Controlled-Release from High-Loaded Reservoir-Type Systems—A Case Study of Ethylene-Vinyl Acetate and Progesterone

Abstract
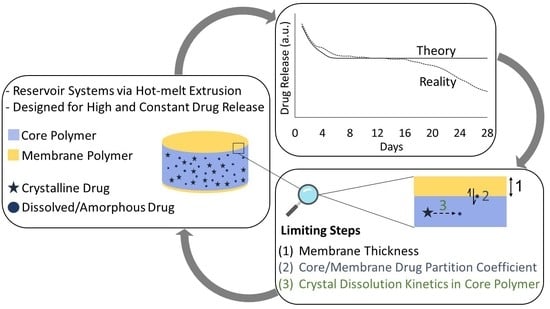
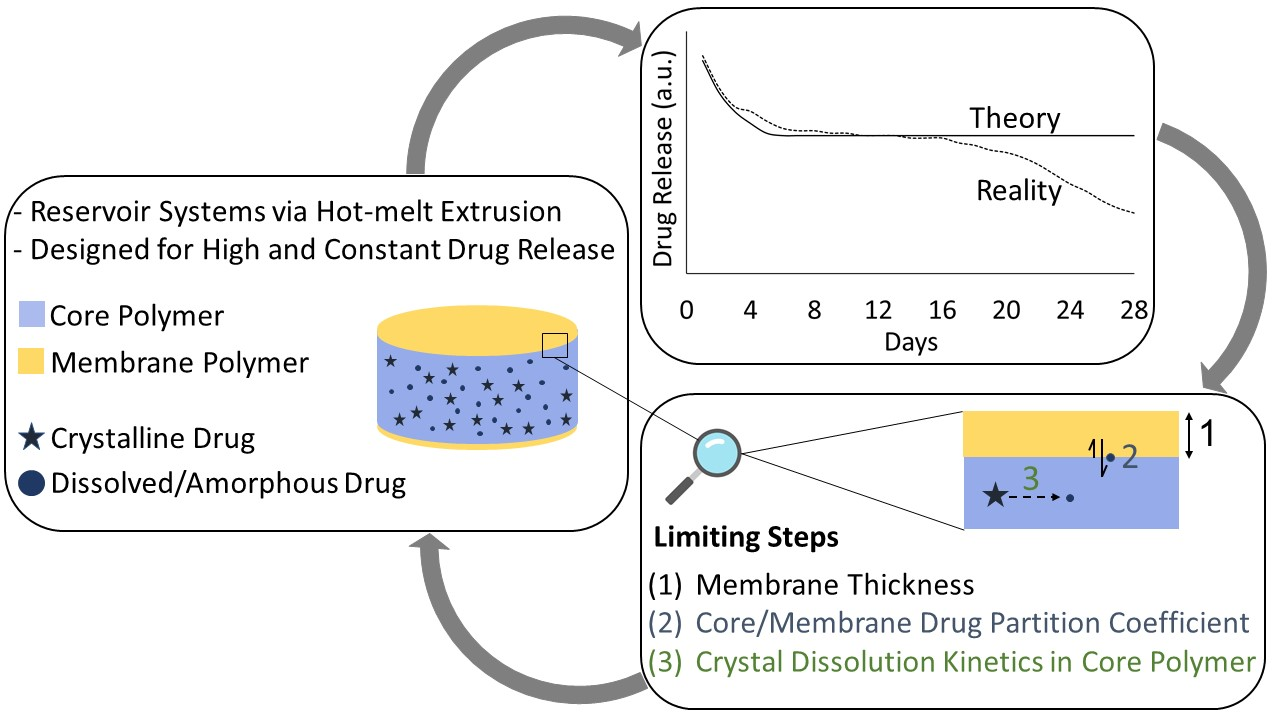
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
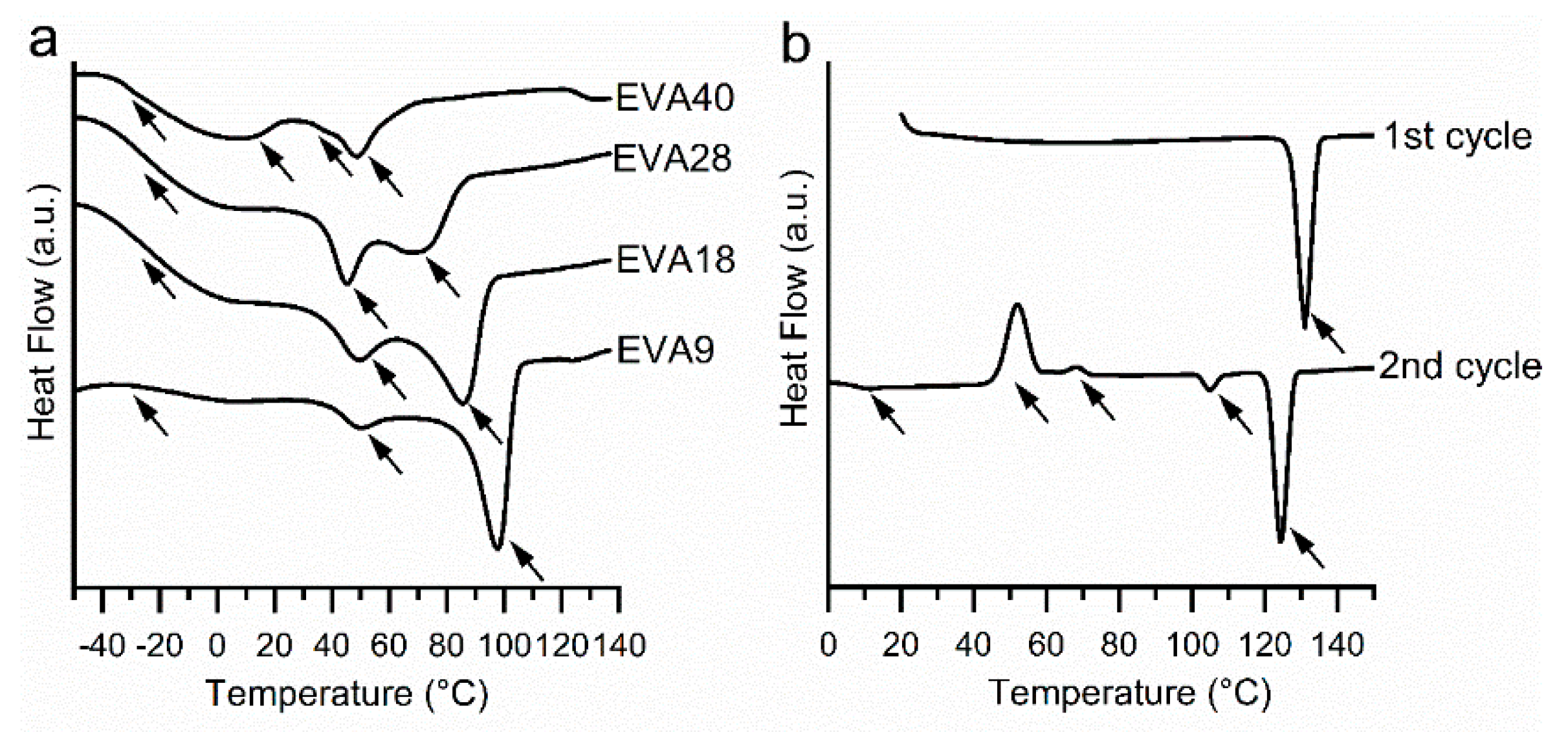
2.2.1. Thermal Analysis of the Pure Components
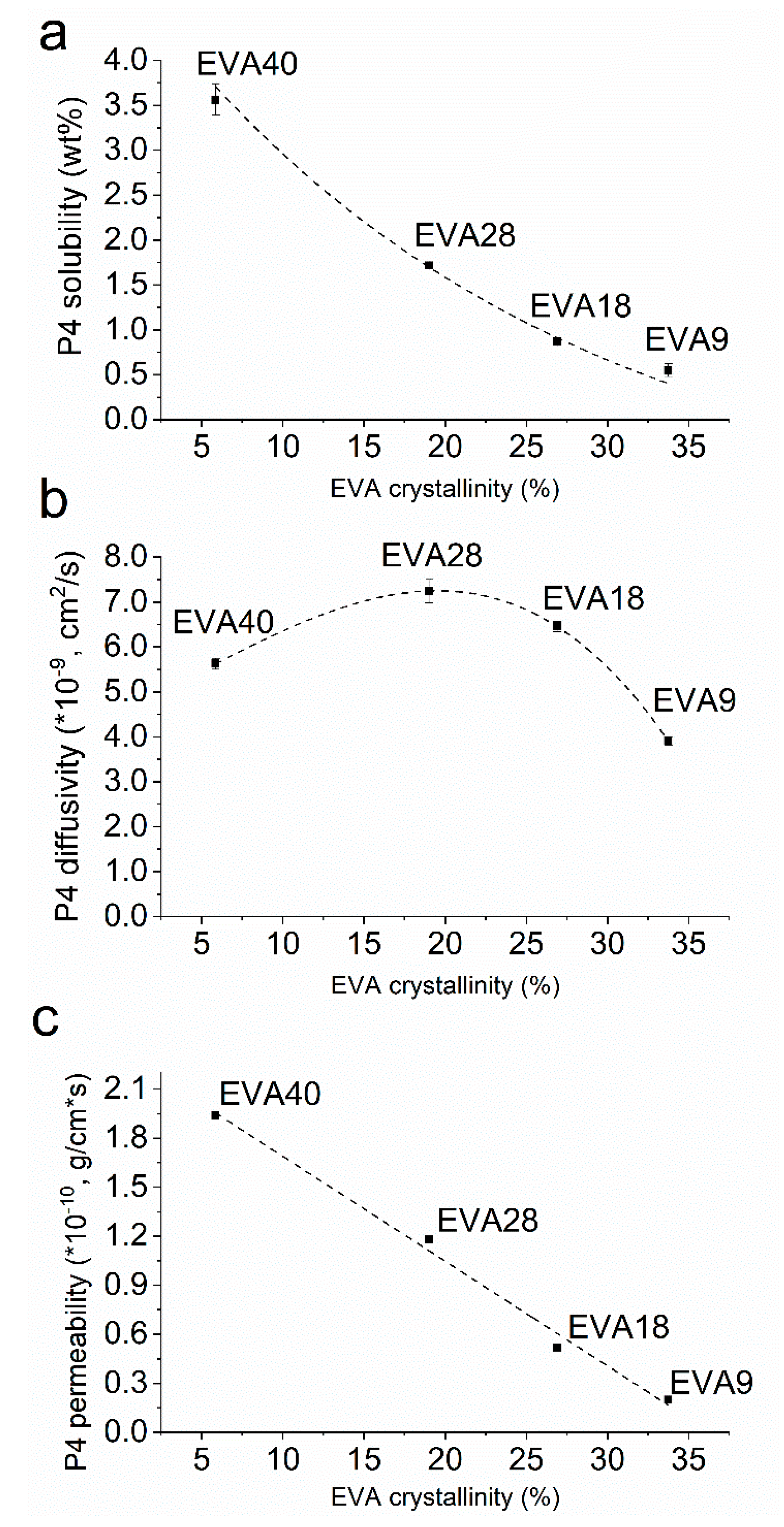
2.2.2. P4 Solubility in EVA Polymers
2.2.3. P4 Diffusivity and Permeability in EVA Polymers
2.2.4. Rational Formulation Design
2.2.5. Preparation of the VCM Systems
2.2.6. Solid State Characterization of the VCM Systems
2.2.7. In-Vitro Drug Dissolution Studies of the VCM Systems
2.2.8. UPLC Method
2.2.9. Statistical Analysis
3. Results
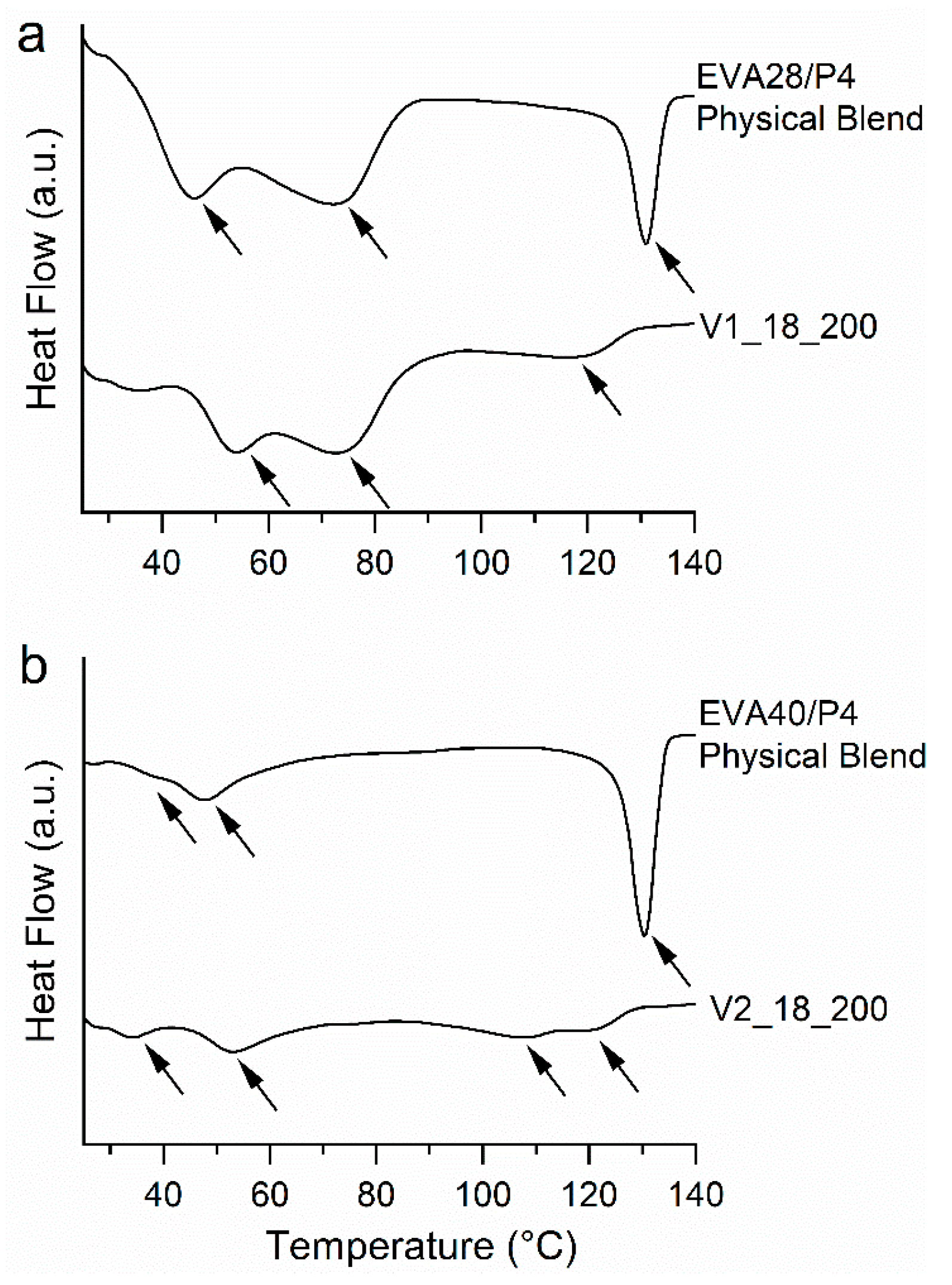
3.1. Thermal Analysis of the Pure Components
3.2. P4 solubility in EVA Polymers
3.3. P4 Diffusivity and Permeability in EVA Polymers
3.4. Rational Formulation Design
3.5. Preparation of the VCM systems
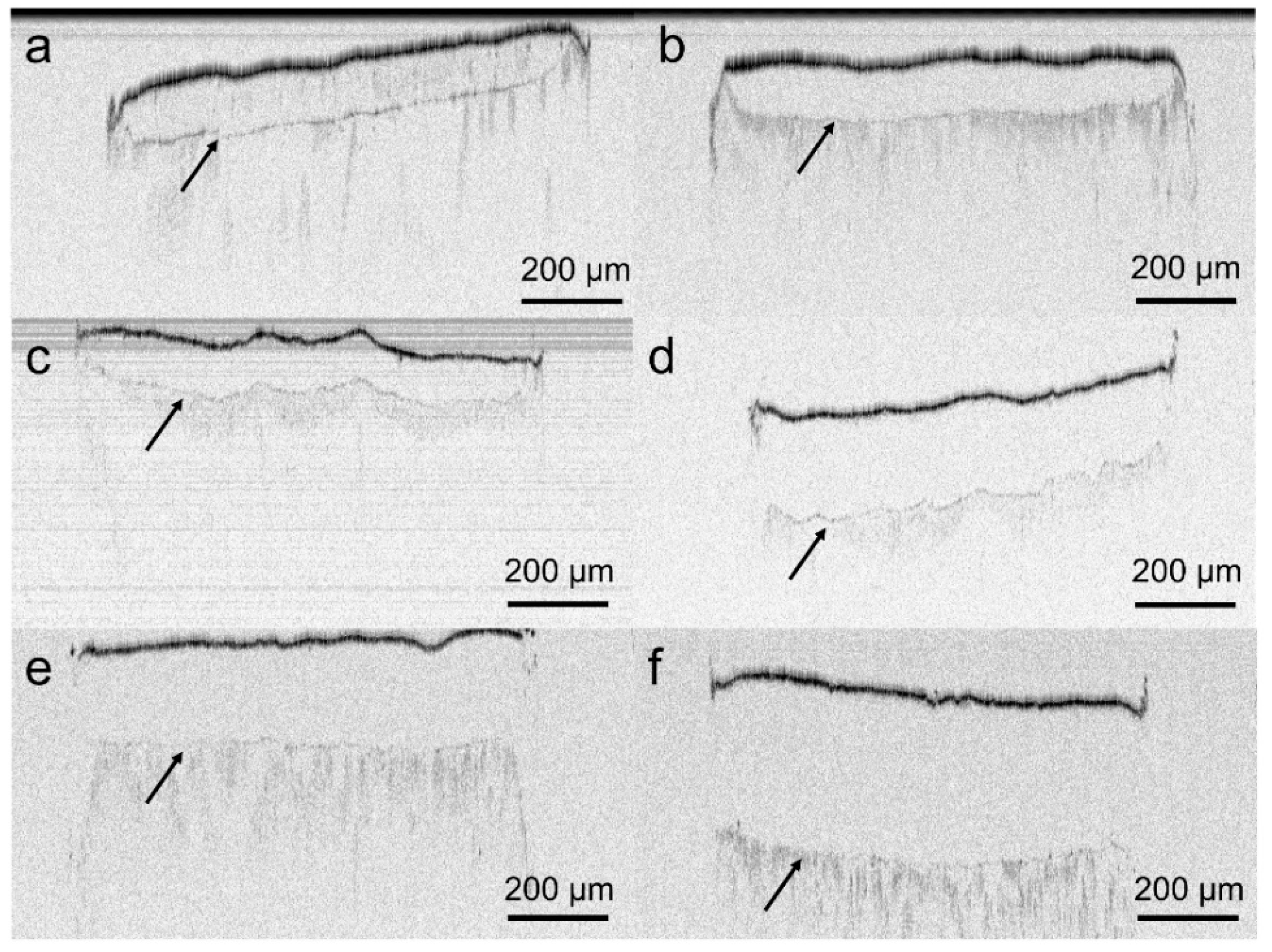
3.6. Solid-State Characterization of the VCM Systems
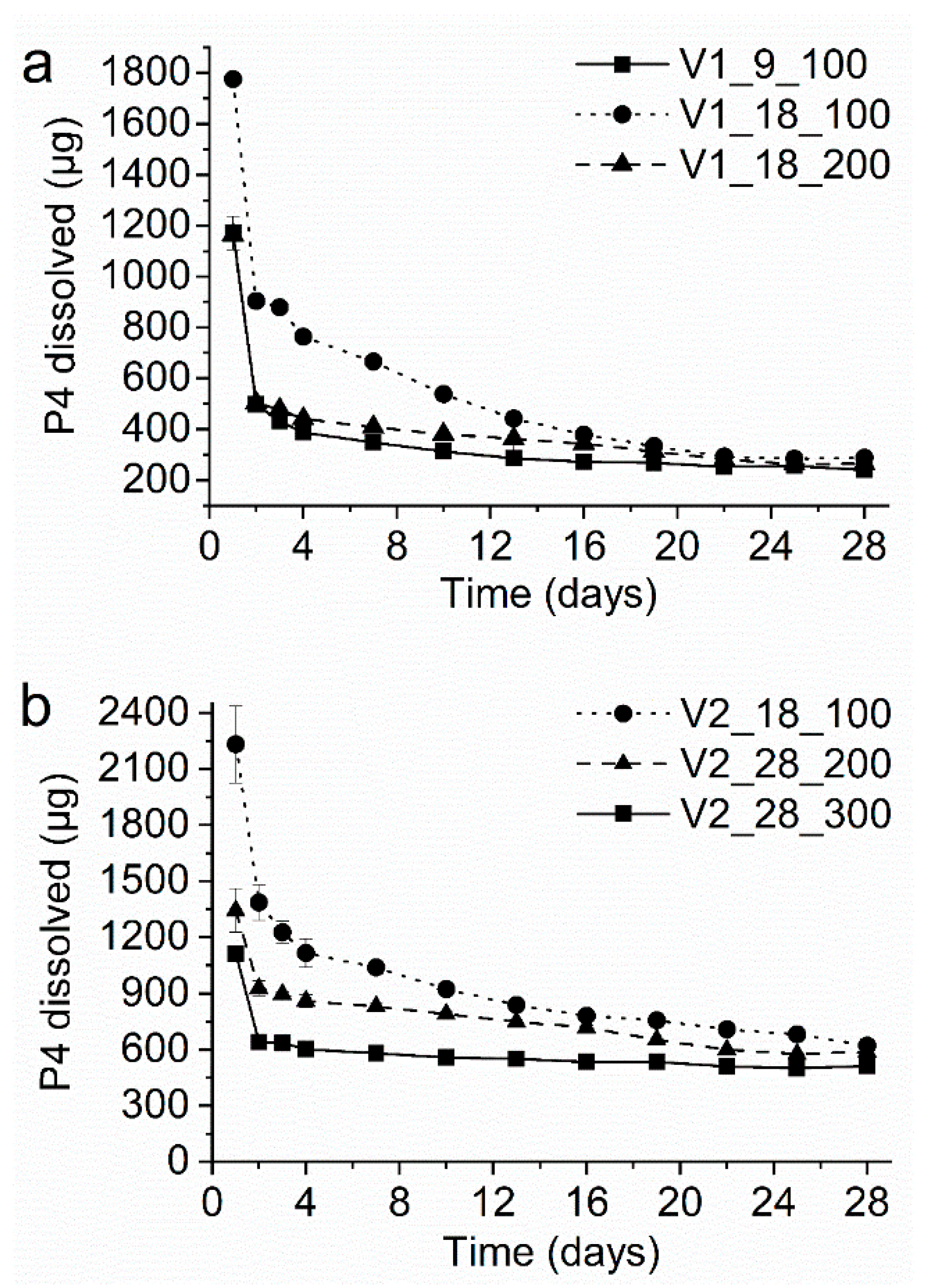
3.7. In-Vitro Drug Dissolution Studies of the VCM Systems
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Jones, D. Pharmaceutical Applications of Polymers for Drug Delivery (Rapra Review Reports), 1st ed.; Smithers Rapra Press: Shawbury, UK, 2004; ISBN 1-85957-479-3. [Google Scholar]
- Yang, W.W.; Pierstorff, E. Reservoir-based polymer drug delivery systems. J. Lab. Autom. 2012, 17, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Fan, L.; Singh, S.K. Controlled Release A Quantitative Treatment; Cantow, H.-J., Harwood, H.J., Kennedy, J.P., Ledwith, A., Meissner, J., Okamura, S., Henrici-Olive, G., Olive, S., Eds.; First; Springer: Heidelberg/Berlin, Germany, 1989; ISBN 978-3-642-74509-6. [Google Scholar]
- Siepmann, J.; Siegel, R.A.; Siepmann, F. Diffusion Controlled Drug Delivery Systems. In Fundamentals and Applications of Controlled Release Drug Delivery; Siepmann, J., Siegel, R.A., Rathbone, M.J., Eds.; Springer: New York, NY, USA, 2012; pp. 127–152. [Google Scholar]
- Nel, A.; Smythe, S.; Young, K.; Malcolm, K.; McCoy, C.; Rosenberg, Z.; Romano, J. Safety and pharmacokinetics of dapivirine delivery from matrix and reservoir intravaginal rings to HIV-negative women. J. Acquir. Immune Defic. Syndr. 2009, 51, 416–423. [Google Scholar] [CrossRef]
- Clark, J.T.; Clark, M.R.; Shelke, N.B.; Johnson, T.J.; Smith, E.M.; Andreasen, A.K.; Nebeker, J.S.; Fabian, J.; Friend, D.R.; Kiser, P.F. Engineering a segmented dual-reservoir polyurethane intravaginal ring for simultaneous prevention of HIV transmission and unwanted pregnancy. PLoS ONE 2014, 9, e88509. [Google Scholar] [CrossRef] [PubMed]
- Brache, V.; Payán, L.J.; Faundes, A. Current status of contraceptive vaginal rings. Contraception 2013, 87, 264–272. [Google Scholar] [CrossRef] [PubMed]
- Johnson, T.J.; Clark, M.R.; Albright, T.H.; Nebeker, J.S.; Tuitupou, A.L.; Clark, J.T.; Fabian, J.; McCabe, R.T.; Chandra, N.; Doncel, G.F.; et al. A 90-day tenofovir reservoir intravaginal ring for mucosal HIV prophylaxis. Antimicrob. Agents Chemother. 2012, 56, 6272–6283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holmgren, P.Å.; Lindskog, M.; von Schoultz, B. Vaginal rings for continuous low-dose release of oestradiol in the treatment of urogenital atrophy. Maturitas 1989, 11, 55–63. [Google Scholar] [CrossRef]
- Croxatto, H.B. Progestin implants. Steroids 2000, 65, 681–685. [Google Scholar] [CrossRef]
- Sivin, I.; Mishell, D.R.; Alvarez, F.; Brache, V.; Elomaa, K.; Lähteenmäki, P.; Massai, R.; Miranda, P.; Croxatto, H.; Dean, C.; et al. Contraceptive vaginal rings releasing Nestorone® and ethinylestradiol: A 1-year dose-finding trial. Contraception 2005, 71, 122–129. [Google Scholar] [CrossRef]
- Schneider, C.; Langer, R.; Loveday, D.; Hair, D. Applications of ethylene vinyl acetate copolymers (EVA) in drug delivery systems. J. Control. Release 2017, 262, 284–295. [Google Scholar] [CrossRef]
- Almeida, A.; Possemiers, S.; Boone, M.N.; De Beer, T.; Quinten, T.; Van Hoorebeke, L.; Remon, J.P.; Vervaet, C. Ethylene vinyl acetate as matrix for oral sustained release dosage forms produced via hot-melt extrusion. Eur. J. Pharm. Biopharm. 2011, 77, 297–305. [Google Scholar] [CrossRef]
- Chen, S.X.; Lostritto, R.T. Diffusion of benzocaine in poly(ethylene-vinyl acetate) membranes: Effects of vehicle ethanol concentration and membrane vinyl acetate content. J. Control. Release 1996, 38, 185–191. [Google Scholar] [CrossRef]
- Hamada, A.L.; Maruo, T.; Samoto, T.; Yoshida, S.; Nash, H.; Spitz, I.M.; Johansson, E. Estradiol/progesterone-releasing vaginal rings for hormone replacement therapy in postmenopausal women. Gynecol. Endocrinol. 2003, 17, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Matlin, S.A.; Beleuguer, A.; Hall, P.E. Progesterone-Releasing Vaginal Rings for Use in Postpartum Contraception. Contraception 1992, 45, 329–341. [Google Scholar] [CrossRef]
- Eder, S.; Beretta, M.; Witschnigg, A.; Koutsamanis, I.; Eggenreich, K.; Khinast, J.G.; Koscher, G.; Paudel, A.; Nickisch, K.; Friedrich, M.; et al. Establishment of a Molding Procedure to Facilitate Formulation Development for Co-extrudates. AAPS PharmSciTech 2017, 18, 2971–2976. [Google Scholar] [CrossRef] [PubMed]
- Koutsamanis, I.; Eder, S.; Beretta, M.; Witschnigg, A.; Paudel, A.; Nickisch, K.; Friedrich, M.; Eggenreich, K.; Roblegg, E. Formulation and processability screening for the rational design of ethylene-vinyl acetate based intra-vaginal rings. Int. J. Pharm. 2019, 564, 90–97. [Google Scholar] [CrossRef] [PubMed]
- Wunderlich, B.; Czornyj, G. A Study of Equilibrium Melting of Polyethylene. Macromolecules 1977, 10, 906–913. [Google Scholar] [CrossRef]
- Helbling, I.M.; Ibarra, J.C.D.; Luna, J.A. The optimization of an intravaginal ring releasing progesterone using a mathematical model. Pharm. Res. 2014, 31, 795–808. [Google Scholar] [CrossRef]
- Lancaster, R.W.; Karamertzanis, P.G.; Hulme, A.T.; Tocher, D.A.; Lewis, T.C.; Price, S.L. The Polymorphism of Progesterone: Stabilization of a ‘Disappearing’ Polymorph by Co-Crystallization. J. Pharm. Sci. 2007, 96, 3419–3431. [Google Scholar] [CrossRef]
- Siepmann, J.; Lecomte, F.; Bodmeier, R. Diffusion-controlled drug delivery systems: Calculation of the required composition to achieve desired release profiles. J. Control. Release 1999, 60, 379–389. [Google Scholar] [CrossRef]
- Van Laarhoven, J.A.H.; Kruft, M.A.B.; Vromans, H. In vitro release properties of etonogestrel and ethinyl estradiol from a contraceptive vaginal ring. Int. J. Pharm. 2002, 232, 163–173. [Google Scholar] [CrossRef]
- Roumen, F.J.M.E.; Dieben, T.O.M. Clinical acceptability of an ethylene-vinyl-acetate nonmedicated vaginal ring. Contraception 1999, 59, 59–62. [Google Scholar] [CrossRef]
- Krier, F.; Mantanus, J.; Sacré, P.Y.; Chavez, P.F.; Thiry, J.; Pestieau, A.; Rozet, E.; Ziemons, E.; Hubert, P.; Evrard, B. PAT tools for the control of co-extrusion implants manufacturing process. Int. J. Pharm. 2013, 458, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Externbrink, A.; Eggenreich, K.; Eder, S.; Mohr, S.; Nickisch, K.; Klein, S. Development and evaluation of accelerated drug release testing methods for a matrix-type intravaginal ring. Eur. J. Pharm. Biopharm. 2017, 110, 1–12. [Google Scholar] [CrossRef] [PubMed]
- LOCTITE® 4011TM Technical Data Sheet. Available online: https://tdsna.henkel.com/NA/UT/HNAUTTDS.nsf/web/40B3DBCA6D797572882571870000D75F/$File/4011-EN.pdf (accessed on 7 February 2019).
- Duclos, R.; Saiter, J.M.; Grenet, J.; Orecchioni, A.M. Polymorphism of progesterone - Influence of the carrier and of the solid dispersion manufacturing processes. A calorimetric and radiocrystallographic study. J. Therm. Anal. 1991, 37, 1869–1875. [Google Scholar] [CrossRef]
- Wang, F.; Wachter, J.A.; Antosz, F.J.; Berglund, K.A. An Investigation of Solvent-Mediated Polymorphic Transformation of Progesterone Using in Situ Raman Spectroscopy. Org. Proc. Res. Dev. 2000, 4, 391–395. [Google Scholar] [CrossRef]
- Legendre, B.; Feutelais, Y.; Defossemont, G. Importance of heat capacity determination in homogeneous nucleation: Application to progesterone. Thermochim. Acta 2003, 400, 213–219. [Google Scholar] [CrossRef]
- Sarkar, A.; Ragab, D.; Rohani, S. Polymorphism of progesterone: A new approach for the formation of form II and the relative stabilities of form i and form II. Cryst. Growth Des. 2014, 14, 4574–4582. [Google Scholar] [CrossRef]
- Wang, L.; Fang, F.; Ye, C.; Feng, J. Solid-State NMR Characterizations on Phase Structures and Molecular Dynamics of Poly(ethylene-co-vinyl acetate). J. Polym. Sci. Part B Polym. Phys. 2004, 44, 2864–2879. [Google Scholar] [CrossRef]
- Shi, X.M.; Zhang, J.; Jin, J.; Chen, S.J. Non-isothermal crystallization and melting of ethylene-vinyl acetate copolymers with different vinyl acetate contents. Express Polym. Lett. 2008, 2, 623–629. [Google Scholar] [CrossRef]
- Brandstaetter-Kuhnert, M.; Kofler, A. Zur mikroskopischen Identitaetspruefung und zur Polymorphie der Sexualhormone. Microchim. Acta 1959, 47, 847–853. [Google Scholar] [CrossRef]
- Mesley, R.J. The infra-red spectra of steroids in the solid state. Spectrochmica Acta 1966, 22, 889–917. [Google Scholar] [CrossRef]
- Araya-Sibaja, A.M.; Paulino, A.S.; Rauber, G.S.; Maduro Campos, C.E.; Cardoso, S.G.; Monti, G.A.; Heredia, V.; Bianco, I.; Beltrano, D.; Cuffini, S.L. Dissolution properties, solid-state transformation and polymorphic crystallization: Progesterone case study. Pharm. Dev. Technol. 2014, 19, 779–788. [Google Scholar] [CrossRef] [PubMed]
- Helbling, I.M.; Ibarra, J.C.D.; Luna, J.A. The Use of Cellulose Membrane to Eliminate Burst Release from Intravaginal Rings. AAPS J. 2016, 18, 960–971. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Brazel, C.S. On the importance and mechanisms of burst release in matrix-controlled drug delivery systems. J. Control. Release 2001, 73, 121–136. [Google Scholar] [CrossRef]
- Maurin, M.B.; Dittert, L.W.; Hussain, A.A. Mechanism of diffusion of monosubstituted benzoic acids through ethylene-vinyl acetate copolymers. J. Pharm. Sci. 1992, 81, 79–84. [Google Scholar] [CrossRef]
- Dlubek, G.; Lüpke, T.; Stejny, J.; Alam, M.A.; Arnold, M. Local free volume in ethylene-vinyl acetate copolymers: A positron lifetime study. Macromolecules 2000, 33, 990–996. [Google Scholar] [CrossRef]
- Van Laarhoven, J.A.H.; Kruft, M.A.B.; Vromans, H. Effect of supersaturation and crystallization phenomena on the release properties of a controlled release device based on EVA copolymer. J. Control. Release 2002, 82, 309–317. [Google Scholar] [CrossRef]
- Boyd, P.; Major, I.; Wang, W.; McConville, C. Development of disulfiram-loaded vaginal rings for the localised treatment of cervical cancer. Eur. J. Pharm. Biopharm. 2014, 88, 945–953. [Google Scholar] [CrossRef]
- Singer, R.; Mawson, P.; Derby, N.; Rodriguez, A.; Kizima, L.; Menon, R.; Goldman, D.; Kenney, J.; Aravantinou, M.; Seidor, S.; et al. An Intravaginal Ring That Releases the NNRTI MIV-150 Reduces SHIV Transmission in Macaques. Sci. Transl. Med. 2012, 4, 241–250. [Google Scholar] [CrossRef] [Green Version]
- Siepmann, J.; Siepmann, F. Modeling of diffusion-controlled drug delivery. J. Control. Release 2012, 161, 351–362. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Abbreviation | Core | Membrane | Cs Is Equilibrium Solubility | Cs is Solubility after Thermal Treatment | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Polymer Type | P4 Loading (wt.%) | Polymer Type | Thickness (µm) | P4 Release Rate (µg/day) | Fraction Released over 28 Days (%) | Remaining P4 Concentration in the Core after 28 Days (wt.%) | P4 release Rate (µg/day) | Fraction Released over 28 Days (%) | Remaining P4 Concentration in the Core after 28 Days (wt.%) | |
| V1_9_100 | EVA28 | 16 | EVA9 | 100 | 273 | 17.0 | 13.3 | 796 | 49.5 | 8.1 |
| V1_18_100 | EVA28 | 16 | EVA18 | 100 | 704 | 43.8 | 9.0 | 2054 | >100 | n.a. |
| V1_18_200 | EVA28 | 16 | EVA18 | 200 | 352 | 21.9 | 12.5 | 1027 | 63.9 | 5.8 |
| V2_18_100 | EVA40 | 32 | EVA18 | 100 | 704 | 20.8 | 25.3 | 1185 | 35.0 | 20.1 |
| V2_28_200 | EVA40 | 32 | EVA28 | 200 | 802 | 23.7 | 24.4 | 1350 | 39.9 | 19.2 |
| V2_28_300 | EVA40 | 32 | EVA28 | 300 | 534 | 15.8 | 26.9 | 899 | 26.6 | 23.5 |
| Abbreviation | Average Experimental Daily P4 Release (µg/day) * (S.D.) | Predicted Daily P4 Release (µg/Day) ** | Deviation Experimental from Predicted (%) | P4 Fraction Released over 28 Days (% of Loading) |
|---|---|---|---|---|
| V1_9_100 | 289.8 (47.5) | 273.2 | +6.1 | 20.6 |
| V1_18_100 | 440.8 (163.7) | 698.4 | -36.9 | 31.4 |
| V1_18_200 | 343.1 (56.5) | 349.9 | -2.0 | 23.5 |
| V2_18_100 | 822.6 (159.9) | 767.7 | +7.2 | 26.4 |
| V2_28_200 | 710.2 (95.0) | 790.9 | -10.2 | 22.2 |
| V2_28_300 | 543.7 (32.0) | 522.0 | +4.2 | 17.1 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koutsamanis, I.; Paudel, A.; Nickisch, K.; Eggenreich, K.; Roblegg, E.; Eder, S. Controlled-Release from High-Loaded Reservoir-Type Systems—A Case Study of Ethylene-Vinyl Acetate and Progesterone. Pharmaceutics 2020, 12, 103. https://doi.org/10.3390/pharmaceutics12020103
Koutsamanis I, Paudel A, Nickisch K, Eggenreich K, Roblegg E, Eder S. Controlled-Release from High-Loaded Reservoir-Type Systems—A Case Study of Ethylene-Vinyl Acetate and Progesterone. Pharmaceutics. 2020; 12(2):103. https://doi.org/10.3390/pharmaceutics12020103
Chicago/Turabian StyleKoutsamanis, Ioannis, Amrit Paudel, Klaus Nickisch, Karin Eggenreich, Eva Roblegg, and Simone Eder. 2020. "Controlled-Release from High-Loaded Reservoir-Type Systems—A Case Study of Ethylene-Vinyl Acetate and Progesterone" Pharmaceutics 12, no. 2: 103. https://doi.org/10.3390/pharmaceutics12020103