Opportunities and Challenges in the Delivery of mRNA-Based Vaccines






Abstract
:1. Introduction
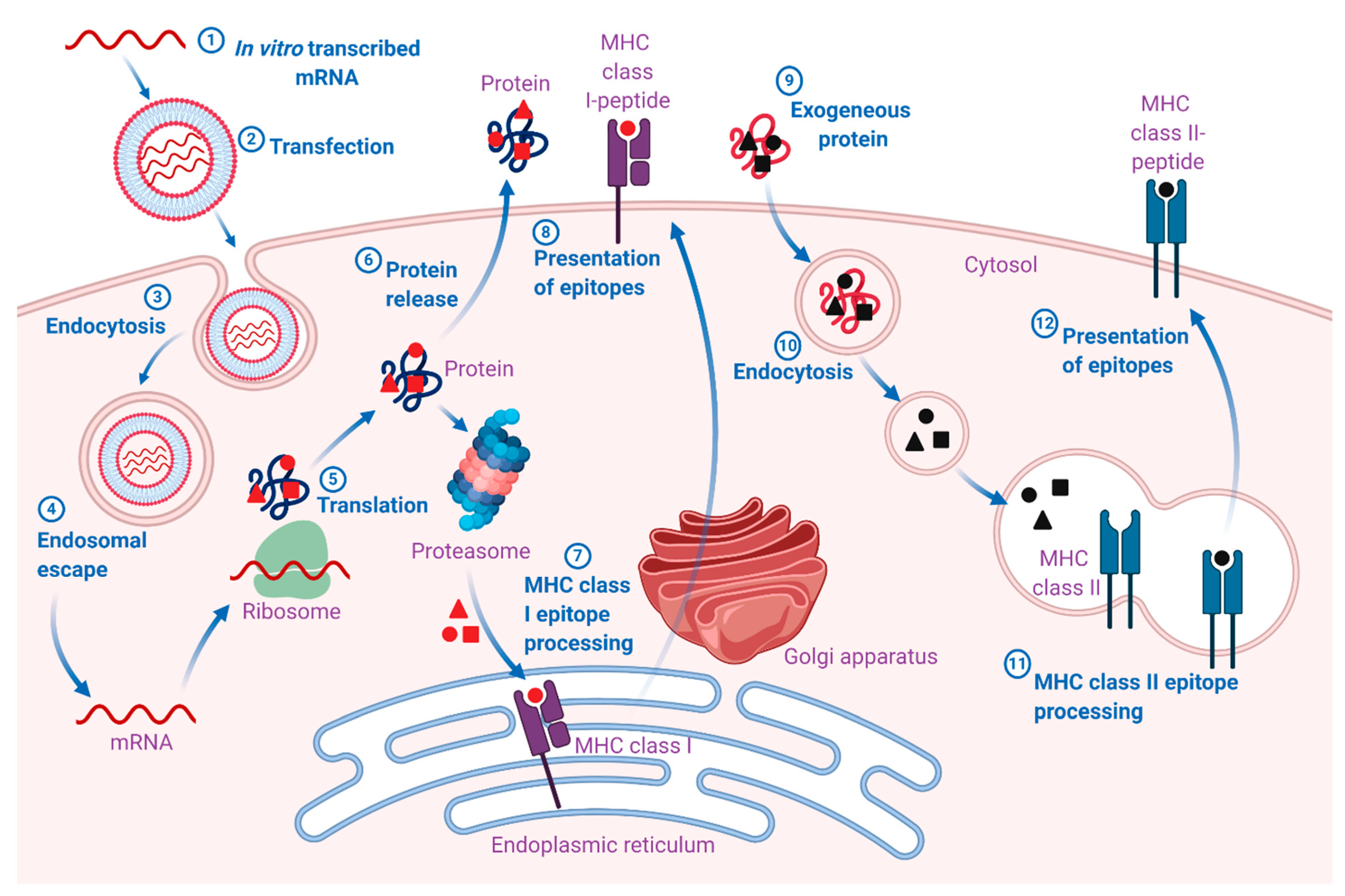
2. mRNA as Vaccines
3. Fundamental Pharmacology of mRNA Vaccines
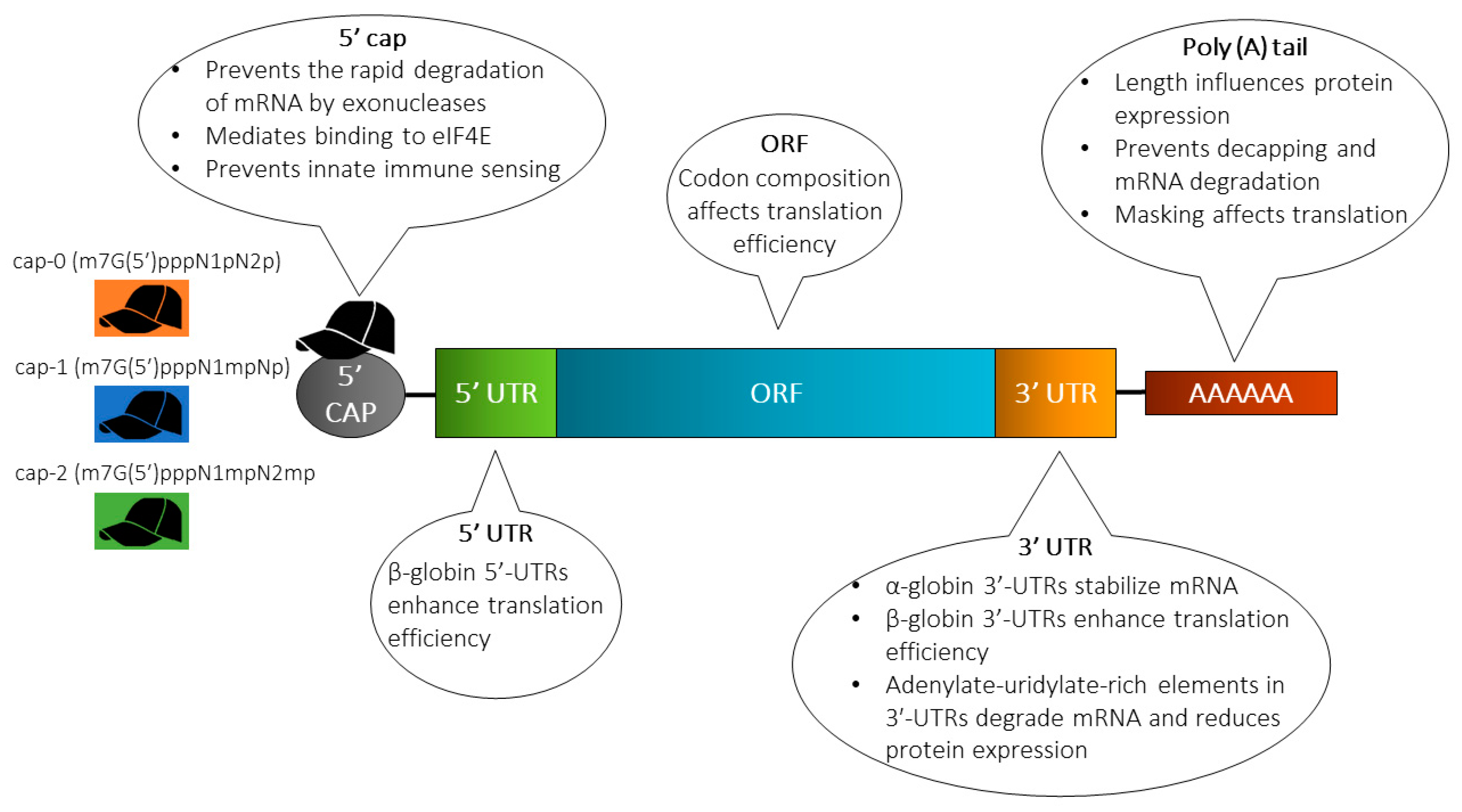
4. Approaches for Enhancing mRNA Stability
4.1. Molecular Stabilization
4.1.1. Cap Analog
4.1.2. 5’ and 3’ Untranslated Regions (UTRs)
4.1.3. Poly(A) Tail
4.2. Formulation Strategies
4.2.1. Naked RNA
4.2.2. Viral Vectors
4.2.3. Polymer-Based Vectors
4.2.4. Lipid-Based Vectors
4.2.5. Lipid-Polymer Hybrid Nanoparticles
4.2.6. Peptide-Based Vectors
4.3. Cell-Specific mRNA Delivery
5. Conclusions and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bragazzi, N.L.; Gianfredi, V.; Villarini, M.; Rosselli, R.; Nasr, A.; Hussein, A.; Martini, M.; Behzadifar, M. Vaccines Meet Big Data: State-of-the-Art and Future Prospects. From the Classical 3Is (“Isolate-Inactivate-Inject”) Vaccinology 1.0 to Vaccinology 3.0, Vaccinomics, and Beyond: A Historical Overview. Front. Pub. Health 2018, 6, 62. [Google Scholar] [CrossRef] [PubMed]
- Chandler, M.; Johnson, M.B.; Panigaj, M.; Afonin, K.A. Innate immune responses triggered by nucleic acids inspire the design of immunomodulatory nucleic acid nanoparticles (NANPs). Curr. Opin. Biotechnol. 2020, 63, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Pardi, N.; Hogan, M.J.; Porter, F.W.; Weissman, D. mRNA vaccines - a new era in vaccinology. Nat. Rev. Drug Discov. 2018, 17, 261–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jahanafrooz, Z.; Baradaran, B.; Mosafer, J.; Hashemzaei, M.; Rezaei, T.; Mokhtarzadeh, A.; Hamblin, M.R. Comparison of DNA and mRNA vaccines against cancer. Drug Discov. Today 2019. [Google Scholar] [CrossRef]
- Iavarone, C.; O’Hagan, D.T.; Yu, D.; Delahaye, N.F.; Ulmer, J.B. Mechanism of action of mRNA-based vaccines. Expert Rev. Vaccines 2017, 16, 871–881. [Google Scholar] [CrossRef]
- Iglesias-Lopez, C.; Agusti, A.; Obach, M.; Vallano, A. Regulatory Framework for Advanced Therapy Medicinal Products in Europe and United States. Front. Pharmacol. 2019, 10, 921. [Google Scholar] [CrossRef] [Green Version]
- Maruggi, G.; Zhang, C.; Li, J.; Ulmer, J.B.; Yu, D. mRNA as a Transformative Technology for Vaccine Development to Control Infectious Diseases. Mol. Ther. 2019, 27, 757–772. [Google Scholar] [CrossRef] [Green Version]
- Hajj, K.A.; Whitehead, K.A. Tools for translation: Non-viral materials for therapeutic mRNA delivery. Nat. Rev. Mat. 2017, 2, 1–17. [Google Scholar] [CrossRef]
- Lin, Y.; Li, Z.; Wang, T.; Wang, X.; Wang, L.; Dong, W.; Jing, C.; Yang, X. MAR characteristic motifs mediate episomal vector in CHO cells. Gene 2015, 559, 137–143. [Google Scholar] [CrossRef]
- Ramamoorth, M.; Narvekar, A. Non viral vectors in gene therapy- an overview. J. Clin. Diagn. Res. 2015, 9, GE01-06. [Google Scholar] [CrossRef]
- Zhong, Z.; Mc Cafferty, S.; Combes, F.; Huysmans, H.; De Temmerman, J.; Gitsels, A.; Vanrompay, D.; Portela Catani, J.; Sanders, N.N. mRNA therapeutics deliver a hopeful message. Nano. Today 2018, 23, 16–39. [Google Scholar] [CrossRef]
- Yamamoto, A.; Kormann, M.; Rosenecker, J.; Rudolph, C. Current prospects for mRNA gene delivery. Eur. J. Pharm. Biopharm. 2009, 71, 484–489. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Hagedorn, C.; Schulz, E.; Lipps, H.J.; Ehrhardt, A. Viral hybrid-vectors for delivery of autonomous replicons. Curr. Gene Ther. 2014, 14, 10–23. [Google Scholar] [CrossRef] [PubMed]
- Vogel, A.B.; Lambert, L.; Kinnear, E.; Busse, D.; Erbar, S.; Reuter, K.C.; Wicke, L.; Perkovic, M.; Beissert, T.; Haas, H.; et al. Self-Amplifying RNA Vaccines Give Equivalent Protection against Influenza to mRNA Vaccines but at Much Lower Doses. Mol. Ther. 2018, 26, 446–455. [Google Scholar] [CrossRef] [Green Version]
- Lundstrom, K. Self-Replicating RNA Viruses for RNA Therapeutics. Molecules 2018, 23. [Google Scholar] [CrossRef] [Green Version]
- Van Hoecke, L.; Roose, K. How mRNA therapeutics are entering the monoclonal antibody field. J. Transl. Med. 2019, 17, 54. [Google Scholar] [CrossRef] [Green Version]
- Sahin, U.; Kariko, K.; Tureci, O. mRNA-based therapeutics--developing a new class of drugs. Nat. Rev. Drug Discov. 2014, 13, 759–780. [Google Scholar] [CrossRef]
- Androulla, M.N.; Lefkothea, P.C. In Vitro-Transcribed (IVT)-mRNA CAR Therapy Development. Methods Mol. Biol. 2020, 2086, 87–117. [Google Scholar] [CrossRef]
- Reichmuth, A.M.; Oberli, M.A.; Jaklenec, A.; Langer, R.; Blankschtein, D. mRNA vaccine delivery using lipid nanoparticles. Ther. Deliv. 2016, 7, 319–334. [Google Scholar] [CrossRef] [Green Version]
- Schlake, T.; Thess, A.; Thran, M.; Jordan, I. mRNA as novel technology for passive immunotherapy. Cell Mol. Life Sci. 2019, 76, 301–328. [Google Scholar] [CrossRef] [Green Version]
- Kowalski, P.S.; Rudra, A.; Miao, L.; Anderson, D.G. Delivering the Messenger: Advances in Technologies for Therapeutic mRNA Delivery. Mol. Ther. 2019, 27, 710–728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohn, L.; Delamarre, L. Dendritic cell-targeted vaccines. Front Immunol. 2014, 5, 255. [Google Scholar] [CrossRef] [PubMed]
- Cu, Y.; Broderick, K.E.; Banerjee, K.; Hickman, J.; Otten, G.; Barnett, S.; Kichaev, G.; Sardesai, N.Y.; Ulmer, J.B.; Geall, A. Enhanced Delivery and Potency of Self-Amplifying mRNA Vaccines by Electroporation in Situ. Vaccines 2013, 1, 367–383. [Google Scholar] [CrossRef] [PubMed]
- Wolff, J.A.; Malone, R.W.; Williams, P.; Chong, W.; Acsadi, G.; Jani, A.; Felgner, P.L. Direct gene transfer into mouse muscle in vivo. Science 1990, 247, 1465–1468. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Krishnan, S.; Lenzen, G.; Magne, R.; Gomard, E.; Guillet, J.G.; Levy, J.P.; Meulien, P. Induction of virus-specific cytotoxic T lymphocytes in vivo by liposome-entrapped mRNA. Eur. J. Immunol. 1993, 23, 1719–1722. [Google Scholar] [CrossRef] [PubMed]
- Kallen, K.J.; Thess, A. A development that may evolve into a revolution in medicine: mRNA as the basis for novel, nucleotide-based vaccines and drugs. Ther. Adv. Vaccines 2014, 2, 10–31. [Google Scholar] [CrossRef] [Green Version]
- Hoerr, I.; Obst, R.; Rammensee, H.G.; Jung, G. In vivo application of RNA leads to induction of specific cytotoxic T lymphocytes and antibodies. Eur. J. Immunol. 2000, 30, 1–7. [Google Scholar] [CrossRef]
- Guan, S.; Rosenecker, J. Nanotechnologies in delivery of mRNA therapeutics using nonviral vector-based delivery systems. Gene Ther. 2017, 24, 133–143. [Google Scholar] [CrossRef]
- Baiersdörfer, M.; Boros, G.; Muramatsu, H.; Mahiny, A.; Vlatkovic, I.; Sahin, U.; Karikó, K. A Facile Method for the Removal of dsRNA Contaminant from In Vitro-Transcribed mRNA. Mol. Ther. Nucleic Acids 2019, 15, 26–35. [Google Scholar] [CrossRef] [Green Version]
- Pardi, N.; Muramatsu, H.; Weissman, D.; Karikó, K. In Vitro Transcription of Long RNA Containing Modified Nucleosides. In Synthetic Messenger RNA and Cell Metabolism Modulation: Methods and Protocols; Rabinovich, P.M., Ed.; Humana Press: Totowa, NJ, USA, 2013; pp. 29–42. [Google Scholar]
- Weissman, D.; Pardi, N.; Muramatsu, H.; Karikó, K. HPLC Purification of In Vitro Transcribed Long RNA. In Synthetic Messenger RNA and Cell Metabolism Modulation: Methods and Protocols; Rabinovich, P.M., Ed.; Humana Press: Totowa, NJ, USA, 2013; pp. 43–54. [Google Scholar]
- Li, Y.; Kiledjian, M. Regulation of mRNA decapping. Wiley Interdiscip. Rev. RNA 2010, 1, 253–265. [Google Scholar] [CrossRef]
- Cowling, V.H.; Cole, M.D. Myc Regulation of mRNA Cap Methylation. Genes Cancer 2010, 1, 576–579. [Google Scholar] [CrossRef] [PubMed]
- Roers, A.; Hiller, B.; Hornung, V. Recognition of Endogenous Nucleic Acids by the Innate Immune System. Immunity 2016, 44, 739–754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wojtczak, B.A.; Sikorski, P.J.; Fac-Dabrowska, K.; Nowicka, A.; Warminski, M.; Kubacka, D.; Nowak, E.; Nowotny, M.; Kowalska, J.; Jemielity, J. 5’-Phosphorothiolate Dinucleotide Cap Analogues: Reagents for Messenger RNA Modification and Potent Small-Molecular Inhibitors of Decapping Enzymes. J. Am. Chem. Soc. 2018, 140, 5987–5999. [Google Scholar] [CrossRef] [PubMed]
- Ramanathan, A.; Robb, G.B.; Chan, S.H. mRNA capping: Biological functions and applications. Nucleic. Acids Res. 2016, 44, 7511–7526. [Google Scholar] [CrossRef]
- Zimmermann, O.; Homann, J.M.; Bangert, A.; Müller, A.M.; Hristov, G.; Goeser, S.; Wiehe, J.M.; Zittrich, S.; Rottbauer, W.; Torzewski, J.; et al. Successful use of mRNA-nucleofection for overexpression of interleukin-10 in murine monocytes/macrophages for anti-inflammatory therapy in a murine model of autoimmune myocarditis. J. Am. Heart Association 2012. [Google Scholar] [CrossRef] [Green Version]
- Hamm, J.; Mattaj, I.W. Monomethylated cap structures facilitate RNA export from the nucleus. Cell 1990, 63, 109–118. [Google Scholar] [CrossRef]
- Rydzik, A.M.; Warminski, M.; Sikorski, P.J.; Baranowski, M.R.; Walczak, S.; Kowalska, J.; Zuberek, J.; Lukaszewicz, M.; Nowak, E.; TD, W.C.; et al. mRNA cap analogues substituted in the tetraphosphate chain with CX2: Identification of O-to-CCl2 as the first bridging modification that confers resistance to decapping without impairing translation. Nucleic Acids Res. 2017, 45, 8661–8675. [Google Scholar] [CrossRef] [Green Version]
- Kariko, K.; Muramatsu, H.; Welsh, F.A.; Ludwig, J.; Kato, H.; Akira, S.; Weissman, D. Incorporation of pseudouridine into mRNA yields superior nonimmunogenic vector with increased translational capacity and biological stability. Mol. Ther. 2008, 16, 1833–1840. [Google Scholar] [CrossRef]
- Kocmik, I.; Piecyk, K.; Rudzinska, M.; Niedzwiecka, A.; Darzynkiewicz, E.; Grzela, R.; Jankowska-Anyszka, M. Modified ARCA analogs providing enhanced translational properties of capped mRNAs. Cell Cycle 2018, 17, 1624–1636. [Google Scholar] [CrossRef] [Green Version]
- Kiriakidou, M.; Tan, G.S.; Lamprinaki, S.; De Planell-Saguer, M.; Nelson, P.T.; Mourelatos, Z. An mRNA m7G cap binding-like motif within human Ago2 represses translation. Cell 2007, 129, 1141–1151. [Google Scholar] [CrossRef] [Green Version]
- Jemielity, J.; Fowler, T.; Zuberek, J.; Stepinski, J.; Lewdorowicz, M.; Niedzwiecka, A.; Stolarski, R.; Darzynkiewicz, E.; Rhoads, R.E. Novel “anti-reverse” cap analogs with superior translational properties. RNA 2003, 9, 1108–1122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grudzien-Nogalska, E.; Jemielity, J.; Kowalska, J.; Darzynkiewicz, E.; Rhoads, R.E. Phosphorothioate cap analogs stabilize mRNA and increase translational efficiency in mammalian cells. RNA 2007, 13, 1745–1755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ziemniak, M.; Kowalska, J.; Lukaszewicz, M.; Zuberek, J.; Wnek, K.; Darzynkiewicz, E.; Jemielity, J. Phosphate-modified analogues of m(7)GTP and m(7)Gppppm(7)G-Synthesis and biochemical properties. Bioorg. Med. Chem. 2015, 23, 5369–5381. [Google Scholar] [CrossRef] [PubMed]
- Kore, A.R.; Shanmugasundaram, M.; Charles, I.; Vlassov, A.V.; Barta, T.J. Locked nucleic acid (LNA)-modified dinucleotide mRNA cap analogue: Synthesis, enzymatic incorporation, and utilization. J. Am. Chem. Soc. 2009, 131, 6364–6365. [Google Scholar] [CrossRef] [PubMed]
- Kowalska, J.; Wypijewska del Nogal, A.; Darzynkiewicz, Z.M.; Buck, J.; Nicola, C.; Kuhn, A.N.; Lukaszewicz, M.; Zuberek, J.; Strenkowska, M.; Ziemniak, M.; et al. Synthesis, properties, and biological activity of boranophosphate analogs of the mRNA cap: Versatile tools for manipulation of therapeutically relevant cap-dependent processes. Nucleic Acids Res. 2014, 42, 10245–10264. [Google Scholar] [CrossRef] [Green Version]
- Grudzien-Nogalska, E.; Kiledjian, M. New insights into decapping enzymes and selective mRNA decay. Wiley Interdiscip Rev RNA 2017, 8. [Google Scholar] [CrossRef] [Green Version]
- Shanmugasundaram, M.; Charles, I.; Kore, A.R. Design, synthesis and biological evaluation of dinucleotide mRNA cap analog containing propargyl moiety. Bioorg. Med. Chem. 2016, 24, 1204–1208. [Google Scholar] [CrossRef]
- Yunus, M.A.; Chung, L.M.; Chaudhry, Y.; Bailey, D.; Goodfellow, I. Development of an optimized RNA-based murine norovirus reverse genetics system. J. Virol. Methods 2010, 169, 112–118. [Google Scholar] [CrossRef] [Green Version]
- Devoldere, J.; Dewitte, H.; De Smedt, S.C.; Remaut, K. Evading innate immunity in nonviral mRNA delivery: don’t shoot the messenger. Drug Discov. Today 2016, 21, 11–25. [Google Scholar] [CrossRef] [Green Version]
- Vaidyanathan, S.; Azizian, K.T.; Haque, A.; Henderson, J.M.; Hendel, A.; Shore, S.; Antony, J.S.; Hogrefe, R.I.; Kormann, M.S.D.; Porteus, M.H.; et al. Uridine Depletion and Chemical Modification Increase Cas9 mRNA Activity and Reduce Immunogenicity without HPLC Purification. Mol. Ther. Nucleic Acids 2018, 12, 530–542. [Google Scholar] [CrossRef]
- Muttach, F.; Muthmann, N.; Rentmeister, A. Synthetic mRNA capping. Beilstein J. Org. Chem. 2017, 13, 2819–2832. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.; Azizian, K.T.; Henderson, J.M.; Lebedev, A.; Hogrefe, R.I.; Houston, M.; McCaffrey, A.P.; BioTechnologies, T. Exploring the Messenger RNA Capping Code: CleanCap Co-Transcriptional Capping Allows Synthesis of Cap 0, Cap 1, Cap 2 and (M6) A (M) Capped RNAs; Cell Press: Cambridge, MA, USA, 2018; p. 91. [Google Scholar]
- Diebold, S.S.; Kaisho, T.; Hemmi, H.; Akira, S.; Reis e Sousa, C. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science 2004, 303, 1529–1531. [Google Scholar] [CrossRef] [PubMed]
- Suknuntha, K.; Tao, L.; Brok-Volchanskaya, V.; D’Souza, S.S.; Kumar, A.; Slukvin, I. Optimization of Synthetic mRNA for Highly Efficient Translation and its Application in the Generation of Endothelial and Hematopoietic Cells from Human and Primate Pluripotent Stem Cells. Stem Cell Rev. Rep. 2018, 14, 525–534. [Google Scholar] [CrossRef] [PubMed]
- Creusot, R.J.; Chang, P.; Healey, D.G.; Tcherepanova, I.Y.; Nicolette, C.A.; Fathman, C.G. A short pulse of IL-4 delivered by DCs electroporated with modified mRNA can both prevent and treat autoimmune diabetes in NOD mice. Mol. Ther. 2010, 18, 2112–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asrani, K.H.; Farelli, J.D.; Stahley, M.R.; Miller, R.L.; Cheng, C.J.; Subramanian, R.R.; Brown, J.M. Optimization of mRNA untranslated regions for improved expression of therapeutic mRNA. RNA Biol. 2018, 15, 756–762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adibzadeh, S.; Fardaei, M.; Takhshid, M.A.; Miri, M.R.; Rafiei Dehbidi, G.; Farhadi, A.; Ranjbaran, R.; Alavi, P.; Nikouyan, N.; Seyyedi, N.; et al. Enhancing Stability of Destabilized Green Fluorescent Protein Using Chimeric mRNA Containing Human Beta-Globin 5’ and 3’ Untranslated Regions. Avicenna J. Med. Biotechnol. 2019, 11, 112–117. [Google Scholar]
- Zhao, Y.; Moon, E.; Carpenito, C.; Paulos, C.M.; Liu, X.; Brennan, A.L.; Chew, A.; Carroll, R.G.; Scholler, J.; Levine, B.L.; et al. Multiple injections of electroporated autologous T cells expressing a chimeric antigen receptor mediate regression of human disseminated tumor. Cancer Res. 2010, 70, 9053–9061. [Google Scholar] [CrossRef] [Green Version]
- Kreiter, S.; Selmi, A.; Diken, M.; Koslowski, M.; Britten, C.M.; Huber, C.; Tureci, O.; Sahin, U. Intranodal vaccination with naked antigen-encoding RNA elicits potent prophylactic and therapeutic antitumoral immunity. Cancer Res. 2010, 70, 9031–9040. [Google Scholar] [CrossRef] [Green Version]
- Kreiter, S.; Selmi, A.; Diken, M.; Sebastian, M.; Osterloh, P.; Schild, H.; Huber, C.; Tureci, O.; Sahin, U. Increased antigen presentation efficiency by coupling antigens to MHC class I trafficking signals. J. Immunol. 2008, 180, 309–318. [Google Scholar] [CrossRef] [Green Version]
- Fotin-Mleczek, M.; Duchardt, K.M.; Lorenz, C.; Pfeiffer, R.; Ojkic-Zrna, S.; Probst, J.; Kallen, K.J. Messenger RNA-based vaccines with dual activity induce balanced TLR-7 dependent adaptive immune responses and provide antitumor activity. J. Immunother. 2011, 34, 1–15. [Google Scholar] [CrossRef]
- Goss, D.J.; Kleiman, F.E. Poly(A) binding proteins: Are they all created equal? Wiley Interdiscip. Rev. RNA 2013, 4, 167–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hershey, J.W. Regulation of protein synthesis and the role of eIF3 in cancer. Braz. J. Med. Biol. Res. 2010, 43, 920–930. [Google Scholar] [CrossRef] [Green Version]
- Anderson, B.R.; Muramatsu, H.; Nallagatla, S.R.; Bevilacqua, P.C.; Sansing, L.H.; Weissman, D.; Kariko, K. Incorporation of pseudouridine into mRNA enhances translation by diminishing PKR activation. Nucleic Acids Res. 2010, 38, 5884–5892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munroe, D.; Jacobson, A. mRNA poly (A) tail, a 3’enhancer of translational initiation. Mol. Cell. Biol. 1990, 10, 3441–3455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kormann, M.S.; Hasenpusch, G.; Aneja, M.K.; Nica, G.; Flemmer, A.W.; Herber-Jonat, S.; Huppmann, M.; Mays, L.E.; Illenyi, M.; Schams, A.; et al. Expression of therapeutic proteins after delivery of chemically modified mRNA in mice. Nat. Biotechnol. 2011, 29, 154–157. [Google Scholar] [CrossRef]
- Holtkamp, S.; Kreiter, S.; Selmi, A.; Simon, P.; Koslowski, M.; Huber, C.; Tureci, O.; Sahin, U. Modification of antigen-encoding RNA increases stability, translational efficacy, and T-cell stimulatory capacity of dendritic cells. Blood 2006, 108, 4009–4017. [Google Scholar] [CrossRef]
- Coller, J.; Parker, R. Eukaryotic mRNA decapping. Annu. Rev. Biochem. 2004, 73, 861–890. [Google Scholar] [CrossRef] [Green Version]
- Mugridge, J.S.; Coller, J.; Gross, J.D. Structural and molecular mechanisms for the control of eukaryotic 5’-3’ mRNA decay. Nat. Struct. Mol. Biol. 2018, 25, 1077–1085. [Google Scholar] [CrossRef]
- Sharova, L.V.; Sharov, A.A.; Nedorezov, T.; Piao, Y.; Shaik, N.; Ko, M.S.H. Database for mRNA half-life of 19 977 genes obtained by DNA microarray analysis of pluripotent and differentiating mouse embryonic stem cells. DNA Res. 2008, 16, 45–58. [Google Scholar] [CrossRef] [Green Version]
- Houseley, J.; Tollervey, D. The many pathways of RNA degradation. Cell 2009, 136, 763–776. [Google Scholar] [CrossRef] [Green Version]
- Deering, R.P.; Kommareddy, S.; Ulmer, J.B.; Brito, L.A.; Geall, A.J. Nucleic acid vaccines: Prospects for non-viral delivery of mRNA vaccines. Expert Opin. Drug Deliv. 2014, 11, 885–899. [Google Scholar] [CrossRef] [PubMed]
- Golombek, S.; Pilz, M.; Steinle, H.; Kochba, E.; Levin, Y.; Lunter, D.; Schlensak, C.; Wendel, H.P.; Avci-Adali, M. Intradermal Delivery of Synthetic mRNA Using Hollow Microneedles for Efficient and Rapid Production of Exogenous Proteins in Skin. Mol. Ther. Nucleic Acids 2018, 11, 382–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koh, K.J.; Liu, Y.; Lim, S.H.; Loh, X.J.; Kang, L.; Lim, C.Y.; Phua, K.K.L. Formulation, characterization and evaluation of mRNA-loaded dissolvable polymeric microneedles (RNApatch). Sci. Rep. 2018, 8, 11842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tavernier, G.; Andries, O.; Demeester, J.; Sanders, N.N.; De Smedt, S.C.; Rejman, J. mRNA as gene therapeutic: How to control protein expression. J. Control. Release 2011, 150, 238–247. [Google Scholar] [CrossRef]
- Zhang, R.; Men, K.; Zhang, X.; Huang, R.; Tian, Y.; Zhou, B.; Yu, C.; Wang, Y.; Ji, X.; Hu, Q.; et al. Delivery of a Modified mRNA Encoding IL-22 Binding Protein (IL-22BP) for Colon Cancer Gene Therapy. J. Biomed. Nanotechnol. 2018, 14, 1239–1251. [Google Scholar] [CrossRef]
- Schlake, T.; Thess, A.; Fotin-Mleczek, M.; Kallen, K.J. Developing mRNA-vaccine technologies. RNA Biol. 2012, 9, 1319–1330. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, A.; Waters, A.K.; Kalyan, P.; Achrol, A.S.; Kesari, S.; Yenugonda, V.M. Lipid-polymer hybrid nanoparticles as a next-generation drug delivery platform: State of the art, emerging technologies, and perspectives. Int. J. Nanomed. 2019, 14, 1937–1952. [Google Scholar] [CrossRef] [Green Version]
- Sahin, U.; Karikó, K.; Türeci, Ö. mRNA-based therapeutics — developing a new class of drugs. Nat. Pub. Gr. 2014. [Google Scholar] [CrossRef]
- Takahashi, M.; Elbarbary, R.A.; Nakashima, A.; Abe, M.; Watanabe, N.; Narita, M.; Takahashi, M.; Tamura, M.; Yoshida, T.; Nashimoto, M. A naked RNA heptamer targeting the human Bcl-2 mRNA induces apoptosis of HL60 leukemia cells. Cancer Lett. 2013, 328, 362–368. [Google Scholar] [CrossRef]
- Weide, B.; Carralot, J.P.; Reese, A.; Scheel, B.; Eigentler, T.K.; Hoerr, I.; Rammensee, H.G.; Garbe, C.; Pascolo, S. Results of the first phase I/II clinical vaccination trial with direct injection of mRNA. J. Immunother. 2008, 31, 180–188. [Google Scholar] [CrossRef]
- Lorenz, C.; Fotin-Mleczek, M.; Roth, G.; Becker, C.; Dam, T.C.; Verdurmen, W.P.; Brock, R.; Probst, J.; Schlake, T. Protein expression from exogenous mRNA: Uptake by receptor-mediated endocytosis and trafficking via the lysosomal pathway. RNA Biol. 2011, 8, 627–636. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.Y.; Kim, M.K.; Kim-Han, T.H.; Indik, Z.K.; Schreiber, A.D. Effect of locally administered Syk siRNA on allergen-induced arthritis and asthma. Mol. Immunol. 2013, 53, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Pardi, N.; Tuyishime, S.; Muramatsu, H.; Kariko, K.; Mui, B.L.; Tam, Y.K.; Madden, T.D.; Hope, M.J.; Weissman, D. Expression kinetics of nucleoside-modified mRNA delivered in lipid nanoparticles to mice by various routes. J. Control. Release 2015, 217, 345–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Maruggi, G.; Shan, H.; Li, J. Advances in mRNA Vaccines for Infectious Diseases. Front. Immunol. 2019, 10, 594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pardi, N.; Hogan, M.J.; Pelc, R.S.; Muramatsu, H.; Andersen, H.; DeMaso, C.R.; Dowd, K.A.; Sutherland, L.L.; Scearce, R.M.; Parks, R.; et al. Zika virus protection by a single low-dose nucleoside-modified mRNA vaccination. Nature 2017, 543, 248–251. [Google Scholar] [CrossRef] [PubMed]
- Zakrewsky, M.; Kumar, S.; Mitragotri, S. Nucleic acid delivery into skin for the treatment of skin disease: Proofs-of-concept, potential impact, and remaining challenges. J. Control. Release 2015, 219, 445–456. [Google Scholar] [CrossRef] [Green Version]
- Phua, K.K.; Leong, K.W.; Nair, S.K. Transfection efficiency and transgene expression kinetics of mRNA delivered in naked and nanoparticle format. J. Control. Release 2013, 166, 227–233. [Google Scholar] [CrossRef] [Green Version]
- Van Lint, S.; Goyvaerts, C.; Maenhout, S.; Goethals, L.; Disy, A.; Benteyn, D.; Pen, J.; Bonehill, A.; Heirman, C.; Breckpot, K.; et al. Preclinical evaluation of TriMix and antigen mRNA-based antitumor therapy. Cancer Res. 2012, 72, 1661–1671. [Google Scholar] [CrossRef] [Green Version]
- Probst, J.; Weide, B.; Scheel, B.; Pichler, B.J.; Hoerr, I.; Rammensee, H.G.; Pascolo, S. Spontaneous cellular uptake of exogenous messenger RNA in vivo is nucleic acid-specific, saturable and ion dependent. Gene Ther. 2007, 14, 1175–1180. [Google Scholar] [CrossRef] [Green Version]
- Sultana, N.; Magadum, A.; Hadas, Y.; Kondrat, J.; Singh, N.; Youssef, E.; Calderon, D.; Chepurko, E.; Dubois, N.; Hajjar, R.J.; et al. Optimizing Cardiac Delivery of Modified mRNA. Mol. Ther. 2017, 25, 1306–1315. [Google Scholar] [CrossRef] [Green Version]
- McNamara, M.A.; Nair, S.K.; Holl, E.K. RNA-Based Vaccines in Cancer Immunotherapy. J. Immunol. Res. 2015, 2015, 794528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selmi, A.; Vascotto, F.; Kautz-Neu, K.; Tureci, O.; Sahin, U.; von Stebut, E.; Diken, M.; Kreiter, S. Uptake of synthetic naked RNA by skin-resident dendritic cells via macropinocytosis allows antigen expression and induction of T-cell responses in mice. Cancer Immunol. Immunother. 2016, 65, 1075–1083. [Google Scholar] [CrossRef] [PubMed]
- De Beuckelaer, A.; Grooten, J.; De Koker, S. Type I Interferons Modulate CD8(+) T Cell Immunity to mRNA Vaccines. Trends Mol. Med. 2017, 23, 216–226. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, N.; Ganguli-Indra, G.; Indra, A.K. Transcriptional control and transcriptomic analysis of lipid metabolism in skin barrier formation and atopic dermatitis (AD). Expert Rev. Proteomics 2019, 16, 627–645. [Google Scholar] [CrossRef]
- Ahlemeyer, B.; Vogt, J.F.; Michel, V.; Hahn-Kohlberger, P.; Baumgart-Vogt, E. Microporation is an efficient method for siRNA-induced knockdown of PEX5 in HepG2 cells: Evaluation of the transfection efficiency, the PEX5 mRNA and protein levels and induction of peroxisomal deficiency. Histochem. Cell Biol. 2014, 142, 577–591. [Google Scholar] [CrossRef]
- Chong, R.H.; Gonzalez-Gonzalez, E.; Lara, M.F.; Speaker, T.J.; Contag, C.H.; Kaspar, R.L.; Coulman, S.A.; Hargest, R.; Birchall, J.C. Gene silencing following siRNA delivery to skin via coated steel microneedles: In vitro and in vivo proof-of-concept. J. Control. Release 2013, 166, 211–219. [Google Scholar] [CrossRef] [Green Version]
- Walther, W.; Stein, U.; Fichtner, I.; Kobelt, D.; Aumann, J.; Arlt, F.; Schlag, P.M. Nonviral jet-injection gene transfer for efficient in vivo cytosine deaminase suicide gene therapy of colon carcinoma. Mol. Ther. 2005, 12, 1176–1184. [Google Scholar] [CrossRef]
- Villemejane, J.; Mir, L.M. Physical methods of nucleic acid transfer: General concepts and applications. Br J. Pharmacol. 2009, 157, 207–219. [Google Scholar] [CrossRef] [Green Version]
- McLenachan, S.; Zhang, D.; Palomo, A.B.; Edel, M.J.; Chen, F.K. mRNA transfection of mouse and human neural stem cell cultures. PLoS ONE 2013, 8, e83596. [Google Scholar] [CrossRef] [Green Version]
- Kigasawa, K.; Kajimoto, K.; Hama, S.; Saito, A.; Kanamura, K.; Kogure, K. Noninvasive delivery of siRNA into the epidermis by iontophoresis using an atopic dermatitis-like model rat. Int. J. Pharm. 2010, 383, 157–160. [Google Scholar] [CrossRef]
- Ryu, Y.C.; Kim, D.I.; Kim, S.H.; Wang, H.-M.D.; Hwang, B.H. Synergistic Transdermal Delivery of Biomacromolecules Using Sonophoresis after Microneedle Treatment. Biotechnol. Bioprocess Eng. 2018, 23, 286–292. [Google Scholar] [CrossRef]
- Su, X.; Fricke, J.; Kavanagh, D.G.; Irvine, D.J. In vitro and in vivo mRNA delivery using lipid-enveloped pH-responsive polymer nanoparticles. Mol. Pharm. 2011, 8, 774–787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michel, T.; Luft, D.; Abraham, M.K.; Reinhardt, S.; Salinas Medina, M.L.; Kurz, J.; Schaller, M.; Avci-Adali, M.; Schlensak, C.; Peter, K.; et al. Cationic Nanoliposomes Meet mRNA: Efficient Delivery of Modified mRNA Using Hemocompatible and Stable Vectors for Therapeutic Applications. Mol. Ther. Nucleic Acids 2017, 8, 459–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dewitte, H.; Van Lint, S.; Heirman, C.; Thielemans, K.; De Smedt, S.C.; Breckpot, K.; Lentacker, I. The potential of antigen and TriMix sonoporation using mRNA-loaded microbubbles for ultrasound-triggered cancer immunotherapy. J. Control. Release 2014, 194, 28–36. [Google Scholar] [CrossRef] [Green Version]
- Gan, L.M.; Lagerstrom-Fermer, M.; Carlsson, L.G.; Arfvidsson, C.; Egnell, A.C.; Rudvik, A.; Kjaer, M.; Collen, A.; Thompson, J.D.; Joyal, J.; et al. Intradermal delivery of modified mRNA encoding VEGF-A in patients with type 2 diabetes. Nat. Commun. 2019, 10, 871. [Google Scholar] [CrossRef]
- Sun, N.; Ning, B.; Hansson, K.M.; Bruce, A.C.; Seaman, S.A.; Zhang, C.; Rikard, M.; DeRosa, C.A.; Fraser, C.L.; Wagberg, M.; et al. Modified VEGF-A mRNA induces sustained multifaceted microvascular response and accelerates diabetic wound healing. Sci. Rep. 2018, 8, 17509. [Google Scholar] [CrossRef] [Green Version]
- Ita, K. Transdermal Delivery of Drugs with Microneedles-Potential and Challenges. Pharmaceutics 2015, 7, 90–105. [Google Scholar] [CrossRef] [Green Version]
- Trepotec, Z.; Lichtenegger, E.; Plank, C.; Aneja, M.K.; Rudolph, C. Delivery of mRNA Therapeutics for the Treatment of Hepatic Diseases. Mol. Ther. 2019, 27, 794–802. [Google Scholar] [CrossRef] [Green Version]
- Chou, J.Y.; Mansfield, B.C. Recombinant AAV-directed gene therapy for type I glycogen storage diseases. Expert Opin. Biol. Ther. 2011, 11, 1011–1024. [Google Scholar] [CrossRef]
- Ehrengruber, M.U.; Lundstrom, K. Alphaviruses: Semliki Forest Virus and Sindbis Virus Vectors for Gene Transfer into Neurons. Curr. Protoc. Neurosci. 2007, 57, 4.22.21–4.22.27. [Google Scholar] [CrossRef]
- Rozovics, J.M.; Chase, A.J.; Cathcart, A.L.; Chou, W.; Gershon, P.D.; Palusa, S.; Wilusz, J.; Semler, B.L. Picornavirus modification of a host mRNA decay protein. MBio 2012, 3, e00431-12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schott, J.W.; Morgan, M.; Galla, M.; Schambach, A. Viral and Synthetic RNA Vector Technologies and Applications. Mol. Ther. 2016, 24, 1513–1527. [Google Scholar] [CrossRef] [Green Version]
- Strauss, J.H.; Strauss, E.G. The alphaviruses: Gene expression, replication, and evolution. Microbiol. Rev. 1994, 58, 491–562. [Google Scholar] [CrossRef]
- Vanlandingham, D.L.; Tsetsarkin, K.; Hong, C.; Klingler, K.; McElroy, K.L.; Lehane, M.J.; Higgs, S. Development and characterization of a double subgenomic chikungunya virus infectious clone to express heterologous genes in Aedes aegypti mosquitoes. Insect Biochem. Mol. Biol. 2005, 35, 1162–1170. [Google Scholar] [CrossRef] [PubMed]
- Lundstrom, K. RNA Viruses as Tools in Gene Therapy and Vaccine Development. Genes 2019, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lundstrom, K. Alphavirus vectors for vaccine production and gene therapy. Expert Rev. Vaccines 2003, 2, 447–459. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, M.; Otsu, M. Development of Sendai virus vectors and their potential applications in gene therapy and regenerative medicine. Curr. Gene Ther. 2012, 12, 410–416. [Google Scholar] [CrossRef] [Green Version]
- Bounds, C.E.; Terry, F.E.; Moise, L.; Hannaman, D.; Martin, W.D.; De Groot, A.S.; Suschak, J.J.; Dupuy, L.C.; Schmaljohn, C.S. An immunoinformatics-derived DNA vaccine encoding human class II T cell epitopes of Ebola virus, Sudan virus, and Venezuelan equine encephalitis virus is immunogenic in HLA transgenic mice. Hum. Vaccin Immunother. 2017, 13, 2824–2836. [Google Scholar] [CrossRef]
- Taylor, K.; Kolokoltsova, O.; Ronca, S.E.; Estes, M.; Paessler, S. Live, Attenuated Venezuelan Equine Encephalitis Virus Vaccine (TC83) Causes Persistent Brain Infection in Mice with Non-functional alphabeta T-Cells. Front. Microbiol. 2017, 8, 81. [Google Scholar] [CrossRef] [Green Version]
- Tezel, A.; Dokka, S.; Kelly, S.; Hardee, G.E.; Mitragotri, S. Topical delivery of anti-sense oligonucleotides using low-frequency sonophoresis. Pharm. Res. 2004, 21, 2219–2225. [Google Scholar] [CrossRef]
- Koch, G. Interaction of poliovirus-specific RNAs with HeLa cells and E. coli. Curr. Top Microbiol. Immunol. 1973, 62, 89–138. [Google Scholar] [CrossRef] [PubMed]
- Malone, R.W.; Felgner, P.L.; Verma, I.M. Cationic liposome-mediated RNA transfection. Proc. Natl. Acad. Sci. USA 1989, 86, 6077–6081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pollard, C.; Rejman, J.; De Haes, W.; Verrier, B.; Van Gulck, E.; Naessens, T.; De Smedt, S.; Bogaert, P.; Grooten, J.; Vanham, G.; et al. Type I IFN counteracts the induction of antigen-specific immune responses by lipid-based delivery of mRNA vaccines. Mol. Ther. 2013, 21, 251–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ulkoski, D.; Bak, A.; Wilson, J.T.; Krishnamurthy, V.R. Recent advances in polymeric materials for the delivery of RNA therapeutics. Expert Opin. Drug Deliv. 2019, 16, 1149–1167. [Google Scholar] [CrossRef]
- Gary, D.J.; Lee, H.; Sharma, R.; Lee, J.S.; Kim, Y.; Cui, Z.Y.; Jia, D.; Bowman, V.D.; Chipman, P.R.; Wan, L.; et al. Influence of nano-carrier architecture on in vitro siRNA delivery performance and in vivo biodistribution: Polyplexes vs micelleplexes. ACS Nano 2011, 5, 3493–3505. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Guo, C.; Feng, B.; Liu, J.; Chen, X.; Wang, D.; Teng, L.; Li, Y.; Yin, Q.; Zhang, Z.; et al. Triple-Layered pH-Responsive Micelleplexes Loaded with siRNA and Cisplatin Prodrug for NF-Kappa B Targeted Treatment of Metastatic Breast Cancer. Theranostics 2016, 6, 14–27. [Google Scholar] [CrossRef]
- Tros de Ilarduya, C.; Sun, Y.; Duzgunes, N. Gene delivery by lipoplexes and polyplexes. Eur. J. Pharm. Sci. 2010, 40, 159–170. [Google Scholar] [CrossRef]
- Rosenkranz, A.A.; Sobolev, A.S. Polyethylenimine-based polyplex nanoparticles and features of their behavior in cells and tissues. Russian Chem. Bull. 2016, 64, 2749–2755. [Google Scholar] [CrossRef]
- Lungwitz, U.; Breunig, M.; Blunk, T.; Gopferich, A. Polyethylenimine-based non-viral gene delivery systems. Eur. J. Pharm. Biopharm. 2005, 60, 247–266. [Google Scholar] [CrossRef]
- Cheng, C.; Convertine, A.J.; Stayton, P.S.; Bryers, J.D. Multifunctional triblock copolymers for intracellular messenger RNA delivery. Biomaterials 2012, 33, 6868–6876. [Google Scholar] [CrossRef] [Green Version]
- Gary, D.J.; Puri, N.; Won, Y.Y. Polymer-based siRNA delivery: Perspectives on the fundamental and phenomenological distinctions from polymer-based DNA delivery. J. Control. Release 2007, 121, 64–73. [Google Scholar] [CrossRef]
- Lu, Y.; Kawakami, S.; Yamashita, F.; Hashida, M. Development of an antigen-presenting cell-targeted DNA vaccine against melanoma by mannosylated liposomes. Biomaterials 2007, 28, 3255–3262. [Google Scholar] [CrossRef] [PubMed]
- Zohra, F.T.; Chowdhury, E.H.; Tada, S.; Hoshiba, T.; Akaike, T. Effective delivery with enhanced translational activity synergistically accelerates mRNA-based transfection. Biochem. Biophys. Res. Commun. 2007, 358, 373–378. [Google Scholar] [CrossRef] [PubMed]
- Michanek, A.; Kristen, N.; Hook, F.; Nylander, T.; Sparr, E. RNA and DNA interactions with zwitterionic and charged lipid membranes - a DSC and QCM-D study. Biochim. Biophys. Acta Biomembr. 2010, 1798, 829–838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, Z.; Munye, M.M.; Tagalakis, A.D.; Manunta, M.D.; Hart, S.L. The role of the helper lipid on the DNA transfection efficiency of lipopolyplex formulations. Sci. Rep. 2014, 4, 7107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arthur, J.F.; Butterfield, L.H.; Roth, M.D.; Bui, L.A.; Kiertscher, S.M.; Lau, R.; Dubinett, S.; Glaspy, J.; McBride, W.H.; Economou, J.S. A comparison of gene transfer methods in human dendritic cells. Cancer Gene Ther. 1997, 4, 17–25. [Google Scholar]
- Granot, Y.; Peer, D. Delivering the right message: Challenges and opportunities in lipid nanoparticles-mediated modified mRNA therapeutics-An innate immune system standpoint. Semin. Immunol. 2017, 34, 68–77. [Google Scholar] [CrossRef]
- Akbarzadeh, A.; Rezaei-Sadabady, R.; Davaran, S.; Joo, S.W.; Zarghami, N.; Hanifehpour, Y.; Samiei, M.; Kouhi, M.; Nejati-Koshki, K. Liposome: Classification, preparation, and applications. Nanoscale Res. Lett. 2013, 8, 102. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Su, H.H.; Yang, Y.; Hu, Y.; Zhang, L.; Blancafort, P.; Huang, L. Systemic delivery of modified mRNA encoding herpes simplex virus 1 thymidine kinase for targeted cancer gene therapy. Mol. Ther. 2013, 21, 358–367. [Google Scholar] [CrossRef]
- Parnaste, L.; Arukuusk, P.; Langel, K.; Tenson, T.; Langel, U. The Formation of Nanoparticles between Small Interfering RNA and Amphipathic Cell-Penetrating Peptides. Mol. Ther. Nucleic Acids 2017, 7, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Viger-Gravel, J.; Schantz, A.; Pinon, A.C.; Rossini, A.J.; Schantz, S.; Emsley, L. Structure of Lipid Nanoparticles Containing siRNA or mRNA by Dynamic Nuclear Polarization-Enhanced NMR Spectroscopy. J. Phys. Chem. B 2018, 122, 2073–2081. [Google Scholar] [CrossRef] [PubMed]
- Filion, M.C.; Phillips, N.C. Toxicity and immunomodulatory activity of liposomal vectors formulated with cationic lipids toward immune effector cells. Biochim. Biophys. Acta Biomembr. 1997, 1329, 345–356. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.N.; Piao, Y.Z.; Zhang, C.M.; Jiang, Y.M.; Liu, A.; Cui, S.H.; Zhi, D.F.; Zhen, Y.H.; Zhang, S.B. Replacement of quaternary ammonium headgroups by tri-ornithine in cationic lipids for the improvement of gene delivery in vitro and in vivo. J. Mater. Chem. B 2017, 5, 7963–7973. [Google Scholar] [CrossRef]
- Landesman-Milo, D.; Peer, D. Toxicity profiling of several common RNAi-based nanomedicines: A comparative study. Drug Deliv. Transl. Res. 2014, 4, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Li, J.; He, F.; Wilson, A.; Pitt, B.; Li, S. Cationic lipids enhance siRNA-mediated interferon response in mice. Biochem. Biophys. Res. Commun. 2005, 330, 755–759. [Google Scholar] [CrossRef]
- Platanias, L.C. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat. Rev. Immunol. 2005, 5, 375–386. [Google Scholar] [CrossRef]
- Lv, H.; Zhang, S.; Wang, B.; Cui, S.; Yan, J. Toxicity of cationic lipids and cationic polymers in gene delivery. J. Control. Release 2006, 114, 100–109. [Google Scholar] [CrossRef]
- Hecker, J.G. Non-Viral, Lipid-Mediated DNA and mRNA Gene Therapy of the Central Nervous System (CNS): Chemical-Based Transfection; Humana Press: New York, NY, USA, 2016; pp. 307–324. [Google Scholar]
- Fenton, O.S.; Kauffman, K.J.; McClellan, R.L.; Appel, E.A.; Dorkin, J.R.; Tibbitt, M.W.; Heartlein, M.W.; DeRosa, F.; Langer, R.; Anderson, D.G. Bioinspired Alkenyl Amino Alcohol Ionizable Lipid Materials for Highly Potent In Vivo mRNA Delivery. Adv. Mater. 2016, 28, 2939–2943. [Google Scholar] [CrossRef]
- de Groot, A.M.; Thanki, K.; Gangloff, M.; Falkenberg, E.; Zeng, X.; van Bijnen, D.C.J.; van Eden, W.; Franzyk, H.; Nielsen, H.M.; Broere, F.; et al. Immunogenicity Testing of Lipidoids In Vitro and In Silico: Modulating Lipidoid-Mediated TLR4 Activation by Nanoparticle Design. Mol. Ther. Nucleic Acids 2018, 11, 159–169. [Google Scholar] [CrossRef] [Green Version]
- McKinlay, C.J.; Benner, N.L.; Haabeth, O.A.; Waymouth, R.M.; Wender, P.A. Enhanced mRNA delivery into lymphocytes enabled by lipid-varied libraries of charge-altering releasable transporters. Proc. Natl. Acad. Sci. USA 2018, 115, E5859–E5866. [Google Scholar] [CrossRef] [Green Version]
- Fenton, O.S.; Kauffman, K.J.; Kaczmarek, J.C.; McClellan, R.L.; Jhunjhunwala, S.; Tibbitt, M.W.; Zeng, M.D.; Appel, E.A.; Dorkin, J.R.; Mir, F.F.; et al. Synthesis and Biological Evaluation of Ionizable Lipid Materials for the In Vivo Delivery of Messenger RNA to B Lymphocytes. Adv. Mater. 2017, 29. [Google Scholar] [CrossRef] [PubMed]
- Phua, K.K.; Nair, S.K.; Leong, K.W. Messenger RNA (mRNA) nanoparticle tumour vaccination. Nanoscale 2014, 6, 7715–7729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kauffman, K.J.; Dorkin, J.R.; Yang, J.H.; Heartlein, M.W.; DeRosa, F.; Mir, F.F.; Fenton, O.S.; Anderson, D.G. Optimization of Lipid Nanoparticle Formulations for mRNA Delivery in Vivo with Fractional Factorial and Definitive Screening Designs. Nano Lett. 2015, 15, 7300–7306. [Google Scholar] [CrossRef]
- Thanki, K.; Zeng, X.; Justesen, S.; Tejlmann, S.; Falkenberg, E.; Van Driessche, E.; Morck Nielsen, H.; Franzyk, H.; Foged, C. Engineering of small interfering RNA-loaded lipidoid-poly(DL-lactic-co-glycolic acid) hybrid nanoparticles for highly efficient and safe gene silencing: A quality by design-based approach. Eur. J. Pharm. Biopharm. 2017, 120, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Jansen, M.A.A.; Klausen, L.H.; Thanki, K.; Lyngso, J.; Skov Pedersen, J.; Franzyk, H.; Nielsen, H.M.; van Eden, W.; Dong, M.; Broere, F.; et al. Lipidoid-polymer hybrid nanoparticles loaded with TNF siRNA suppress inflammation after intra-articular administration in a murine experimental arthritis model. Eur. J. Pharm. Biopharm. 2019, 142, 38–48. [Google Scholar] [CrossRef]
- Zhao, W.; Zhang, C.; Li, B.; Zhang, X.; Luo, X.; Zeng, C.; Li, W.; Gao, M.; Dong, Y. Lipid Polymer Hybrid Nanomaterials for mRNA Delivery. Cell. Mol. Bioeng. 2018, 11, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Kaczmarek, J.C.; Patel, A.K.; Kauffman, K.J.; Fenton, O.S.; Webber, M.J.; Heartlein, M.W.; DeRosa, F.; Anderson, D.G. Polymer-Lipid Nanoparticles for Systemic Delivery of mRNA to the Lungs. Angew Chem. Int. Ed. Engl. 2016, 55, 13808–13812. [Google Scholar] [CrossRef] [Green Version]
- Ball, R.L.; Hajj, K.A.; Vizelman, J.; Bajaj, P.; Whitehead, K.A. Lipid Nanoparticle Formulations for Enhanced Co-delivery of siRNA and mRNA. Nano Lett. 2018, 18, 3814–3822. [Google Scholar] [CrossRef]
- Colombo, S.; Cun, D.; Remaut, K.; Bunker, M.; Zhang, J.; Martin-Bertelsen, B.; Yaghmur, A.; Braeckmans, K.; Nielsen, H.M.; Foged, C. Mechanistic profiling of the siRNA delivery dynamics of lipid-polymer hybrid nanoparticles. J. Control. Release 2015, 201, 22–31. [Google Scholar] [CrossRef]
- van den Brand, D.; Gorris, M.A.J.; van Asbeck, A.H.; Palmen, E.; Ebisch, I.; Dolstra, H.; Hällbrink, M.; Massuger, L.F.A.G.; Brock, R. Peptide-mediated delivery of therapeutic mRNA in ovarian cancer. Eur. J. Pharm. Biopharm. 2019, 141, 180–190. [Google Scholar] [CrossRef]
- Udhayakumar, V.K.; De Beuckelaer, A.; McCaffrey, J.; McCrudden, C.M.; Kirschman, J.L.; Vanover, D.; Van Hoecke, L.; Roose, K.; Deswarte, K.; De Geest, B.G.; et al. Arginine-Rich Peptide-Based mRNA Nanocomplexes Efficiently Instigate Cytotoxic T Cell Immunity Dependent on the Amphipathic Organization of the Peptide. Adv. Healthc. Mater. 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Kang, Z.; Meng, Q.; Liu, K. Peptide-based gene delivery vectors. J. Mater. Chem. B 2019, 7, 1824–1841. [Google Scholar] [CrossRef]
- Lacroix, C.; Humanes, A.; Coiffier, C.; Gigmes, D.; Verrier, B.; Trimaille, T. Polylactide-Based Reactive Micelles as a Robust Platform for mRNA Delivery. Pharm. Res. 2020, 37, 30. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Dorkin, J.R.; Wang, W.; Chang, P.H.; Webber, M.J.; Tang, B.C.; Yang, J.; Abutbul-Ionita, I.; Danino, D.; DeRosa, F.; et al. Poly(glycoamidoamine) Brushes Formulated Nanomaterials for Systemic siRNA and mRNA Delivery in Vivo. Nano Lett. 2016, 16, 842–848. [Google Scholar] [CrossRef] [Green Version]
- Zhao, M.; Li, M.; Zhang, Z.; Gong, T.; Sun, X. Induction of HIV-1 gag specific immune responses by cationic micelles mediated delivery of gag mRNA. Drug Deliv. 2016, 23, 2596–2607. [Google Scholar] [CrossRef]
- Sunshine, J.C.; Sunshine, S.B.; Bhutto, I.; Handa, J.T.; Green, J.J. Poly(beta-amino ester)-nanoparticle mediated transfection of retinal pigment epithelial cells in vitro and in vivo. PLoS ONE 2012, 7, e37543. [Google Scholar] [CrossRef]
- Islam, M.A.; Reesor, E.K.; Xu, Y.; Zope, H.R.; Zetter, B.R.; Shi, J. Biomaterials for mRNA delivery. Biomater. Sci. 2015, 3, 1519–1533. [Google Scholar] [CrossRef] [Green Version]
- Siewert, C.; Haas, H.; Nawroth, T.; Ziller, A.; Nogueira, S.S.; Schroer, M.A.; Blanchet, C.E.; Svergun, D.I.; Radulescu, A.; Bates, F.; et al. Investigation of charge ratio variation in mRNA - DEAE-dextran polyplex delivery systems. Biomaterials 2019, 192, 612–620. [Google Scholar] [CrossRef]
- Miao, L.; Li, L.; Huang, Y.; Delcassian, D.; Chahal, J.; Han, J.; Shi, Y.; Sadtler, K.; Gao, W.; Lin, J.; et al. Delivery of mRNA vaccines with heterocyclic lipids increases anti-tumor efficacy by STING-mediated immune cell activation. Nat. Biotechnol. 2019, 37, 1174–1185. [Google Scholar] [CrossRef]
- Rybakova, Y.; Kowalski, P.S.; Huang, Y.; Gonzalez, J.T.; Heartlein, M.W.; DeRosa, F.; Delcassian, D.; Anderson, D.G. mRNA Delivery for Therapeutic Anti-HER2 Antibody Expression In Vivo. Mol. Ther. 2019, 27, 1415–1423. [Google Scholar] [CrossRef]
- Sedic, M.; Senn, J.J.; Lynn, A.; Laska, M.; Smith, M.; Platz, S.J.; Bolen, J.; Hoge, S.; Bulychev, A.; Jacquinet, E.; et al. Safety Evaluation of Lipid Nanoparticle-Formulated Modified mRNA in the Sprague-Dawley Rat and Cynomolgus Monkey. Vet. Pathol. 2018, 55, 341–354. [Google Scholar] [CrossRef] [PubMed]
- Persano, S.; Guevara, M.L.; Li, Z.; Mai, J.; Ferrari, M.; Pompa, P.P.; Shen, H. Lipopolyplex potentiates anti-tumor immunity of mRNA-based vaccination. Biomaterials 2017, 125, 81–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kranz, L.M.; Diken, M.; Haas, H.; Kreiter, S.; Loquai, C.; Reuter, K.C.; Meng, M.; Fritz, D.; Vascotto, F.; Hefesha, H.; et al. Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immunotherapy. Nature 2016, 534, 396–401. [Google Scholar] [CrossRef]
- Veiga, N.; Goldsmith, M.; Granot, Y.; Rosenblum, D.; Dammes, N.; Kedmi, R.; Ramishetti, S.; Peer, D. Cell specific delivery of modified mRNA expressing therapeutic proteins to leukocytes. Nat. Comm. 2018, 9, 4493. [Google Scholar] [CrossRef]
- Hossain, M.K.; Wall, K.A. Use of Dendritic Cell Receptors as Targets for Enhancing Anti-Cancer Immune Responses. Cancers 2019, 11, 418. [Google Scholar] [CrossRef] [Green Version]
- Midoux, P.; Pichon, C. Lipid-based mRNA vaccine delivery systems. Expert Rev. Vaccines 2015, 14, 221–234. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Xiao, W.; Elbahnasawy, M.A.; Bao, X.; Zheng, Q.; Gong, L.; Zhou, Y.; Yang, S.; Fang, A.; Farag, M.M.S.; et al. Optimization of the Linker Length of Mannose-Cholesterol Conjugates for Enhanced mRNA Delivery to Dendritic Cells by Liposomes. Front. Pharmacol. 2018, 9, 980. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| mRNA | Mechanism of Action | Disease/Condition | Administration Route | Study Phase | Sponsor/Collaborator | National Clinical Trial Identifier |
|---|---|---|---|---|---|---|
| Therapeutic mRNA | ||||||
| W_ova1 vaccine | Induction of an anti-tumor immune response | Ovarian cancer | Intravenous | Phase I | University Medical Center Groningen/BioNTech | NCT04163094 |
| CT7, MAGE-A3, and WT1 mRNA-electroporated Langerhans cells (LCs) | Electroporation of dendritic cells with antigen mRNA | Multiple Myeloma | Subcutaneous | Phase I | Memorial Sloan Kettering Cancer Center | NCT01995708 |
| Personalized Cellular mRNA | Immunization with DCs pulsed with mRNA encoded tumor antigens | Brain cancer/Neoplasm Metastases | Not specified | Phase I | Guangdong 999 Brain Hospital/Beijing, Tricision, Trinomab, Jinan University Guangzhou | NCT02808416 |
| Personalized mRNA | Immunization with DCs pulsed with personalized mRNA | Glioblastoma | Not specified | Phase I | Guangdong 999 Brain Hospital/Beijing Tricision, Trinomab, Jinan University Guangzhou | NCT02808364 |
| MiHA mRNA | Immunization with DCs loaded with MiHA mRNA | Hematological malignancies | Intravenous | Phase I Phase II | Radboud University/ZonMw: The Netherlands Organization for Health Research and Development Dutch Cancer Society | NCT02528682 |
| WT1-mRNA | Immunization with DCs electroporated with WT1-mRNA | Acute myeloid leukemia | Not Specified | Phase II | Zwi Berneman/Kom Op Tegen Kanker stichting tegen kanker Research Foundation - Flanders (FWO: Fonds Wetenschappelijk Onderzoek) | NCT01686334 |
| Human CMV pp65-LAMP mRNA | Immunization with DCs pulsed with CMV pp65-LAMP mRNA | Glioblastoma | Intradermal | Phase II | Gary Archer Ph.D./Duke University | NCT03927222 |
| Personalized mRNA | Personalized mRNA tumor vaccine encoding neoantigen | Advanced esophageal squamous carcinoma, gastric adenocarcinoma, pancreatic adenocarcinoma, colorectal adenocarcinoma | Subcutaneous | Enrolling | Changhai Hospital/Stemirna Therapeutics | NCT03468244 |
| mRNA[BI 1361849 (formerly CV9202)] | Not specified | Metastatic non-small cell lung cancer | Not specified | Phase I Phase II | Ludwig Institute for Cancer Research/Cancer Research Institute, New York City, Boehringer Ingelheim MedImmune, CureVac, PharmaJet | NCT03164772 |
| mRNA-5671/V941 | Not specified | Neoplasms, carcinoma, non-small-cell lung, pancreatic neoplasms, colorectal neoplasms | Intramuscular | Phase I | Merck Sharp & Dohme | NCT03948763 |
| mRNA-4157 | Immunostimulants | Solid tumors | Not specified | Phase I | Moderna/Merck Sharp & Dohme | NCT03313778 |
| mRNA-4157 | Immunotherapy with the personalized cancer vaccine | Cutaneous melanoma | Not specified | Phase II | Moderna/Merck Sharp & Dohme | NCT03897881 |
| Personalized mRNA | Encoding neoantigen | Esophageal cancer Non-small cell lung cancer | Subcutaneous | Enrolling | Stemirna Therapeutics/The First Affiliated Hospital of Zhengzhou University | NCT03908671 |
| mRNA-3704 | Alpha-galactosidase stimulants; Methylmalonyl CoA mutase stimulants; Protein synthesis stimulants | Methylmalonic acidemia, metabolism, inborn errors | Intravenous | Phase I Phase II | Moderna | NCT03810690 |
| mRNA-2416 | OX40 ligand modulators | Relapsed/Refractory solid tumor malignancies or lymphoma | Intratumoral | Phase I | Moderna | NCT03323398 |
| mRNA-2752 | IL36G protein stimulants; Interleukin 23 stimulants; OX40 ligand modulators | Relapsed/Refractory solid tumor malignancies or lymphoma | Intratumoral | Phase I | Moderna/AstraZeneca | NCT03739931 |
| Prophylactic mRNA | ||||||
| mRNA-1647, mRNA-1443 | Not specified | Cytomegalovirus | Not specified | Phase I | Moderna | NCT03382405 |
| mRNA-1893 | Not specified | Zika virus | Not specified | Phase I | Moderna/Biomedical Advanced Research and Development Authority | NCT04064905 |
| mRNA-1653 | A combined human metapneumovirus and human parainfluenza virus type 3 vaccine | Human metapneumovirus and Human Parainfluenzavirus | Not specified | Phase I | Moderna. | NCT03392389 NCT04144348 |
| mRNA-1944 | Encoding for an anti-Chikungunya virus monoclonal antibody | Chikungunya virus | Parenteral | Phase I | Moderna | NCT03829384 |
| mRNA-1653 | Immunostimulants | Metapneumovirus and Parainfluenza virus | Parenteral | Phase I | Moderna | NCT03392389 |
| CV7202 | Immunostimulants | Rabies | Intramuscular | Phase I | CureVac | NCT03713086 |
| Drug Delivery Systems | Composition | RNA | Disease/Condition | References |
|---|---|---|---|---|
| Polymers | Poly(glycoamidoamine) | Erythropoietin (EPO) mRNA | Anemia and myelodysplasia | [168] |
| Polyethyleneimine | HIV-1 gag mRNA | HIV | [169] | |
| Poly(β-amino ester) (PBAE) | eGFP mRNA | N/A | [170] | |
| Triblock copolymer (comprising DMAEMA, PEGMA, DEAEMA and BMA) | eGFP and ovalbumin | N/A | [171] | |
| DEAE-Dextran | Luciferase-encoding mRNA | N/A | [172] | |
| Lipids | DOTAP/DOPE | HxB-2 HIV-1 Gag antigen mRNA | HIV | [126] |
| DOPE/DC-Cholesterol [2:1] | eGFP mRNA | N/A | [106] | |
| DOTAP/Cholesterol [1:1] liposome with DSPE-PEG and DSPE-PEG-AA | HSV I Thymidine kinase mRNA | Cancer | [142] | |
| C12-200:Cholesterol: DOPE:C14-PEG2000 | EPO mRNA | N/A | [157] | |
| A18 | Ovalbumin mRNA | Melanoma | [173] | |
| cKK-E12 | HER2 antibody mRNA | Cancer | [174] | |
| (6Z,9Z,28Z,31Z)-heptatriaconta-6,9,28,31-tetraen-19-yl 4-(dimethylamino)butanoate (MC3), DSPC, cholesterol, and 1,2-dimyristoyl-rac-glycerol, methoxypolyethylene glycol (PEG2000-DMG) | Human erythropoietin | [175] | ||
| DOTAP/DOPE [1:1] | HIV-1 antigen Gag mRNA | HIV | [126] | |
| 3β-[N-(N’,N’-dimethylaminoethane) carbamoyl](DC-Cholesterol)/DOPE (1:2) | eGFP mRNA | N/A | [106] | |
| Lipid polymer hybrid NPs | TT3:DOPE:Cholesterol:DMG-PEG2000 with PLGA core | Firefly luciferase (FLuc) mRNA and eGFP mRNA | N/A | [160] |
| PBAE:C14-PEG2000 | FLuc mRNA | N/A | [161] | |
| PBAE:EDOPC/DOPE/DSPE-PEG | Ovalbumin mRNA | N/A | [176] | |
| PBAE: DOPC, DOTAP, and DSPE-PEG | eGFP mRNA | N/A | [105] | |
| Peptides and peptide-polymer hybrids | PepFect14 | eGFP mRNA | Ovarian cancer | [164] |
| RALA | eGFP mRNA OVA mRNA | N/A | [165] | |
| RALA-PLA | eGFPmRNA FLuc mRNA | N/A | [167] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wadhwa, A.; Aljabbari, A.; Lokras, A.; Foged, C.; Thakur, A. Opportunities and Challenges in the Delivery of mRNA-Based Vaccines. Pharmaceutics 2020, 12, 102. https://doi.org/10.3390/pharmaceutics12020102
Wadhwa A, Aljabbari A, Lokras A, Foged C, Thakur A. Opportunities and Challenges in the Delivery of mRNA-Based Vaccines. Pharmaceutics. 2020; 12(2):102. https://doi.org/10.3390/pharmaceutics12020102
Chicago/Turabian StyleWadhwa, Abishek, Anas Aljabbari, Abhijeet Lokras, Camilla Foged, and Aneesh Thakur. 2020. "Opportunities and Challenges in the Delivery of mRNA-Based Vaccines" Pharmaceutics 12, no. 2: 102. https://doi.org/10.3390/pharmaceutics12020102