Nuclear Medicine in Times of COVID-19: How Radiopharmaceuticals Could Help to Fight the Current and Future Pandemics


Abstract
:1. Introduction
2. SARS-CoV-2 Structure, Infection, Replication and Treatment
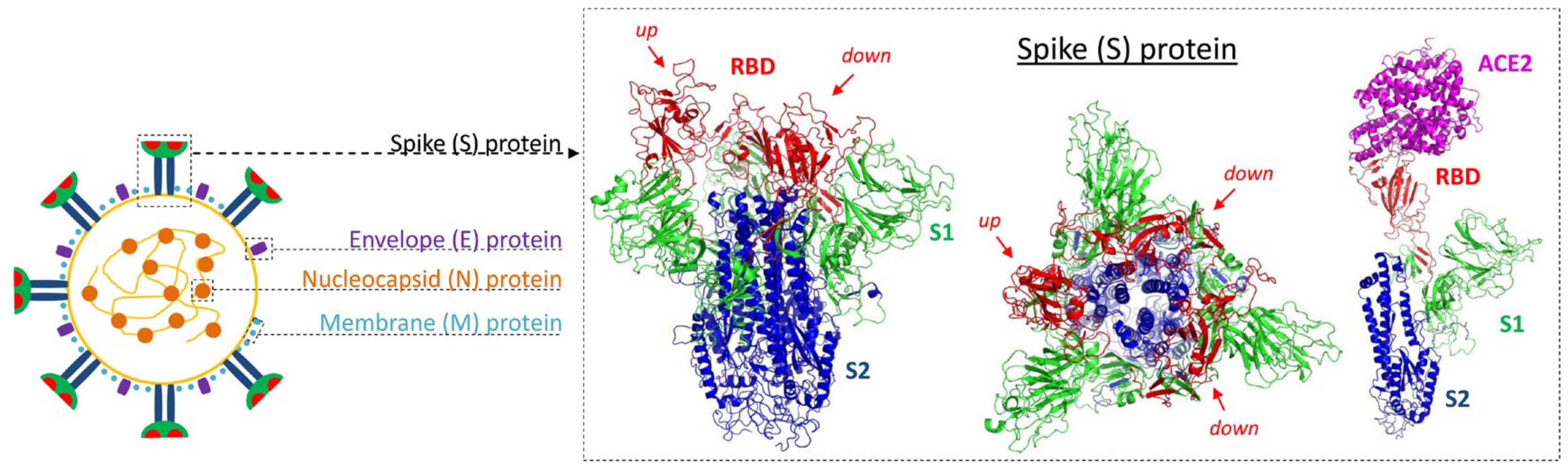
2.1. Virion Structure
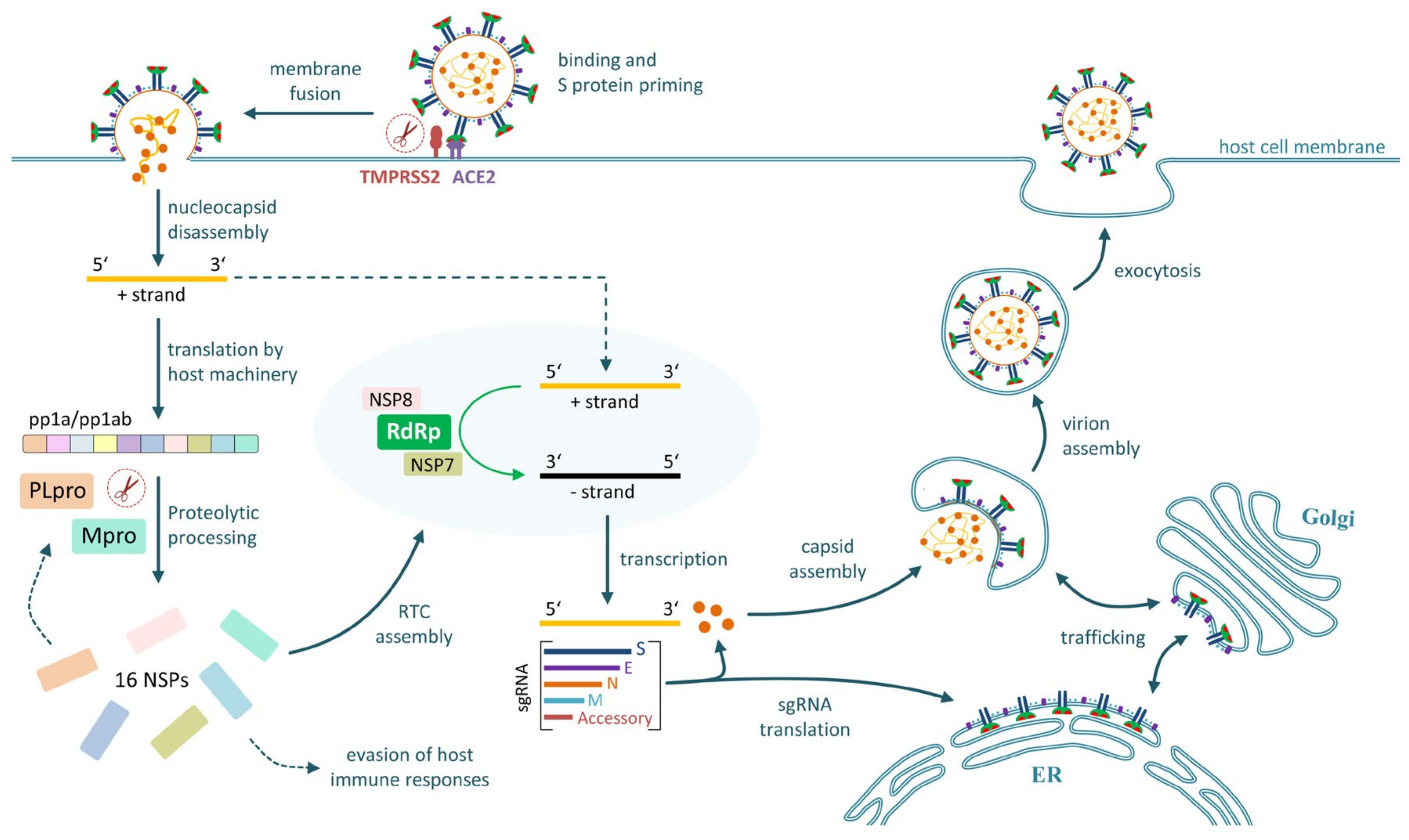
2.2. Cell Entry and Tissue Tropism
2.3. Replication in Infected Cells
2.4. Experimental Drugs Targeting SARS-CoV-2 Cell Entry
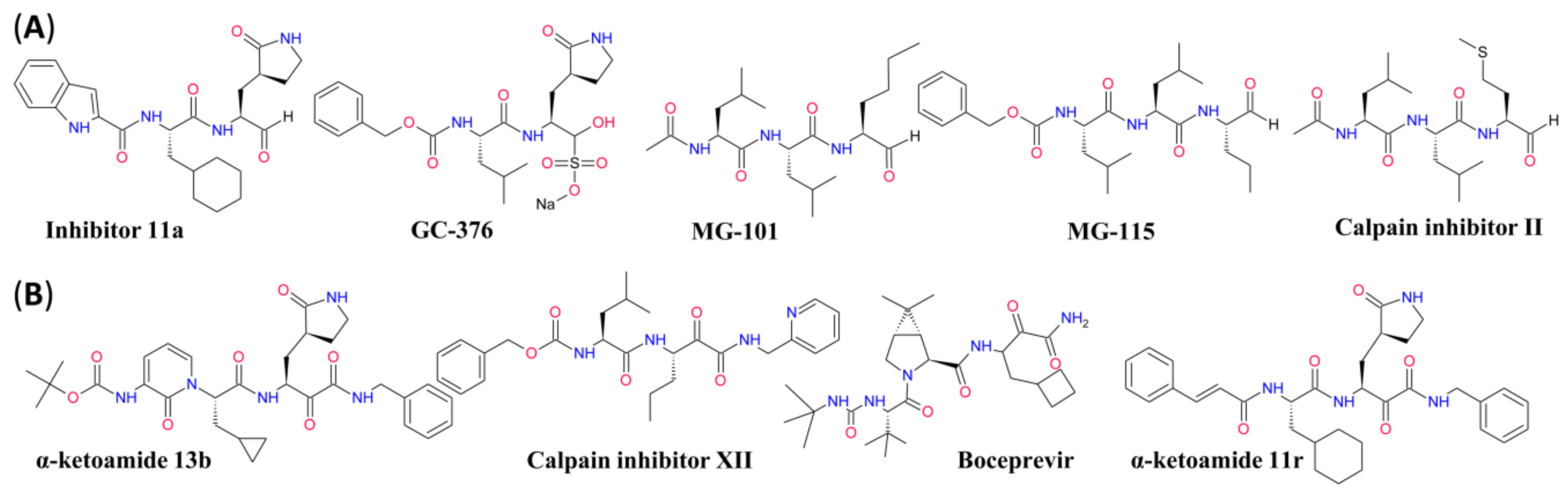
2.5. Experimental Drugs Targeting SARS-CoV-2 Replication
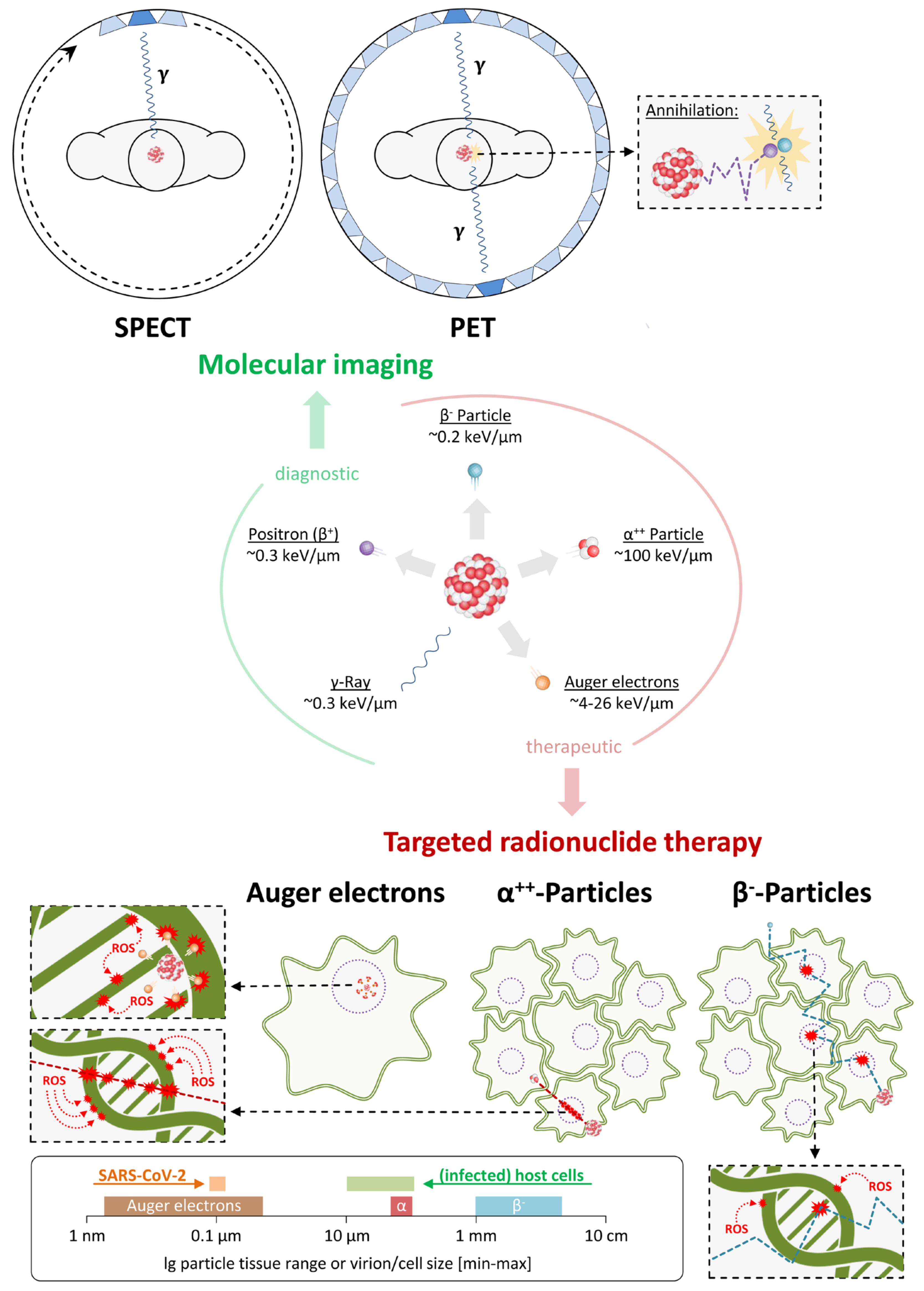
3. Application of Nuclear Imaging in the Context of SARS-CoV-2
3.1. Imaging of Host Responses to SARS-CoV-2 Infection
3.2. Imaging of Host Molecules Involved in SARS-CoV-2 Infection
3.3. PET- or SPECT-Based Antiviral Drug Development
3.4. SARS-CoV-2-Specific Nuclear Imaging
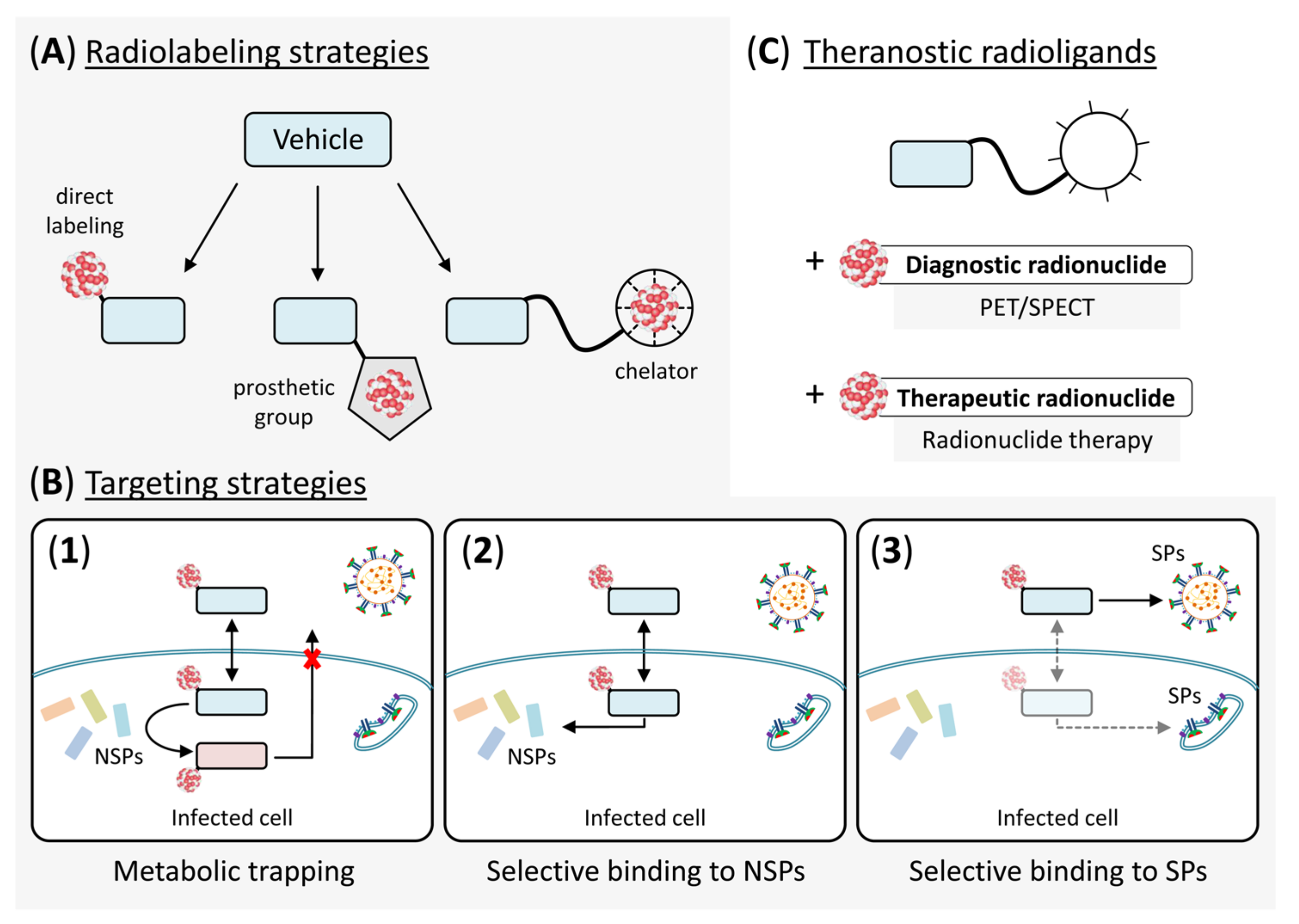
4. Use of Radiopharmaceuticals for the Treatment of COVID-19
4.1. Radionuclide Therapy Targeting SARS-CoV-2 Virions
4.2. Radionuclide Therapy Targeting SARS-CoV-2-Infected Cells
5. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
Appendix A

References
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Alderman, T.S.; Frothingham, R.; Sempowski, G.D. Validation of an Animal Isolation Imaging Chamber for Use in Animal Biosafety Level-3 Containment. Appl. Biosaf. 2010, 15, 62–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, S.L.; Nuermberger, E.L.; Um, P.K.; Vidal, C.; Jedynak, B.; Pomper, M.G.; Bishai, W.R.; Jain, S.K. Noninvasive Pulmonary [18F]-2-Fluoro-Deoxy-d-Glucose Positron Emission Tomography Correlates with Bactericidal Activity of Tuberculosis Drug Treatment. Antimicrob. Agents Chemother. 2009, 53, 4879–4884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jahrling, P.B.; Keith, L.; Claire, M.S.; Johnson, R.F.; Bollinger, L.; Lackemeyer, M.G.; Hensley, L.E.; Kindrachuk, J.; Kuhn, J.H. The NIAID Integrated Research Facility at Frederick, Maryland: A unique international resource to facilitate medical countermeasure development for BSL-4 pathogens. Pathog. Dis. 2014, 71, 213–219. [Google Scholar] [CrossRef] [Green Version]
- Hartman, A.L.; Nambulli, S.; McMillen, C.M.; White, A.G.; Tilston-Lunel, N.L.; Albe, J.R.; Cottle, E.; Dunn, M.D.; Frye, L.J.; Gilliland, T.H.; et al. SARS-CoV-2 infection of African green monkeys results in mild respiratory disease discernible by PET/CT imaging and shedding of infectious virus from both respiratory and gastrointestinal tracts. PLoS Pathog. 2020, 16, e1008903. [Google Scholar] [CrossRef]
- Keith, L.; Chefer, S.; Bollinger, L.; Solomon, J.; Yellayi, S.; Seidel, J.; Thomasson, D.; Jahrling, P. Preclinical Imaging in BSL-3 and BSL-4 Environments: Imaging Pathophysiology of Highly Pathogenic Infectious Diseases. In Continuous Pharmaceutical Processing; Springer Science and Business Media LLC: Berlin/Heidelberg, Germany, 2014; pp. 271–290. [Google Scholar]
- Yu, F.; Du, L.; Ojcius, D.M.; Pan, C.; Jiang, S. Measures for diagnosing and treating infections by a novel coronavirus responsible for a pneumonia outbreak originating in Wuhan, China. Microbes Infect. 2020, 22, 74–79. [Google Scholar] [CrossRef]
- De Haan, C.A.; Rottier, P.J. Molecular Interactions in the Assembly of Coronaviruses. In Advances in Clinical Chemistry; Elsevier BV: Amsterdam, The Netherlands, 2005; Volume 64, pp. 165–230. [Google Scholar]
- Schoeman, D.; Fielding, B.C. Coronavirus envelope protein: Current knowledge. Virol. J. 2019, 16, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Walls, A.C.; Park, Y.-J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281–292.e6. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef]
- Sungnak, W.; Network, H.L.B.; Huang, N.; Bécavin, C.; Berg, M.; Queen, R.; Litvinukova, M.; Talavera-López, C.; Maatz, H.; Reichart, D.; et al. SARS-CoV-2 entry factors are highly expressed in nasal epithelial cells together with innate immune genes. Nat. Med. 2020, 26, 681–687. [Google Scholar] [CrossRef] [Green Version]
- Bertram, S.; Heurich, A.; Lavender, H.; Gierer, S.; Danisch, S.; Perin, P.; Lucas, J.M.; Nelson, P.S.; Pöhlmann, S.; Soilleux, E.J. Influenza and SARS-Coronavirus Activating Proteases TMPRSS2 and HAT Are Expressed at Multiple Sites in Human Respiratory and Gastrointestinal Tracts. PLoS ONE 2012, 7, e35876. [Google Scholar] [CrossRef] [PubMed]
- Zou, X.; Chen, K.; Zou, J.; Han, P.; Hao, J.; Han, Z. Single-cell RNA-seq data analysis on the receptor ACE2 expression reveals the potential risk of different human organs vulnerable to 2019-nCoV infection. Front. Med. 2020, 14, 185–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, F.; Qian, S.; Zhang, S.; Zhang, Z. Single cell RNA sequencing of 13 human tissues identify cell types and receptors of human coronaviruses. Biochem. Biophys. Res. Commun. 2020, 526, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Hamming, I.; Timens, W.; Bulthuis, M.L.C.; Lely, A.T.; Navis, G.J.; Van Goor, H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J. Pathol. 2004, 203, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Cantuti-Castelvetri, L.; Ojha, R.; Pedro, L.D.; Djannatian, M.; Franz, J.; Kuivanen, S.; Van Der Meer, F.; Kallio, K.; Kaya, T.; Anastasina, M.; et al. Neuropilin-1 facilitates SARS-CoV-2 cell entry and infectivity. Science 2020, eabd2985. [Google Scholar] [CrossRef] [PubMed]
- Varga, Z.; Flammer, A.J.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A.S.; Mehra, M.R.; Schuepbach, R.A.; Ruschitzka, F.; Moch, H. Endothelial cell infection and endotheliitis in COVID-19. Lancet 2020, 395, 1417–1418. [Google Scholar] [CrossRef]
- Robba, C.; Battaglini, D.; Pelosi, P.; Rocco, P.R.M. Multiple organ dysfunction in SARS-CoV-2: MODS-CoV-2. Expert Rev. Respir. Med. 2020, 1–4. [Google Scholar] [CrossRef]
- Xiong, T.-Y.; Redwood, S.R.; Prendergast, B.; Chen, M. Coronaviruses and the cardiovascular system: Acute and long-term implications. Eur. Heart J. 2020, 41, 1798–1800. [Google Scholar] [CrossRef] [Green Version]
- Zhu, G.; Zhu, C.; Zhu, Y.; Sun, F. Minireview of progress in the structural study of SARS-CoV-2 proteins. Curr. Res. Microb. Sci. 2020, 1, 53–61. [Google Scholar] [CrossRef]
- Dai, W.; Zhang, B.; Jiang, X.-M.; Su, H.; Li, J.; Zhao, Y.; Xie, X.; Jin, Z.; Peng, J.; Liu, F.; et al. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science 2020, 368, 1331–1335. [Google Scholar] [CrossRef] [Green Version]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, D.; Mukherjee, R.; Grewe, D.; Bojkova, D.; Baek, K.; Bhattacharya, A.; Schulz, L.; Widera, M.; Mehdipour, A.R.; Tascher, G.; et al. Papain-like protease regulates SARS-CoV-2 viral spread and innate immunity. Nature 2020, 1–10. [Google Scholar] [CrossRef]
- Santerre, M.; Arjona, S.P.; Allen, C.N.; Shcherbik, N.; Sawaya, B.E. Why do SARS-CoV-2 NSPs rush to the ER? J. Neurol. 2020, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Yan, L.; Huang, Y.; Liu, F.; Zhao, Y.; Cao, L.; Wang, T.; Sun, Q.; Ming, Z.; Zhang, L.; et al. Structure of the RNA-dependent RNA polymerase from COVID-19 virus. Science 2020, 368, 779–782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Li, R.; Pan, Z.; Qian, C.; Yang, Y.; You, R.; Zhao, J.; Liu, P.; Gao, L.; Li, Z.; et al. Human monoclonal antibodies block the binding of SARS-CoV-2 spike protein to angiotensin converting enzyme 2 receptor. Cell. Mol. Immunol. 2020, 17, 647–649. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Li, W.; Drabek, D.; Okba, N.; Van Haperen, R.; Osterhaus, A.D.M.E.; Van Kuppeveld, F.J.M.; Haagmans, B.L.; Grosveld, F.; Bosch, B.J. A human monoclonal antibody blocking SARS-CoV-2 infection. Nat. Commun. 2020, 11, 1–6. [Google Scholar] [CrossRef]
- Brouwer, P.J.M.; Caniels, T.G.; Van Der Straten, K.; Snitselaar, J.L.; Aldon, Y.; Bangaru, S.; Torres, J.L.; Okba, N.; Claireaux, M.; Kerster, G.; et al. Potent neutralizing antibodies from COVID-19 patients define multiple targets of vulnerability. Science 2020, 369, 643–650. [Google Scholar] [CrossRef]
- Wang, X.; Cao, R.; Zhang, H.; Liu, J.; Xu, M.; Hu, H.; Li, Y.; Zhao, L.; Li, W.; Sun, X.; et al. The anti-influenza virus drug, arbidol is an efficient inhibitor of SARS-CoV-2 in vitro. Cell Discov. 2020, 6, 1–5. [Google Scholar] [CrossRef]
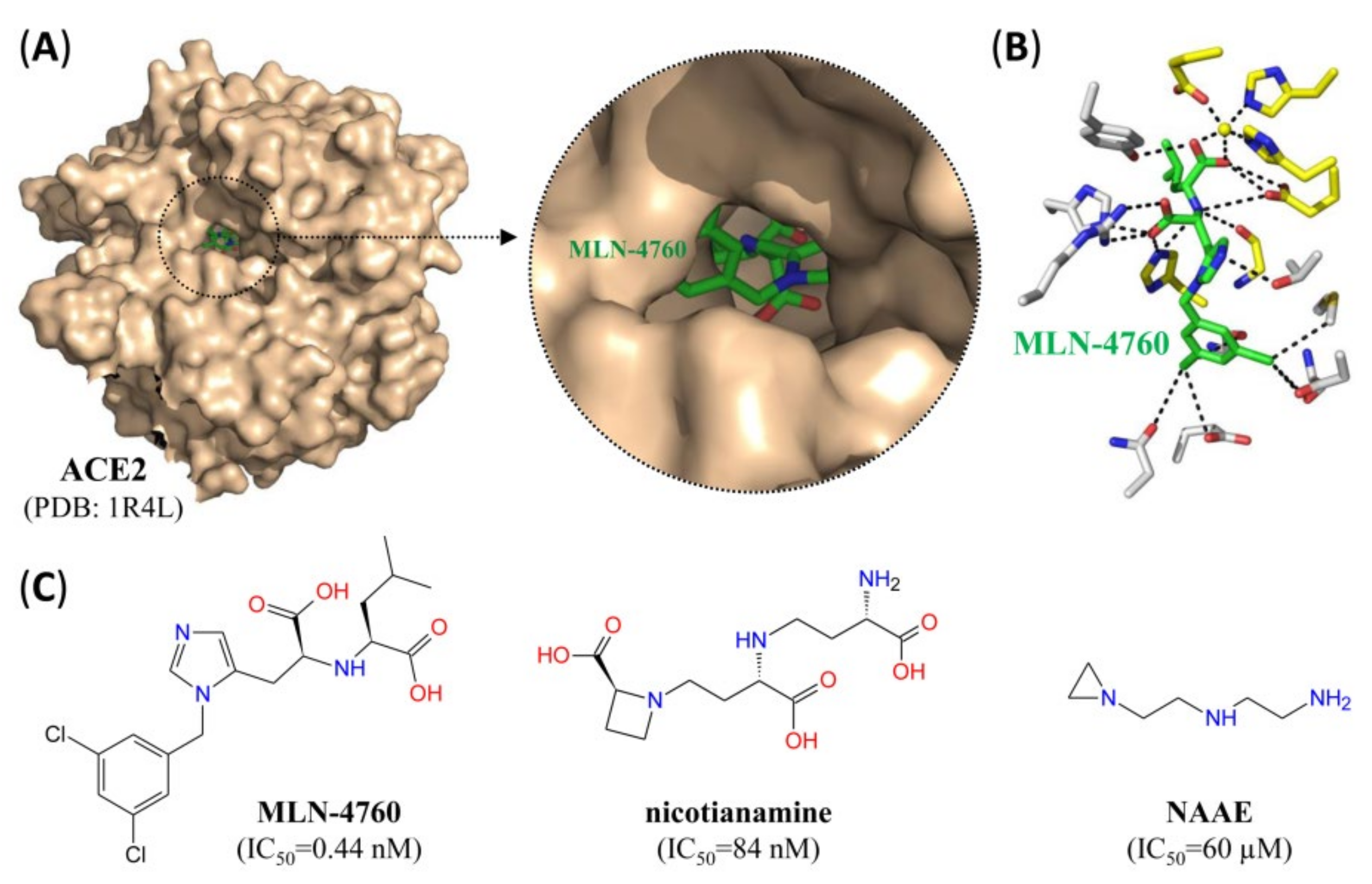
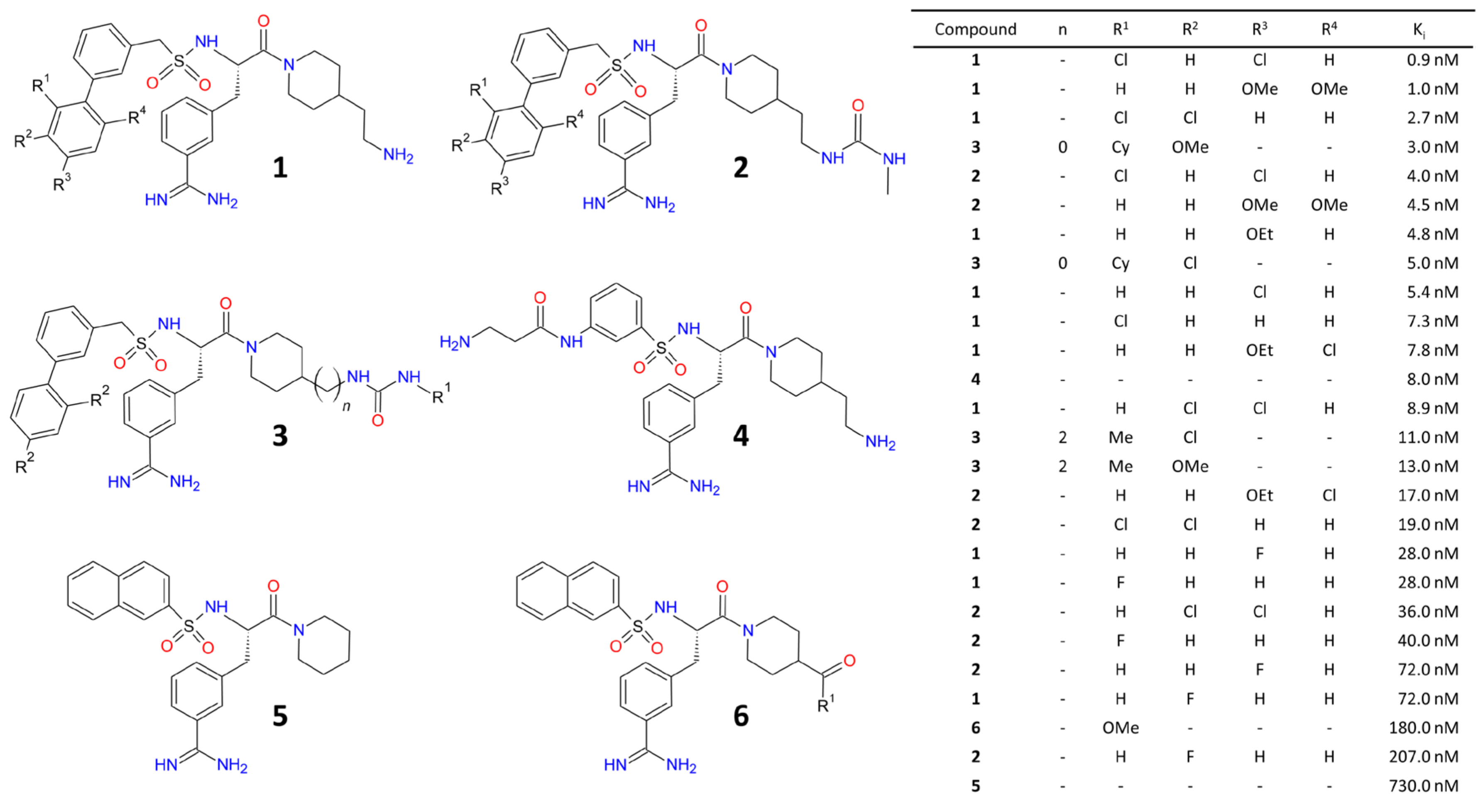
- Dales, N.A.; Gould, A.E.; Brown, J.A.; Calderwood, E.F.; Guan, B.; Minor, C.A.; Gavin, J.M.; Hales, P.; Kaushik, V.K.; Stewart, M.; et al. Substrate-Based Design of the First Class of Angiotensin-Converting Enzyme-Related Carboxypeptidase (ACE2) Inhibitors. J. Am. Chem. Soc. 2002, 124, 11852–11853. [Google Scholar] [CrossRef]
- Takahashi, S.; Yoshiya, T.; Yoshizawa-Kumagaye, K.; Sugiyama, T. Nicotianamine is a novel angiotensin-converting enzyme 2 inhibitor in soybean. Biomed. Res. 2015, 36, 219–224. [Google Scholar] [CrossRef] [Green Version]
- Huentelman, M.J.; Zubcevic, J.; Prada, J.A.H.; Xiao, X.; Dimitrov, D.S.; Raizada, M.K.; Ostrov, D.A. Structure-Based Discovery of a Novel Angiotensin-Converting Enzyme 2 Inhibitor. Hypertension 2004, 44, 903–906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, L.; Sexton, D.J.; Skogerson, K.; Devlin, M.; Smith, R.; Sanyal, I.; Parry, T.; Kent, R.; Enright, J.; Wu, Q.-L.; et al. Novel Peptide Inhibitors of Angiotensin-converting Enzyme 2. J. Biol. Chem. 2003, 278, 15532–15540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedersen, K.B.; Sriramula, S.; Chhabra, K.H.; Xia, H.; Lazartigues, E. Species-specific inhibitor sensitivity of angiotensin-converting enzyme 2 (ACE2) and its implication for ACE2 activity assays. Am. J. Physiol. Integr. Comp. Physiol. 2011, 301, R1293–R1299. [Google Scholar] [CrossRef] [PubMed]
- Ye, M.; Wysocki, J.; Gonzalez-Pacheco, F.R.; Salem, M.; Evora, K.; Garcia-Halpin, L.; Poglitsch, M.; Schuster, M.; Batlle, D. Murine Recombinant Angiotensin-Converting Enzyme 2. Hypertension 2012, 60, 730–740. [Google Scholar] [CrossRef] [Green Version]
- Tian, X.; Li, C.; Huang, A.; Xia, S.; Lu, S.; Shi, Z.; Lu, L.; Jiang, S.; Yang, Z.; Wu, Y.; et al. Potent binding of 2019 novel coronavirus spike protein by a SARS coronavirus-specific human monoclonal antibody. Emerg. Microbes Infect. 2020, 9, 382–385. [Google Scholar] [CrossRef] [Green Version]
- Yuan, M.; Wu, N.C.; Zhu, X.; Lee, C.-C.D.; So, R.T.Y.; Lv, H.; Mok, C.K.P.; Wilson, I.A. A highly conserved cryptic epitope in the receptor binding domains of SARS-CoV-2 and SARS-CoV. Science 2020, 368, 630–633. [Google Scholar] [CrossRef] [Green Version]
- Kawase, M.; Shirato, K.; Van Der Hoek, L.; Taguchi, F.; Matsuyama, S. Simultaneous Treatment of Human Bronchial Epithelial Cells with Serine and Cysteine Protease Inhibitors Prevents Severe Acute Respiratory Syndrome Coronavirus Entry. J. Virol. 2012, 86, 6537–6545. [Google Scholar] [CrossRef] [Green Version]
- Dana, D.; Kumar, S. A Review of Small Molecule Inhibitors and Functional Probes of Human Cathepsin L. Molecules 2020, 25, 698. [Google Scholar] [CrossRef] [Green Version]
- Kos, J.; Mitrović, A.; Mirković, B. The current stage of cathepsin B inhibitors as potential anticancer agents. Futur. Med. Chem. 2014, 6, 1355–1371. [Google Scholar] [CrossRef]
- Meyer, D.; Sielaff, F.; Hammami, M.; Böttcher-Friebertshäuser, E.; Garten, W.; Steinmetzer, T. Identification of the first synthetic inhibitors of the type II transmembrane serine protease TMPRSS2 suitable for inhibition of influenza virus activation. Biochem. J. 2013, 452, 331–343. [Google Scholar] [CrossRef] [Green Version]
- Ma, C.; Sacco, M.D.; Hurst, B.; Townsend, J.A.; Hu, Y.; Szeto, T.; Zhang, X.; Tarbet, B.; Marty, M.T.; Chen, Y.; et al. Boceprevir, GC-376, and calpain inhibitors II, XII inhibit SARS-CoV-2 viral replication by targeting the viral main protease. Cell Res. 2020, 30, 678–692. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 2020, 368, 409–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Xie, W.; Xue, X.; Yang, K.; Ma, J.; Liang, W.; Zhao, Q.; Zhou, Z.; Pei, D.; Ziebuhr, J.; et al. Design of Wide-Spectrum Inhibitors Targeting Coronavirus Main Proteases. PLoS Biol. 2005, 3, e324. [Google Scholar] [CrossRef]
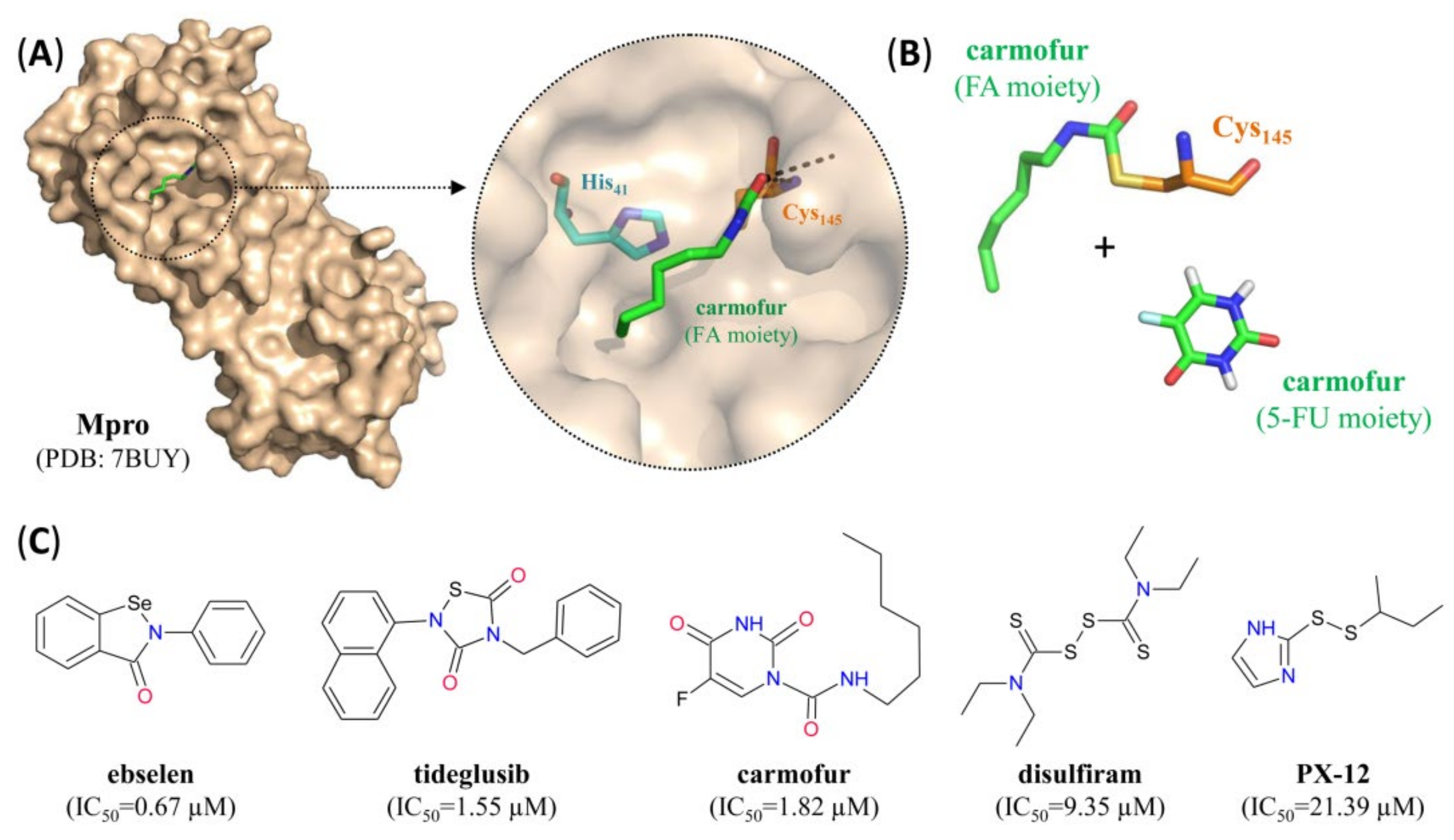
- Jin, Z.; Zhao, Y.; Sun, Y.; Zhang, B.; Wang, H.; Wu, Y.; Zhu, Y.; Zhu, C.; Hu, T.; Du, X.; et al. Structural basis for the inhibition of SARS-CoV-2 main protease by antineoplastic drug carmofur. Nat. Struct. Mol. Biol. 2020, 27, 529–532. [Google Scholar] [CrossRef] [PubMed]
- Weglarz-Tomczak, E.; Tomczak, J.M.; Talma, M.; Brul, S. Ebselen as a highly active inhibitor of PLproCoV2. bioRxiv Prepr. 2020. [Google Scholar] [CrossRef]
- Sargsyan, K.; Lin, C.-C.; Chen, T.; Grauffel, C.; Chen, Y.-P.; Yang, W.-Z.; Yuan, H.S.; Lim, C. Multi-targeting of functional cysteines in multiple conserved SARS-CoV-2 domains by clinically safe Zn-ejectors. ChemRxiv Prepr. 2020. [Google Scholar] [CrossRef]
- Su, H.X.; Yao, S.; Zhao, W.F.; Li, M.J.; Liu, J.; Shang, W.J.; Xie, H.; Ke, C.Q.; Hu, H.C.; Gao, M.N.; et al. Anti-SARS-CoV-2 activities in vitro of Shuanghuanglian preparations and bioactive ingredients. Acta Pharmacol. Sin. 2020, 41, 1167–1177. [Google Scholar] [CrossRef]
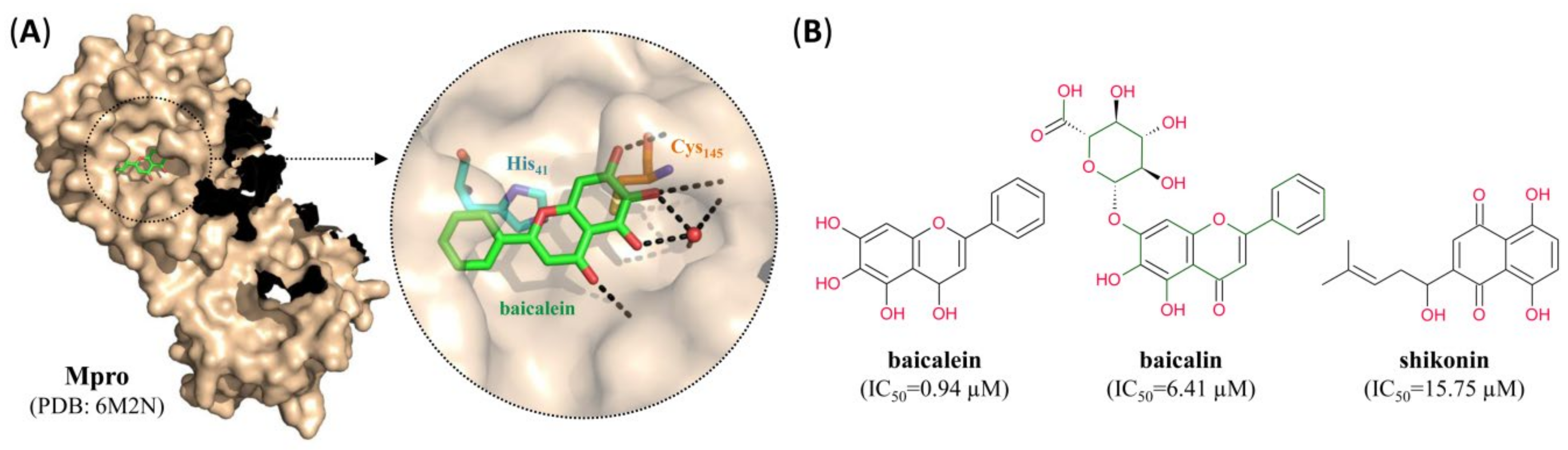
- Jo, S.; Kim, S.; Shin, D.H.; Kim, M.-S. Inhibition of SARS-CoV 3CL protease by flavonoids. J. Enzym. Inhib. Med. Chem. 2020, 35, 145–151. [Google Scholar] [CrossRef] [Green Version]
- Jo, S.; Kim, H.; Kim, S.; Shin, D.H.; Kim, M.-S. Characteristics of flavonoids as potent MERS-CoV 3C-like protease inhibitors. Chem. Biol. Drug Des. 2019, 94, 2023–2030. [Google Scholar] [CrossRef] [Green Version]
- Freitas, B.T.; Durie, I.A.; Murray, J.; Longo, J.E.; Miller, H.C.; Crich, D.; Hogan, R.J.; Tripp, R.A.; Pegan, S.D. Characterization and Noncovalent Inhibition of the Deubiquitinase and deISGylase Activity of SARS-CoV-2 Papain-Like Protease. ACS Infect. Dis. 2020, 6, 2099–2109. [Google Scholar] [CrossRef]
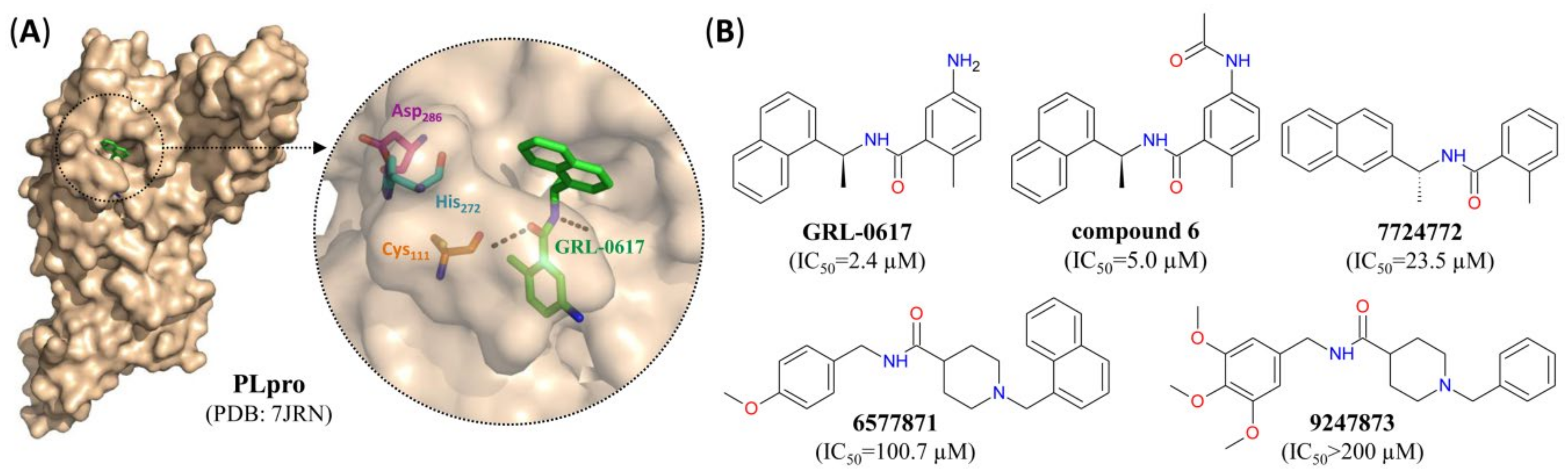
- Rut, W.; Lv, Z.; Zmudzinski, M.; Patchett, S.; Nayak, D.; Snipas, S.J.; El Oualid, F.; Huang, T.T.; Bekes, M.; Drag, M.; et al. Activity profiling and structures of inhibitor-bound SARS-CoV-2-PLpro protease provides a framework for anti-COVID-19 drug design. bioRxiv Prepr. 2020. [Google Scholar] [CrossRef]
- Gordon, C.J.; Tchesnokov, E.P.; Woolner, E.; Perry, J.K.; Feng, J.Y.; Porter, D.P.; Götte, M. Remdesivir is a direct-acting antiviral that inhibits RNA-dependent RNA polymerase from severe acute respiratory syndrome coronavirus 2 with high potency. J. Biol. Chem. 2020, 295, 6785–6797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shu, T.; Huang, M.; Wu, D.; Ren, Y.; Zhang, X.; Han, Y.; Mu, J.; Wang, R.; Qiu, Y.; Zhang, D.-Y.; et al. SARS-Coronavirus-2 Nsp13 Possesses NTPase and RNA Helicase Activities That Can Be Inhibited by Bismuth Salts. Virol. Sin. 2020, 35, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.; Wang, R.; Chan, J.F.-W.; Zhang, A.J.; Cheng, T.; Chik, K.K.-H.; Ye, Z.-W.; Wang, S.; Lee, A.C.-Y.; Jin, L.; et al. Metallodrug ranitidine bismuth citrate suppresses SARS-CoV-2 replication and relieves virus-associated pneumonia in Syrian hamsters. Nat. Microbiol. 2020, 5, 1439–1448. [Google Scholar] [CrossRef] [PubMed]
- PET and SPECT Scanners. Molecular Imaging; Springer: Berlin/Heidelberg, Germany, 2009; pp. 59–82. [Google Scholar]
- Herzog, H. In vivo functional imaging with SPECT and PET. Radiochim. Acta 2001, 89, 203–214. [Google Scholar] [CrossRef] [Green Version]
- Kostelnik, T.I.; Orvig, C. Radioactive Main Group and Rare Earth Metals for Imaging and Therapy. Chem. Rev. 2019, 119, 902–956. [Google Scholar] [CrossRef]
- Ametamey, S.M.; Honer, M.; Schubiger, P.A. Molecular Imaging with PET. Chem. Rev. 2008, 108, 1501–1516. [Google Scholar] [CrossRef]
- Hatori, A.; Arai, T.; Yanamoto, K.; Yamasaki, T.; Kawamura, K.; Yui, J.; Konno, F.; Nakao, R.; Suzuki, K.; Zhang, M.-R. Biodistribution and metabolism of the anti-influenza drug [11C]oseltamivir and its active metabolite [11C]Ro 64-0802 in mice. Nucl. Med. Biol. 2009, 36, 47–55. [Google Scholar] [CrossRef]
- Bergström, M.; Cass, L.M.; Valind, S.; Westerberg, G.; Lundberg, E.-L.; Gray, S.; Bye, A.; Långström, B. Deposition and Disposition of [C]Zanamivir Following Administration as an Intranasal Spray. Clin. Pharmacokinet. 1999, 36, 33–39. [Google Scholar] [CrossRef]
- Saleem, A.; Harte, R.J.; Matthews, J.C.; Osman, S.; Brady, F.; Luthra, S.K.; Brown, G.D.; Bleehen, N.; Connors, T.; Jones, T.; et al. Pharmacokinetic Evaluation of N-[2-(Dimethylamino) Ethyl]Acridine-4-Carboxamide in Patients by Positron Emission Tomography. J. Clin. Oncol. 2001, 19, 1421–1429. [Google Scholar] [CrossRef]
- Aboagye, E.; Price, P.M.; Jones, T. In vivo pharmacokinetics and pharmacodynamics in drug development using positron-emission tomography. Drug Discov. Today 2001, 6, 293–302. [Google Scholar] [CrossRef]
- Rahmim, A.; Zaidi, H. PET versus SPECT: Strengths, limitations and challenges. Nucl. Med. Commun. 2008, 29, 193–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- George, N.; Gean, E.G.; Nandi, A.; Frolov, B.; Zaidi, E.; Lee, H.; Brašić, J.R.; Wong, D.F. Advances in CNS Imaging Agents: Focus on PET and SPECT Tracers in Experimental and Clinical Use. CNS Drugs 2015, 29, 313–330. [Google Scholar] [CrossRef] [PubMed]
- Mariani, G.; Bruselli, L.; Kuwert, T.; Kim, E.E.; Flotats, A.; Israel, O.; Dondi, M.; Watanabe, N. A review on the clinical uses of SPECT/CT. Eur. J. Nucl. Med. Mol. Imaging 2010, 37, 1959–1985. [Google Scholar] [CrossRef]
- Phelps, M.E.; Huang, S.C.; Hoffman, E.J.; Selin, C.; Sokoloff, L.; Kuhl, D.E. Tomographic measurement of local cerebral glucose metabolic rate in humans with (F-18)2-fluoro-2-deoxy-D-glucose: Validation of method. Ann. Neurol. 1979, 6, 371–388. [Google Scholar] [CrossRef]
- Verger, A.; Guedj, E. The renaissance of functional 18F-FDG PET brain activation imaging. Eur. J. Nucl. Med. Mol. Imaging 2018, 45, 2338–2341. [Google Scholar] [CrossRef] [Green Version]
- Fletcher, J.W.; Djulbegovic, B.; Soares, H.P.; Siegel, B.A.; Lowe, V.J.; Lyman, G.H.; Coleman, R.E.; Wahl, R.; Paschold, J.C.; Avril, N.; et al. Recommendations on the Use of 18F-FDG PET in Oncology. J. Nucl. Med. 2008, 49, 480–508. [Google Scholar] [CrossRef] [Green Version]
- Mackie, G.C. F-18 Fluorodeoxyglucose Positron Emission Tomographic Imaging of Cytomegalovirus Pneumonia. Clin. Nucl. Med. 2004, 29, 569–571. [Google Scholar] [CrossRef]
- Love, C.; Tomas, M.B.; Tronco, G.G.; Palestro, C.J. FDG PET of Infection and Inflammation. Radiographics 2005, 25, 1357–1368. [Google Scholar] [CrossRef] [Green Version]
- Bellani, G.; Messa, C.; Guerra, L.; Spagnolli, E.; Foti, G.; Patroniti, N.; Fumagalli, R.; Musch, G.; Fazio, F.; Pesenti, A. Lungs of patients with acute respiratory distress syndrome show diffuse inflammation in normally aerated regions: A [18F]-fluoro-2-deoxy-D-glucose PET/CT study. Crit. Care Med. 2009, 37, 2216–2222. [Google Scholar] [CrossRef]
- Setti, L.; Kirienko, M.; Dalto, S.C.; Bonacina, M.; Bombardieri, E. FDG-PET/CT findings highly suspicious for COVID-19 in an Italian case series of asymptomatic patients. Eur. J. Nucl. Med. Mol. Imaging 2020, 47, 1649–1656. [Google Scholar] [CrossRef] [PubMed]
- Olivari, L.; Riccardi, N.; Rodari, P.; Buonfrate, D.; Diodato, S.; Formenti, F.; Angheben, A.; Salgarello, M. Accidental diagnosis of COVID-19 pneumonia after 18F FDG PET/CT: A case series. Clin. Transl. Imaging 2020, 8, 393–400. [Google Scholar] [CrossRef] [PubMed]
- Lütje, S.; Marinova, M.; Kütting, D.; Attenberger, U.; Essler, M.; Bundschuh, R.A. Nuclear medicine in SARS-CoV-2 pandemia: 18F-FDG-PET/CT to visualize COVID-19. Nuklearmedizin 2020, 59, 276–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dyall, J.; Johnson, R.F.; Chen, D.-Y.; Huzella, L.; Ragland, D.R.; Mollura, D.J.; Byrum, R.; Reba, R.C.; Jennings, G.; Jahrling, P.B.; et al. Evaluation of Monkeypox Disease Progression by Molecular Imaging. J. Infect. Dis. 2011, 204, 1902–1911. [Google Scholar] [CrossRef] [Green Version]
- Dyall, J.; Johnson, R.F.; Chefer, S.; Leyson, C.; Thomasson, D.; Seidel, J.; Ragland, D.R.; Byrum, R.; Jett, C.; Cann, J.A.; et al. [18F]-Fluorodeoxyglucose Uptake in Lymphoid Tissue Serves as a Predictor of Disease Outcome in the Nonhuman Primate Model of Monkeypox Virus Infection. J. Virol. 2017, 91, e00897-17. [Google Scholar] [CrossRef] [Green Version]
- Jonsson, C.B.; Camp, J.V.; Wu, A.; Zheng, H.; Kraenzle, J.L.; Biller, A.E.; Vanover, C.D.; Chu, Y.-K.; Ng, C.K.; Proctor, M.; et al. Molecular Imaging Reveals a Progressive Pulmonary Inflammation in Lower Airways in Ferrets Infected with 2009 H1N1 Pandemic Influenza Virus. PLoS ONE 2012, 7, e40094. [Google Scholar] [CrossRef] [Green Version]
- Schniering, J.; Benešová, M.; Brunner, M.; Haller, S.; Cohrs, S.; Frauenfelder, T.; Vrugt, B.; Feghali-Bostwick, C.; Schibli, R.; Distler, O.; et al. 18F-AzaFol for Detection of Folate Receptor-β Positive Macrophages in Experimental Interstitial Lung Disease—A Proof-of-Concept Study. Front. Immunol. 2019, 10, 2724. [Google Scholar] [CrossRef] [Green Version]
- Dorward, D.A.; Lucas, C.D.; Rossi, A.; Haslett, C.; Dhaliwal, K. Imaging inflammation: Molecular strategies to visualize key components of the inflammatory cascade, from initiation to resolution. Pharmacol. Ther. 2012, 135, 182–199. [Google Scholar] [CrossRef]
- Venneti, S.; Lopresti, B.J.; Wang, G.; Bissel, S.J.; Mathis, C.A.; Meltzer, C.C.; Boada, F.; Capuano, S.; Kress, G.J.; Davis, D.K.; et al. PET imaging of brain macrophages using the peripheral benzodiazepine receptor in a macaque model of neuroAIDS. J. Clin. Investig. 2004, 113, 981–989. [Google Scholar] [CrossRef] [Green Version]
- Rubin, R.H.; Fischman, A.J. The use of radiolabeled, nonspecific immunoglobulin in the detection of focal inflammation. Semin. Nucl. Med. 1994, 24, 169–179. [Google Scholar] [CrossRef]
- Buscombe, J.R.; Oyen, W.J.; Corstens, F.H. Use of polyclonal IgG in HIV infection and AIDS. Q. J. Nucl. Med. Off. Publ. Ital. Assoc. Nucl. Med. (AIMN) Int. Assoc. Radiopharm. (IAR) 1995, 39, 212–220. [Google Scholar]
- Kumar, V. Radiolabeled white blood cells and direct targeting of micro-organisms for infection imaging. Q. J. Nucl. Med. Mol. Imaging 2005, 49, 325–338. [Google Scholar] [PubMed]
- Wanahita, A.; Villeda, C.; Kutka, N.; Ramírez, J.A.; Musher, D. Diagnostic sensitivity and specificity of the radionuclide (indium)-labeled leukocyte scan. J. Infect. 2007, 55, 214–219. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Liu, Y. 99mTc-Leukocyte Scintigraphy Revealed Viral Pulmonary Infection in a COVID-19 Patient. Clin. Nucl. Med. 2020, 45, 821–823. [Google Scholar] [CrossRef]
- Carfì, A.; Bernabei, R.; Landi, F.; for the Gemelli Against COVID-19 Post-Acute Care Study Group. Persistent Symptoms in Patients After Acute COVID-19. JAMA 2020, 324, 603. [Google Scholar] [CrossRef]
- Zhao, Y.-M.; Shang, Y.-M.; Song, W.-B.; Li, Q.-Q.; Xie, H.; Xu, Q.-F.; Jia, J.-L.; Li, L.-M.; Mao, H.-L.; Zhou, X.-M.; et al. Follow-up study of the pulmonary function and related physiological characteristics of COVID-19 survivors three months after recovery. EClinicalMedicine 2020, 25, 100463. [Google Scholar] [CrossRef]
- Lyons, D.; Frampton, M.; Naqvi, S.; Donohoe, D.; Adams, G.; Glynn, K. Fallout from the COVID-19 pandemic—should we prepare for a tsunami of post viral depression? Ir. J. Psychol. Med. 2020, 1–6. [Google Scholar] [CrossRef]
- Perrotta, F.; Matera, M.G.; Cazzola, M.; Bianco, A. Severe respiratory SARS-CoV2 infection: Does ACE2 receptor matter? Respir. Med. 2020, 168, 105996. [Google Scholar] [CrossRef]
- Fang, L.; Karakiulakis, G.; Roth, M. Are patients with hypertension and diabetes mellitus at increased risk for COVID-19 infection? Lancet Respir. Med. 2020, 8, e21. [Google Scholar] [CrossRef]
- Ehlerding, E.B.; Sun, L.; Lan, X.; Zeng, D.; Cai, W. Dual-Targeted Molecular Imaging of Cancer. J. Nucl. Med. 2018, 59, 390–395. [Google Scholar] [CrossRef]
- Liu, Z.; Wang, F. Dual-targeted molecular probes for cancer imaging. Curr. Pharm. Biotechnol. 2010, 11, 610–619. [Google Scholar] [CrossRef] [PubMed]
- Di Mascio, M.; Srinivasula, S.; Bhattacharjee, A.; Cheng, L.; Martiniova, L.; Herscovitch, P.; Lertora, J.; Kiesewetter, D. Antiretroviral Tissue Kinetics: In Vivo Imaging Using Positron Emission Tomography. Antimicrob. Agents Chemother. 2009, 53, 4086–4095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konno, F.; Arai, T.; Zhang, M.-R.; Hatori, A.; Yanamoto, K.; Ogawa, M.; Ito, G.; Odawara, C.; Yamasaki, T.; Kato, K.; et al. Radiosyntheses of two positron emission tomography probes: [11C]Oseltamivir and its active metabolite [11C]Ro 64-0802. Bioorganic Med. Chem. Lett. 2008, 18, 1260–1263. [Google Scholar] [CrossRef] [PubMed]
- Cass, L.M.R.; Brown, J.; Pickford, M.; Fayinka, S.; Newman, S.P.; Johansson, C.J.; Bye, A. Pharmacoscintigraphic Evaluation of Lung Deposition of Inhaled Zanamivir in Healthy Volunteers. Clin. Pharmacokinet. 1999, 36, 21–31. [Google Scholar] [CrossRef]
- Brader, P.; Kelly, K.; Gang, S.; Shah, J.P.; Wong, R.J.; Hricak, H.; Blasberg, R.G.; Fong, Y.; Gil, Z. Imaging of Lymph Node Micrometastases Using an Oncolytic Herpes Virus and [18F] FEAU PET. PLoS ONE 2009, 4, e4789. [Google Scholar] [CrossRef] [Green Version]
- Kuruppu, D.; Brownell, A.-L.; Zhu, A.; Yu, M.; Wang, X.; Kulu, Y.; Fuchs, B.C.; Kawasaki, H.; Tanabe, K.K. Positron Emission Tomography of Herpes Simplex Virus 1 Oncolysis. Cancer Res. 2007, 67, 3295–3300. [Google Scholar] [CrossRef] [Green Version]
- Diaz, L.A.; Foss, C.A.; Thornton, K.; Nimmagadda, S.; Endres, C.J.; Uzuner, O.; Seyler, T.M.; Ulrich, S.D.; Conway, J.; Bettegowda, C.; et al. Imaging of Musculoskeletal Bacterial Infections by [124I]FIAU-PET/CT. PLoS ONE 2007, 2, e1007. [Google Scholar] [CrossRef]
- Gowrishankar, G.; Hardy, J.; Wardak, M.; Namavari, M.; Reeves, R.E.; Neofytou, E.; Srinivasan, A.; Wu, J.C.; Contag, C.H.; Gambhir, S.S. Specific Imaging of Bacterial Infection Using 6″-18F-Fluoromaltotriose: A Second-Generation PET Tracer Targeting the Maltodextrin Transporter in Bacteria. J. Nucl. Med. 2017, 58, 1679–1684. [Google Scholar] [CrossRef] [Green Version]
- Bray, M.; Di Mascio, M.; De Kok-Mercado, F.; Mollura, D.J.; Jagoda, E. Radiolabeled antiviral drugs and antibodies as virus-specific imaging probes. Antivir. Res. 2010, 88, 129–142. [Google Scholar] [CrossRef]
- Bray, M.; Lawler, J.V.; Paragas, J.; Jahrling, P.B.; Mollura, D.J. Molecular Imaging of Influenza and Other Emerging Respiratory Viral Infections. J. Infect. Dis. 2011, 203, 1348–1359. [Google Scholar] [CrossRef] [Green Version]
- Bocan, T.M.; Panchal, R.G.; Bavari, S. Applications of In Vivo Imaging in the Evaluation of the Pathophysiology of Viral and Bacterial Infections and in Development of Countermeasures to BSL3/4 Pathogens. Mol. Imaging Biol. 2014, 17, 4–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santangelo, P.J.; A Rogers, K.; Zurla, C.; Blanchard, E.L.; Gumber, S.; Strait, K.; Connor-Stroud, F.; Schuster, D.M.; Amancha, P.K.; Hong, J.J.; et al. Whole-body immunoPET reveals active SIV dynamics in viremic and antiretroviral therapy–treated macaques. Nat. Chem. Biol. 2015, 12, 427–432. [Google Scholar] [CrossRef] [Green Version]
- Sharma, S.K.; Lyashchenko, S.K.; Park, H.A.; Pillarsetty, N.; Roux, Y.; Wu, J.; Poty, S.; Tully, K.M.; Poirier, J.T.; Lewis, J.S. A rapid bead-based radioligand binding assay for the determination of target-binding fraction and quality control of radiopharmaceuticals. Nucl. Med. Biol. 2019, 71, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Pillarsetty, N.; Carter, L.M.; Lewis, J.S.; Reiner, T. Oncology-Inspired Treatment Options for COVID-19. J. Nucl. Med. 2020, 61, 1720–1723. [Google Scholar] [CrossRef]
- Nie, J.; Li, Q.; Wu, J.; Zhao, C.; Hao, H.; Liu, H.; Zhang, L.; Nie, L.; Qin, H.; Wang, M.; et al. Establishment and validation of a pseudovirus neutralization assay for SARS-CoV-2. Emerg. Microbes Infect. 2020, 9, 680–686. [Google Scholar] [CrossRef] [Green Version]
- Ou, X.; Liu, Y.; Lei, X.; Li, P.; Mi, D.; Ren, L.; Guo, L.; Guo, R.; Chen, T.; Hu, J.; et al. Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat. Commun. 2020, 11, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.-W.; Tai, C.-H.; Hsu, Y.-M.; Cheng, D.; Hung, S.-J.; Chai, K.M.; Wang, Y.-F.; Wang, J.-R. Assessing the application of a pseudovirus system for emerging SARS-CoV-2 and re-emerging avian influenza virus H5 subtypes in vaccine development. Biomed. J. 2020, 43, 375–387. [Google Scholar] [CrossRef]
- Scandella, E.; Eriksson, K.K.; Hertzig, T.; Drosten, C.; Chen, L.; Gui, C.; Luo, X.; Shen, J.; Shen, X.; Siddell, S.G.; et al. Identification and Evaluation of Coronavirus Replicase Inhibitors Using a Replicon Cell Line. In Retinal Degenerative Diseases; Springer Science and Business Media LLC: Berlin/Heidelberg, Germany, 2006; Volume 581, pp. 609–613. [Google Scholar]
- Wang, J.-M.; Wang, L.-F.; Shi, Z.-L. Construction of a non-infectious SARS coronavirus replicon for application in drug screening and analysis of viral protein function. Biochem. Biophys. Res. Commun. 2008, 374, 138–142. [Google Scholar] [CrossRef]
- Oppenheimer, A. Roentgen therapy of “virus” pneumonia. Am. J. Roentgenol. Rad. Ther. 1943, 49, 635–638. [Google Scholar]
- Dubin, I.N.; Baylin, G.J.; Gobble, W.G. The effect of roentgen therapy on experimental virus pneumonia; on pneumonia produced in white mice by swine influenza virus. Am. J. Roentgenol. Rad. Ther. 1946, 55, 478–481. [Google Scholar]
- Seegenschmiedt, M.H.; Micke, O.; Muecke, R.; The German Cooperative Group on Radiotherapy for Non-malignant Diseases (GCG-BD). Radiotherapy for non-malignant disorders: State of the art and update of the evidence-based practice guidelines. Br. J. Radiol. 2015, 88, 20150080. [Google Scholar] [CrossRef] [PubMed]
- Trott, K.R. Therapeutic effects of low radiation doses. Strahlenther. Onkol. 1994, 170, 1–12. [Google Scholar] [PubMed]
- Available online: https://clinicaltrials.gov/ct2/show/NCT04380818 (accessed on 20 December 2020).
- Available online: https://clinicaltrials.gov/ct2/show/NCT04377477 (accessed on 20 December 2020).
- Available online: https://clinicaltrials.gov/ct2/show/NCT04366791 (accessed on 20 December 2020).
- Tharmalingam, H.; Díez, P.; Tsang, Y.; Hawksley, A.; Conibear, J.; Thiruthaneeswaran, N. Personal View: Low-dose Lung Radiotherapy for COVID-19 Pneumonia—The Atypical Science and the Unknown Collateral Consequence. Clin. Oncol. 2020, 32, 497–500. [Google Scholar] [CrossRef] [PubMed]
- Salomaa, S.; Bouffler, S.D.; Atkinson, M.J.; Cardis, E.; Hamada, N. Is there any supportive evidence for low dose radiotherapy for COVID-19 pneumonia? Int. J. Radiat. Biol. 2020, 2020, 1–8. [Google Scholar] [CrossRef]
- Dash, A.; Knapp, F.F.R.; Pillai, M.R.A. Targeted Radionuclide Therapy—An Overview. Curr. Radiopharm. 2013, 6, 152–180. [Google Scholar] [CrossRef]
- Sgouros, G.; Bodei, L.; McDevitt, M.R.; Nedrow, J.R. Radiopharmaceutical therapy in cancer: Clinical advances and challenges. Nat. Rev. Drug Discov. 2020, 19, 589–608. [Google Scholar] [CrossRef]
- Sugiura, G.; Kühn, H.; Sauter, M.; Haberkorn, U.; Mier, W. Radiolabeling Strategies for Tumor-Targeting Proteinaceous Drugs. Molecules 2014, 19, 2135–2165. [Google Scholar] [CrossRef]
- Zoller, F.; Eisenhut, M.; Haberkorn, U.; Mier, W. Endoradiotherapy in cancer treatment—Basic concepts and future trends. Eur. J. Pharmacol. 2009, 625, 55–62. [Google Scholar] [CrossRef]
- Price, E.W.; Orvig, C. Matching chelators to radiometals for radiopharmaceuticals. Chem. Soc. Rev. 2014, 43, 260–290. [Google Scholar] [CrossRef]
- Boros, E.; Packard, A.B. Radioactive Transition Metals for Imaging and Therapy. Chem. Rev. 2019, 119, 870–901. [Google Scholar] [CrossRef]
- Cutler, C.S.; Hennkens, H.M.; Sisay, N.; Huclier-Markai, S.; Jurisson, S.S. Radiometals for Combined Imaging and Therapy. Chem. Rev. 2013, 113, 858–883. [Google Scholar] [CrossRef] [PubMed]
- Kassis, A.I. Therapeutic Radionuclides: Biophysical and Radiobiologic Principles. Semin. Nucl. Med. 2008, 38, 358–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walicka, M.A.; Vaidyanathan, G.; Zalutsky, M.R.; Adelstein, S.J.; Kassis, A.I. Survival and DNA damage in Chinese hamster V79 cells exposed to alpha particles emitted by DNA-incorporated astatine-211. Radiat. Res. 1998, 150, 263. [Google Scholar] [CrossRef] [PubMed]
- Kassis, A.I.; Harris, C.R.; Adelstein, S.J. The in Vitro Radiobiology of Astatine-211 Decay. Radiat. Res. 1986, 105, 27. [Google Scholar] [CrossRef]
- Charlton, D.; Kassis, A.; Adelstein, S. A Comparison of Experimental and Calculated Survival Curves for V79 Cells Grown as Monolayers or in Suspension Exposed to Alpha Irradiation from 212Bi Distributed in the Growth Medium. Radiat. Prot. Dosim. 1994, 52, 311–315. [Google Scholar] [CrossRef]
- Chan, P.C.; Lisco, E.; Lisco, H.; Adelstein, S.J. The radiotoxicity of iodine-125 in mammalian cells II. A comparative study on cell survival and cytogenetic responses to 125IUdR, 131TUdR, and 3HTdR. Radiat. Res. 1976, 67, 332–343. [Google Scholar] [CrossRef]
- Cornelissen, B.; A Vallis, K. Targeting the nucleus: An overview of Auger-electron radionuclide therapy. Curr. Drug Discov. Technol. 2010, 7, 263–279. [Google Scholar] [CrossRef]
- Le, D. Radiopharmaceuticals for Therapy. J. Nucl. Med. 2017, 58, 1526. [Google Scholar] [CrossRef] [Green Version]
- Helal, M.; Dadachova, E. Radioimmunotherapy as a Novel Approach in HIV, Bacterial, and Fungal Infectious Diseases. Cancer Biother. Radiopharm. 2018, 33, 330–335. [Google Scholar] [CrossRef]
- Dadachova, E.; Patel, M.C.; Toussi, S.; Apostolidis, C.; Morgenstern, A.; Brechbiel, M.W.; Gorny, M.K.; Zolla-Pazner, S.; Casadevall, A.; Goldstein, H. Targeted Killing of Virally Infected Cells by Radiolabeled Antibodies to Viral Proteins. PLoS Med. 2006, 3, e427. [Google Scholar] [CrossRef]
- Dadachova, E. Future Vistas in Alpha Therapy of Infectious Diseases. J. Med. Imaging Radiat. Sci. 2019, 50, S49–S52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casadevall, A.; Goldstein, H.; Dadachova, E. Targeting host cells harbouring viruses with radiolabeled antibodies. Expert Opin. Biol. Ther. 2007, 7, 595–597. [Google Scholar] [CrossRef] [PubMed]
- Dadachova, E.; Burns, T.; Bryan, R.A.; Apostolidis, C.; Brechbiel, M.W.; Nosanchuk, J.D.; Casadevall, A.; Pirofski, L. Feasibility of Radioimmunotherapy of Experimental Pneumococcal Infection. Antimicrob. Agents Chemother. 2004, 48, 1624–1629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dadachova, E.; Nakouzi, A.; Bryan, R.A.; Casadevall, A. Ionizing radiation delivered by specific antibody is therapeutic against a fungal infection. Proc. Natl. Acad. Sci. USA 2003, 100, 10942–10947. [Google Scholar] [CrossRef] [Green Version]
- Vogg, A.T.J.; Drude, N.; Mottaghy, F.M.; Morgenroth, A.; Miran, T. Modulation of glutathione promotes apoptosis in triple-negative breast cancer cells. FASEB J. 2018, 32, 2803–2813. [Google Scholar] [CrossRef] [Green Version]
- Miran, T.; Vogg, A.T.J.; El Moussaoui, L.; Kaiser, H.-J.; Drude, N.; Von Felbert, V.; Mottaghy, F.M.; Morgenroth, A. Dual addressing of thymidine synthesis pathways for effective targeting of proliferating melanoma. Cancer Med. 2017, 6, 1639–1651. [Google Scholar] [CrossRef]
- Morgenroth, A.; Deisenhofer, S.; Glatting, G.; Kunkel, F.H.-G.; Dinger, C.; Zlatopolskiy, B.D.; Vogg, A.T.; Kull, T.; Reske, S.N. Preferential Tumor Targeting and Selective Tumor Cell Cytotoxicity of 5-[131/125I]Iodo-4′-Thio-2′-Deoxyuridine. Clin. Cancer Res. 2008, 14, 7311–7319. [Google Scholar] [CrossRef] [Green Version]
- Morgenroth, A.; Dinger, C.; Zlatopolskiy, B.D.; Al-Momani, E.; Glatting, G.; Mottaghy, F.M.; Reske, S.N. Auger electron emitter against multiple myeloma—targeted endo-radio-therapy with 125I-labeled thymidine analogue 5-iodo-4′-thio-2′-deoxyuridine. Nucl. Med. Biol. 2011, 38, 1067–1077. [Google Scholar] [CrossRef]
- Morgenroth, A.; Vogg, A.T.J.; Ermert, K.; Zlatopolskiy, B.D.; Mottaghy, F.M. Hedgehog signaling sensitizes Glioma stem cells to endogenous nano-irradiation. Oncotarget 2014, 5, 5483–5493. [Google Scholar] [CrossRef] [Green Version]
- Morgenroth, A.; Vogg, A.; Mottaghy, F.M.; Schmaljohann, J. Targeted endoradiotherapy using nucleotides. Methods 2011, 55, 203–214. [Google Scholar] [CrossRef]
- Morgenroth, A.; Vogg, A.T.J.; Zlatopolskiy, B.D.; Siluschek, M.; Oedekoven, C.A.; Mottaghy, F.M. Breaking the Invulnerability of Cancer Stem Cells: Two-Step Strategy to Kill the Stem-like Cell Subpopulation of Multiple Myeloma. Mol. Cancer Ther. 2013, 13, 144–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arvin, A.M.; Fink, K.; Schmid, M.A.; Cathcart, A.; Spreafico, R.; Havenar-Daughton, C.; Lanzavecchia, A.; Corti, D.; Virgin, H.W. A perspective on potential antibody-dependent enhancement of SARS-CoV-2. Nat. Cell Biol. 2020, 584, 353–363. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.S.; Wheatley, A.; Kent, S.J.; DeKosky, B.J. Antibody-dependent enhancement and SARS-CoV-2 vaccines and therapies. Nat. Microbiol. 2020, 5, 1185–1191. [Google Scholar] [CrossRef] [PubMed]
- Dadachova, E.; Kitchen, S.G.; Bristol, G.; Baldwin, G.C.; Revskaya, E.; Empig, C.; Thornton, G.B.; Gorny, M.K.; Zolla-Pazner, S.; Casadevall, A. Pre-Clinical Evaluation of a 213Bi-Labeled 2556 Antibody to HIV-1 gp41 Glycoprotein in HIV-1 Mouse Models as a Reagent for HIV Eradication. PLoS ONE 2012, 7, e31866. [Google Scholar] [CrossRef]
- Dingli, D.; Peng, K.-W.; Harvey, M.E.; Vongpunsawad, S.; Bergert, E.R.; Kyle, R.A.; Cattaneo, R.; Morris, J.C.; Russell, S.J. Interaction of measles virus vectors with Auger electron emitting radioisotopes. Biochem. Biophys. Res. Commun. 2005, 337, 22–29. [Google Scholar] [CrossRef]
- De Kruijff, R.M.; Wolterbeek, H.T.; Denkova, A.G. A Critical Review of Alpha Radionuclide Therapy—How to Deal with Recoiling Daughters? Pharmaceuticals 2015, 8, 321–336. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inhibitor | IC50 [µM] | CC50 [µM] | Class | Target | Ref. |
|---|---|---|---|---|---|
| 311mab-31B5 | 0.0338 | NA | nAB | S protein (RBD) | [27] |
| 311mab-32D4 | 0.0698 | NA | nAB | S protein (RBD) | [27] |
| 47D11 | 0.57 | NA | nAB | S protein (RBD) | [28] |
| COVA1-18 | NA | NA | nAB | S protein (RBD) | [29] |
| COVA2-15 | NA | NA | nAB | S protein (RBD) | [29] |
| CR3022 | - | NA | nnAB | S protein (RBD) | [37,38] |
| Arbidol | 4.11 µM | 31.79 µM | SM | S protein (S2) | [30] |
| Inhibitor | IC50 [µM] | EC50 [µM] | CC50 [µM] | Class | MOA | Ref. |
|---|---|---|---|---|---|---|
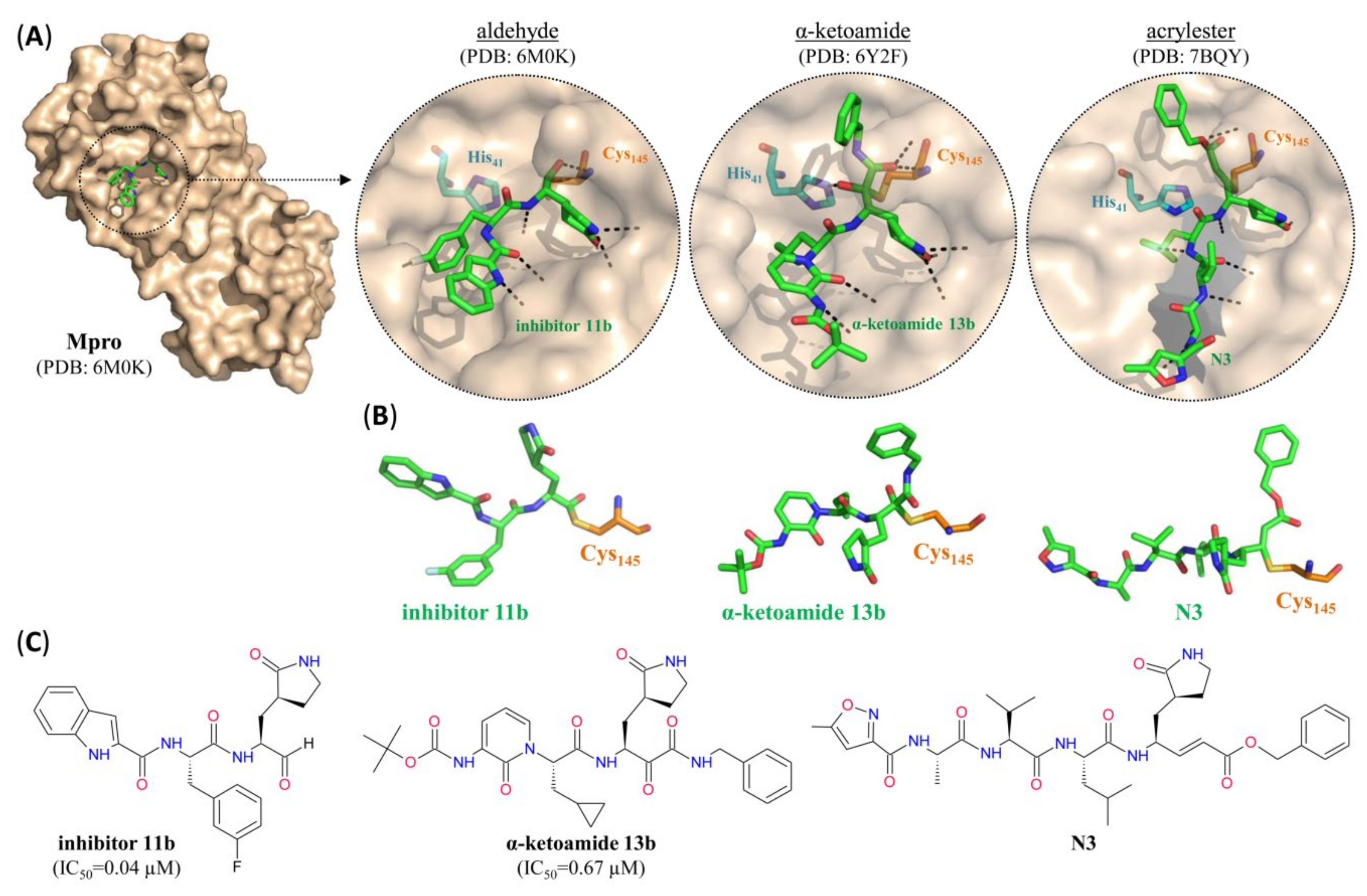
| GC-376 | 0.03 | 3.14–3.37 | >100 | PA1 | C | [43] |
| Inhibitor 11b | 0.04 | 0.72 | NA | PA | C | [22] |
| Inhibitor 11a | 0.05 | 0.53 | NA | PA | C | [22] |
| alpha-ketoamide 11r | 0.18 | NA | NA | PAK | C | [44] |
| Calpain inhibitor XII | 0.45 | 0.49–0.78 | >50 | PAK | C | [43] |
| Ebselen | 0.67 | 4.67 | NA | SM | C | [23] |
| alpha-ketoamide 13b | 0.67 | 4–5 | NA | PAK | C | [44] |
| Baicalein | 0.94 | 2.94 | >200 | SM | NC | [49] |
| Calpain inhibitor II (ALLM) | 0.97 | 2.07–3.70 | >100 | PA | C | [43] |
| Tideglusib | 1.55 | NA | NA | SM | C | [23] |
| Carmofur | 1.82 | 24.30 | NA | SM | C | [23,46] |
| alpha-ketoamide 13a | 2.39 | NA | NA | PAK | C | [44] |
| MG-115 (Proteasome inhibitor) | 3.14 | NA | NA | PA | C | [43] |
| MG-132 (Proteasome inhibitor) | 3.90 | NA | <1 | PA | C | [43] |
| Boceprevir | 4.13 | 1.31–1.95 | >100 | PAK | C | [43] |
| Narlaprevir | 4.73 | NA | NA | PAK | C | [43] |
| Baicalin | 6.41 | 27.87 | >200 | SM | NC | [49] |
| Calpain inhibitor I (ALLN/MG-101) | 8.60 | NA | NA | PA | C | [43] |
| Disulfiram | 9.35 | NA | NA | SM | C | [23] |
| Proteasome inhibitor I (PSI) | 10.38 | NA | NA | PA | C | [43] |
| Calpeptin | 10.69 | NA | NA | PA | C | [43] |
| Simeprevir | 13.75 | NA | NA | PMC | C | [43] |
| Shikonin | 15.75 | NA | NA | SM | NC | [23] |
| PX-12 | 21.39 | NA | NA | SM | C | [23] |
| N3 | NA | 16.77 | >100 | PAE | C | [23,45] |
| Inhibitor | IC50 [µM] | EC50 [µM] | CC50 [µM] | Class | MOA | Ref. |
|---|---|---|---|---|---|---|
| Ebselen | 2.0–2.4 | 4.67 | NA | SM | C+ZE | [23,47,48] |
| GRL-0617 | 2.4 | 27.6 | NA | SM | NC | [24,52] |
| Compound 6 | 5.0 | 21.0 | NA | SM | NC | [52] |
| Disulfiram | 7.5 | NA | NA | SM | C+ZE | [48] |
| 7724772 | 23.5 | NA | NA | SM | NC | [52] |
| 6577871 | 100.7 | NA | NA | SM | NC | [52] |
| 9247873 | >200 | NA | NA | SM | NC | [52] |
| VIR251 | NA | NA | NA | PVME | C | [53] |
| VIR250 | NA | NA | NA | PVME | C | [53] |
| Radionuclide | Half-Life | Daughters | Decay Type (Probability) | Use |
|---|---|---|---|---|
| 15O | 2 min | 15N (stable) | β+ (99.9%), EC (0.1%) | PET |
| 13N | 10 min | 13C (stable) | β+ (99.8%), EC (0.2%) | PET |
| 11C | 20 min | 11B (stable) | β+ (99.7%), EC (0.3%) | PET |
| 68Ga | 67 min | 68Zn (stable) | β+ (88.9%), EC (11.1%) | PET |
| 18F | 110 min | 18O (stable) | β+ (97.0%), EC (3.0%) | PET |
| 64Cu | 12.7 h | 64Ni/64Zn (stable) | β+ (17.9%), EC (43.1%), β- (39.0%) | PET |
| 99mTc | 6 h | 99Tc a | [γ-Ray (88%), IC (12%)] | SPECT |
| 123I | 13.2 h | 123Te b | EC (100%) [γ-Ray (84%), IC (16%)] | SPECT |
| 111In | 67 h | 111Cd (stable) | EC (100%) [γ-Ray (100%)] | SPECT |
| α -Particle Emission | β -Particle Emission | Auger Emission | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Radio- | Pa | Emax | Range | Pa | Emax | Range | Pa | Emax | Range | ||
| Nuclide | Daughters | Half-Life | [%] | [MeV] | [µm] | [%] | [MeV] | [mm] | [%] | [keV] | [µm] |
| 213Bi | 46 min | 2.2 | 5.9 | 50 | 97.8 | 0.5 | 1.7 | - | - | - | |
| 213 Po→209Pb | 4.2 µs | 100 | 8.4 | 90 | - | - | - | - | - | - | |
| 209 TI→209Pb | 2.2 min | - | - | - | 100 | 0.6 | 2.3 | - | - | - | |
| 209 Pb→209Bi (stable) | 3.3 h | - | - | - | 100 | 0.2 | 0.5 | - | - | - | |
| 212Bi | 1 h | 35.9 | 6.1 | 50 | 64.1 | 0.8 | 3.3 | - | - | - | |
| 212 Po→208Pb (stable) | 299 ns | 100 | 8.8 | 100 | - | - | - | - | - | - | |
| 208 TI→208Pb (stable) | 3.1 min | - | 100 | 0.3–0.6 | 0.9–2.3 | - | - | - | |||
| 211At | 7.2 h | 41.8 | 5.9 | 50 | - | - | - | 58.2 | 93 | <0.5 | |
| 211 Po→207Pb (stable) | 516 ms | 100 | 7.4 | 70 | - | - | - | - | - | - | |
| 207 Bi→207Pb (stable) | 33 yrsb | - | - | - | - | - | - | 100 | 88 | <0.5 | |
| 188Re | 188Os (stable) | 17.0 h | - | - | - | 100 | 2.0 | 10 | - | - | - |
| 166Ho | 166Er (stable) | 28.8 h | - | - | - | 100 | 1.8 | 9 | - | - | - |
| 103mRh | 103Rh (stable) | 56 min | - | - | - | - | - | - | 100 | 39.7 | <0.5 |
| 161Ho | 161Dy (stable) | 2.48 h | - | - | - | - | - | - | 100 | NA | <0.5 |
| 123I | 123Te (stable) | 13.2 h | - | - | - | - | - | - | 100 | 32 | <0.5 |
| 111In | 111Cd (stable) | 67 h | - | - | - | - | - | - | 100 | 26 | <0.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Neumaier, F.; Zlatopolskiy, B.D.; Neumaier, B. Nuclear Medicine in Times of COVID-19: How Radiopharmaceuticals Could Help to Fight the Current and Future Pandemics. Pharmaceutics 2020, 12, 1247. https://doi.org/10.3390/pharmaceutics12121247
Neumaier F, Zlatopolskiy BD, Neumaier B. Nuclear Medicine in Times of COVID-19: How Radiopharmaceuticals Could Help to Fight the Current and Future Pandemics. Pharmaceutics. 2020; 12(12):1247. https://doi.org/10.3390/pharmaceutics12121247
Chicago/Turabian StyleNeumaier, Felix, Boris D. Zlatopolskiy, and Bernd Neumaier. 2020. "Nuclear Medicine in Times of COVID-19: How Radiopharmaceuticals Could Help to Fight the Current and Future Pandemics" Pharmaceutics 12, no. 12: 1247. https://doi.org/10.3390/pharmaceutics12121247