibiotic-impregnated polymethylmethacrylate hip spacers. Antimicrob. Agents Chemother. 2006, 50, 332–335. [Google Scholar] [CrossRef] [Green Version]
- Liu, K.-S.; Chen, W.-H.; Lee, C.-H.; Su, Y.-F.; Liu, S.-J. Extended pain relief achieved by analgesic-eluting biodegradable nanofibers in the Nuss procedure: In vitro and in vivo studies. Int. J. Nanomedicine 2018, 13, 8355–8364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leung, D.H.; Kapoor, Y.; Alleyne, C.; Walsh, E.; Leithead, A.; Habulihaz, B.; Salituro, G.M.; Bak, A.; Rhodes, T. Development of a convenient in vitro gel diffusion model for predicting the in vivo performance of subcutaneous parenteral formulations of large and small molecules. AAPS PharmSciTech 2017, 18, 2203–2213. [Google Scholar] [CrossRef] [PubMed]
- Pernodet, N.; Maaloum, M.; Tinland, B. Pore size of agarose gels by atomic force microscopy. Electrophoresis 1997, 18, 55–58. [Google Scholar] [CrossRef] [PubMed]
- Ye, F.; Larsen, S.W.; Yaghmur, A.; Jensen, H.; Larsen, C.; Østergaard, J. Drug release into hydrogel-based subcutaneous surrogates studied by UV imaging. J. Pharm. Biomed. Anal. 2012, 71, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Astary, G.W.; Sepulveda, H.; Mareci, T.H.; Sarntinoranont, M. Quantitative assessment of macromolecular concentration during direct infusion into an agarose hydrogel phantom using contrast-enhanced MRI. Magn. Reson. Imaging 2008, 26, 1433–1441. [Google Scholar] [CrossRef] [Green Version]
- McCabe, M. The diffusion coefficient of caffeine through agar gels containing a hyaluronic acid–protein complex. A model system for the study of the permeability of connective tissues. Biochem. J. 1972, 127, 249–253. [Google Scholar] [CrossRef] [Green Version]
- Salloum, M.; Ma, R.H.; Weeks, D.; Zhu, L. Controlling nanoparticle delivery in magnetic nanoparticle hyperthermia for cancer treatment: Experimental study in agarose gel. Int. J. Hyperth. 2008, 24, 337–345. [Google Scholar] [CrossRef]
- Salmoria, G.V.; Ghizoni, G.B.; Gindri, I.M.; Marques, M.S.; Kanis, L.A. Hot extrusion of PE/fluorouracil implantable rods for targeted drug delivery in cancer treatment. Polym. Bull. 2019, 76, 1825–1838. [Google Scholar] [CrossRef]
- Launonen, V.; Vierimaa, O.; Kiuru, M.; Isola, J.; Roth, S.; Pukkala, E.; Sistonen, P.; Herva, R.; Aaltonen, L.A. Inherited susceptibility to uterine leiomyomas and renal cell cancer. Proc. Natl. Acad. Sci. USA 2001, 98, 3387–3392. [Google Scholar] [CrossRef] [Green Version]
- Johnson, L.M.; Krovi, S.A.; Li, L.; Girouard, N.; Demkovich, Z.R.; Myers, D.; Creelman, B.; van der Straten, A. Characterization of a reservoir-style implant for sustained release of tenofovir alafenamide (TAF) for HIV pre-exposure prophylaxis (PrEP). Pharmaceutics 2019, 11, 315. [Google Scholar] [CrossRef] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Curve 1 | Curve 2 | F1 | F2 |
|---|---|---|---|
| Implant A | Implant B | 60.06 | 33.00 |
| Implant A | Implant C | 73.89 | 13.58 |
| Implant B | Implant C | 28.93 | 32.12 |
| Implant D | Implant E | 19.61 | 34.75 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stewart, S.A.; Domínguez-Robles, J.; McIlorum, V.J.; Mancuso, E.; Lamprou, D.A.; Donnelly, R.F.; Larrañeta, E. Development of a Biodegradable Subcutaneous Implant for Prolonged Drug Delivery Using 3D Printing. Pharmaceutics 2020, 12, 105. https://doi.org/10.3390/pharmaceutics12020105
Stewart SA, Domínguez-Robles J, McIlorum VJ, Mancuso E, Lamprou DA, Donnelly RF, Larrañeta E. Development of a Biodegradable Subcutaneous Implant for Prolonged Drug Delivery Using 3D Printing. Pharmaceutics. 2020; 12(2):105. https://doi.org/10.3390/pharmaceutics12020105
Chicago/Turabian StyleStewart, Sarah A., Juan Domínguez-Robles, Victoria J. McIlorum, Elena Mancuso, Dimitrios A. Lamprou, Ryan F. Donnelly, and Eneko Larrañeta. 2020. "Development of a Biodegradable Subcutaneous Implant for Prolonged Drug Delivery Using 3D Printing" Pharmaceutics 12, no. 2: 105. https://doi.org/10.3390/pharmaceutics12020105