1. Introduction
Male sexual dysfunction (MSD) is a mixed group of complaints that are typically related to a person’s inability to respond sexually or to achieve sexual stimulation. Male sexual dysfunction is a common problem mainly associated with erectile dysfunction (ED) and/or premature ejaculation (PE). It was reported that by 2025 ED is expected to affect about 322 million men around the world. ED is not life-threatening, but it can affect the quality of life of sexual partners and lead to some emotional problems such as anxiety and depression. ED clearly can lead to isolation and prevention between the couple and sometimes these problems can be extended to their jobs and cause negative interaction with others [
1]. In addition, the prevalence of PE ranged from 19% to 30% in the general population [
2,
3]. Regardless of the type of PE, it is usually associated with psychological difficulties that may happen to a person and this will reduce his sexual desire. It affects the quality of a partner’s life that affects his sexual satisfaction, confidence, and interpersonal relationships [
4,
5,
6]. About one-hundred million men worldwide cannot reach satisfactory sexual performance related to the failure of an adequate erection. Patient numbers with male sexual dysfunction will increase to double their current amount in the next twenty-five years [
7]. Therefore, it is very important to treat a person that suffers from sexual dysfunction because this issue will lead to a deteriorating partner’s life quality [
8].
It is worth mentioning that PE is present in up to 30% of men with ED [
9]. ED and PE are not separate things but should be considered as an interconnected case of sexual dysfunction. This point of view supports sexual health care professionals in providing the most suitable therapeutic tactic to improve patient-related results in sexual medicine [
10]. It was reported that ED was strongly associated with PE among men with diabetes [
11]. PE and ED often coexist and are connected with psychosocial anguish, which tourist attractions the vital role of screening for their co-existence and the need for combined therapy [
12].
A potent and selective phosphodiesterase-5 inhibitor (PDE5-I), tadalafil (TDL) is one of the most efficient medicines for the treatment of ED [
13]. On the other hand, a selective serotonin reuptake inhibitor (SSRI), dapoxetine (DPX) is newly approved for the treatment of PE [
14,
15]. It is the first and unique oral pharmacological agent used, and the only SSRI approved in more than 60 countries to treat men with PE [
16]. On comparing the on-demand dosing of DPX alone and combined with PDE5-I in subjects with PE and without ED, it was found that a low dose of DPX combined with PDE5-I showed better outcomes compared with that of DPX only. This finding supports the recommendation that the PDE5-Is have a potential role in the treatment of PE without ED [
17]. Besides, the combined use of SSRIs and PDE5-Is provided additive favorable effects in men with PE compared with SSRIs or PDE5-I monotherapy [
18]. Moreover, DPX provided a remarkable treatment benefit in men with PE and comorbid ED on a stable regimen of PDE5-I [
19]. Finally, it was reported that the combination of DPX with TDL is well tolerated and the concomitant administration of TDL and DPX did not affect the pharmacokinetics of both APIs [
20].
However, TDL undergoes low bioavailability due to its inherent poor aqueous solubility [
21,
22]. In addition, DPX was suffering from low and variable oral bioavailability that ranges from 15 to 76% [
23]. This low and variable drug concentration in the blood may lead to a decrease in their efficacy and/or exaggerated side effects.
To overwhelm this hurdle that encounters the formulators of the oral solid dosage form, many researchers have developed various approaches to improve drug water-solubility. From these approaches, adjustment of the pH, the addition of cosolvent, particle size reduction, solid dispersion [
24], salt formation or formulation of the drug in lipid-based nanovesicles such as liposomes [
25], nanosuspension development [
26,
27], prodrug synthesis [
28], the formation of micro- and nano-particles [
29], or incorporation of drugs into the porous structure and nanoemulsion formulations [
30,
31]. However, the liquisolid (LS) technique is a promising approach used to enhance the solubility of drugs. When the patient takes drugs orally in a state of a solid dosage form such as capsule or tablet, it should be released and dissolved in gastrointestinal fluids before absorbing to give its desired therapeutic effects. Many poorly water-soluble drugs have a limitation of bioavailability related to their dissolution rate. The technique is used to convert a liquid into easily compressible, non-adherent, and free-flowing dry powder by mixing with selected appropriate excipients known as carrier materials (such as cellulose, starch, lactose, and Avicel PH 101) and coating materials (such as silica powder). It is used to increase drug release properties, hence the bioavailability of water-insoluble drugs due to the observed increase in the surface area of the drug and the wetting properties available for dissolution. The LS technique has contributed to diminishing the effect of pH changes on drug release [
23,
25,
32].
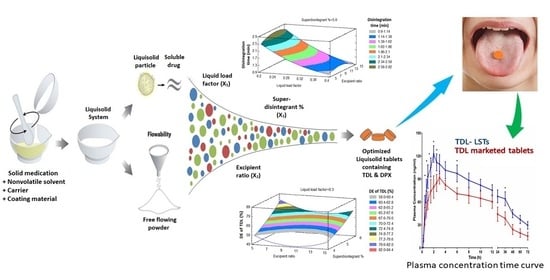
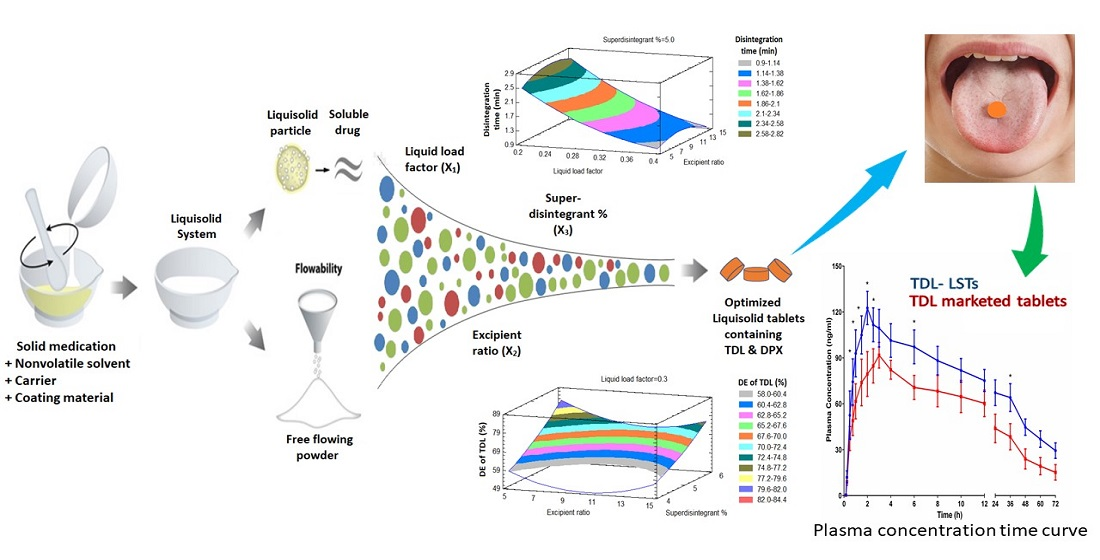
Therefore, the objective of the present study was to develop an optimized combined-dose medication containing TDL and DPX as an effective therapy for male sexual dysfunction. To achieve this aim, the BBD will be used to investigate the effect of different formulation factors on the quality of LSTs that subsequently affect the dissolution and the bioavailability of TDL and DPX.
2. Materials and Methods
TDL was gifted from SAJA Pharmaceutical Co. Ltd. (Jeddah, Saudi Arabia). DPX was kindly gifted from Spimaco Addwaeih (Riyadh, Saudi Arabia). Glycerine was supplied by Crescent Diagnostics (Jeddah, Saudi Arabia). Cellulose microcrystalline (Avicel® PH-101), sorbitan monooleate 80 (Span® 80), Macrogolglycerol ricinoleate; Kolliphor® EL (Cremophor® EL), and Polysorbate 80 (Tween® 80) were purchased from Sigma Aldrich (Steinheim, Germany). Silica fumed anhydride, amorphous silicon, and silicon dioxide was obtained from Sigma-Aldrich (St. Louis, MO, USA). Polyethylene glycol (PEG) 200 was purchased from BDH Limited (Poole, England). Polyethylene glycol 400 was purchased from Across Organics (Morris Plains, NJ, USA). Caprylocaproyl macrogol-8/polyoxyl-8 glyceride (Labrasol®) was supplied by Gattefosse’ (Saint-Priest Cedex, France). Crospovidone NF (Polyplasdone XL-10) was supplied by ISP Technologies (Ashland, KY, USA). Methanol was purchased from Honeywell (Seelze, Germany). Magnesium stearate was purchased from (Prolabo, France). Magnesium Trisilicate was purchased from Loba Chemie Pvt. Ltd. (Mumbai, India). Talc powder was purchased from Qualigens fine chemicals (Mumbai, India).
2.1. Pre-Formulation Studies
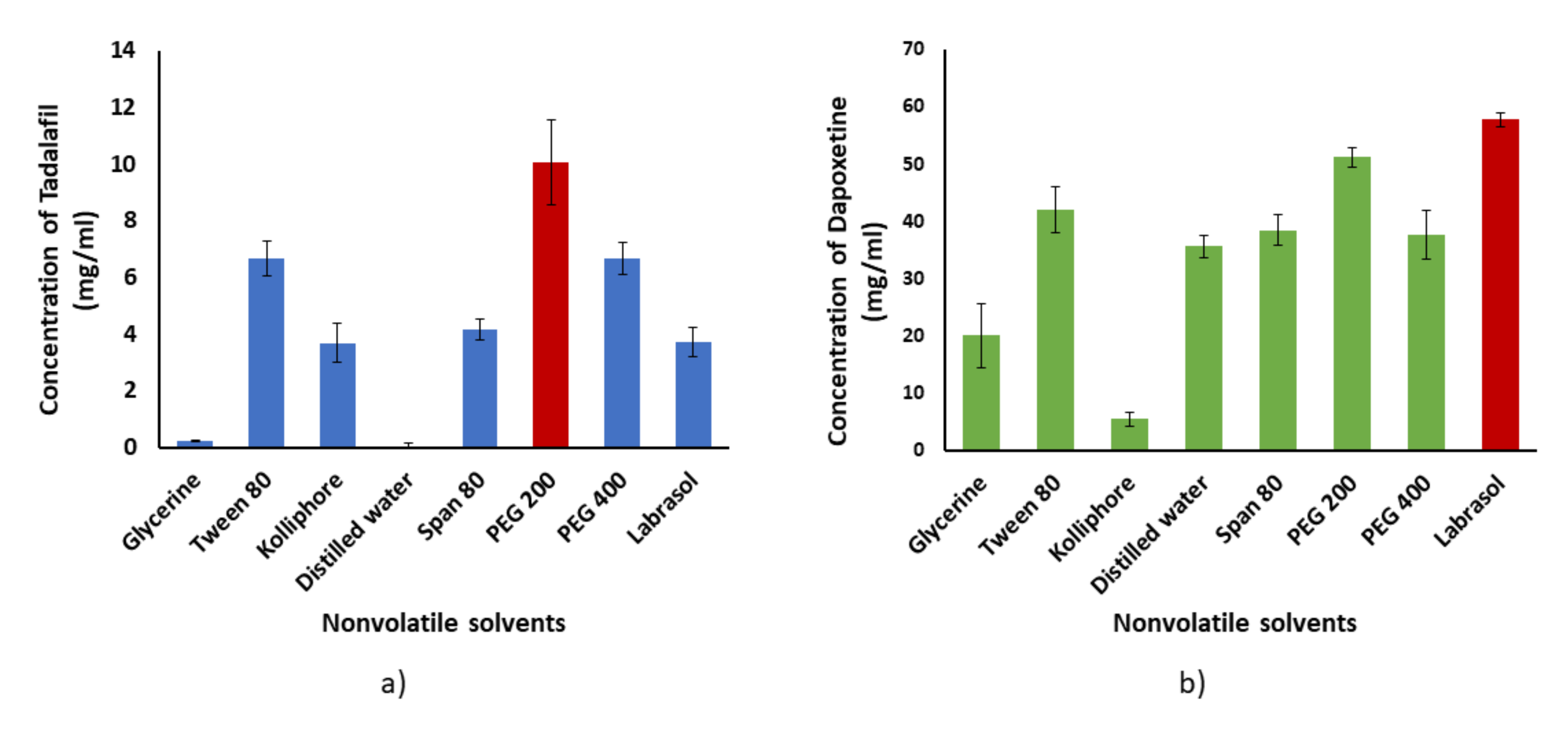
2.1.1. Solubility Studies
Solubility studies of TDL and DPX were determined in various nonvolatile solvents separately, as we described previously with full details published [
23,
33,
34]. The solvents used in this study were Span 80, Tween 80, PEG 400, PEG 200, Labrasol
®, Kolliphor
® EL, glycerin, and distilled water. The solubility of both drugs in all solvents was determined three times and the data were expressed as the average ± the standard deviation.
2.1.2. Holding Capacity and Determination of the Liquid Load Factor (Lf)
The holding capacity of the liquisolid excipients is the maximum amount of a non-volatile liquid that can be held inside the bulk of the carrier and coating powders while maintaining an acceptable flowability that was obtained by using the previously reported method [
23]. Briefly, different weights (0.5, 1.0, 1.5, 2.0 and 2.5 g) of solvents; PEG 200 and Labrasol
®; were added to different mortars containing 5 g of Avicel PH-101 and triturated well. Then 0.5 g that gradually increased to 1.0 g of silica was added and triturated to give good distribution for the liquid through the powder blend. Powder addition and trituration were sustained up until mortar contents began to appear as a dry powder. This procedure was repeated by increasing the weight of silica in the powder to 1.0 g to evaluate if there is an improvement in the flowability of powder blends achieved or not. Finally, the liquid load factor (L
f) which possesses an acceptable flowable and compressible blend, was determined. The liquid load factor (L
f) was calculated by dividing the liquid medication weight (W) by the carrier powder weight (Q) in the system (i.e., L
f = W/Q). The flowability of the prepared mixtures was examined according to the U.S. Pharmacopeia as described in the General Chapters: <1174> POWDER FLOW either by direct technique through the determination of the angle of repose, or with the indirect method by calculation of both Hausner ratio and Carr’s index after measuring the bulk and tap densities of the LS powder blends as described in Equations (1)–(3).
2.1.3. Solid-State Characterization Studies
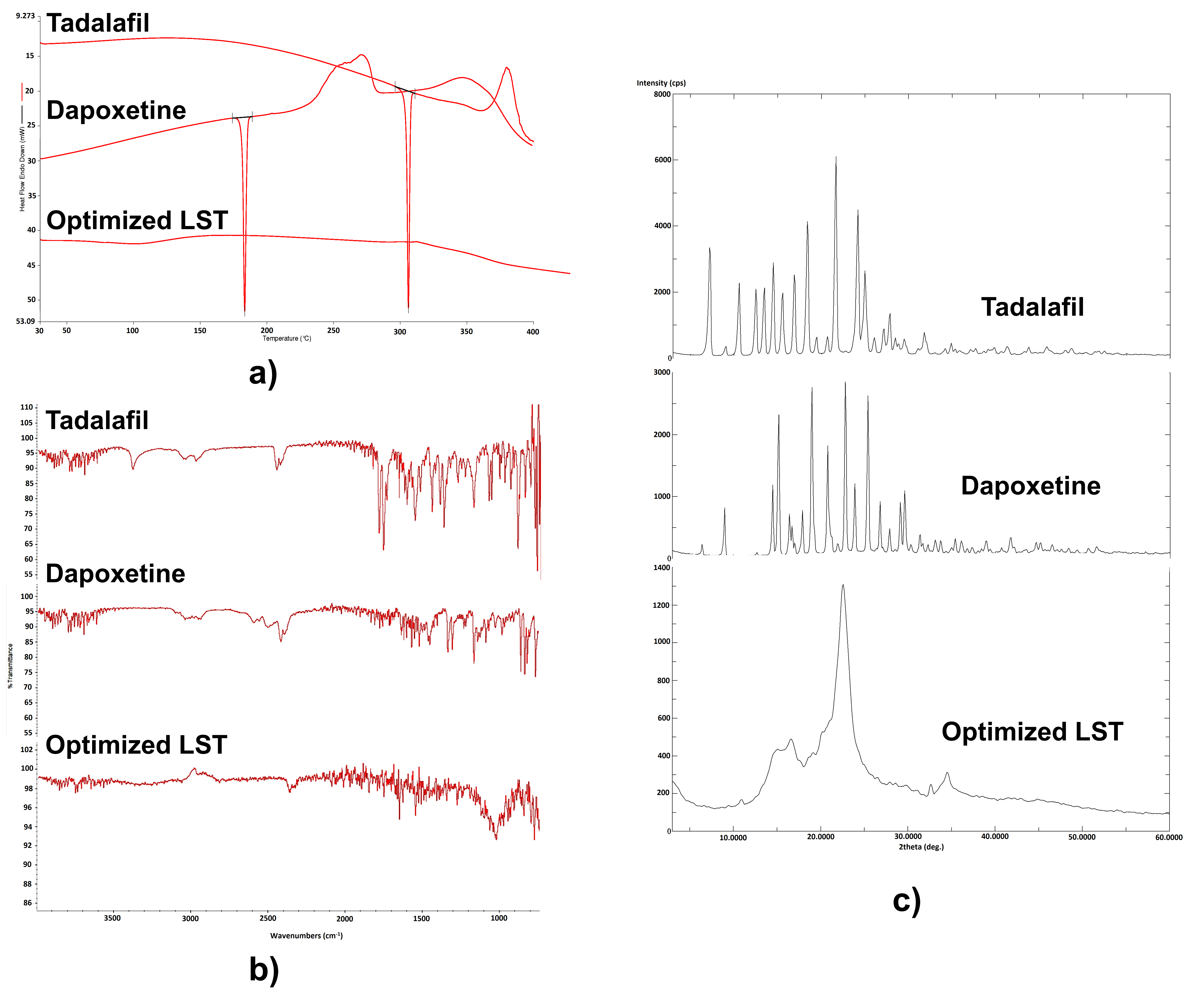
Differential Scanning Calorimetry (DSC)
DSC was carried out to evaluate the thermotropic characteristics and thermal performance of TDL and DPX and the LS compacts using a DSC 8000, PerkinElmer, Inc. (Waltham, MA, USA). About 5 mg of the sample was sealed in aluminum pans and heated at the rate of 10 °C/min, covering a temperature range of 25–400 °C under a nitrogen atmosphere at a flow rate of 100 mL/min.
Powder X-ray Diffraction (PXRD)
The crystallinity of LS powder formulations was considered using PXRD. PXRD diffractograms for TDL and DPX and the prepared LS system were determined using the Ultima IV diffractometer (Rigaku Inc., Tokyo, Japan).
2.2. Formulation of TDL and DPX Liquisolid Tablets
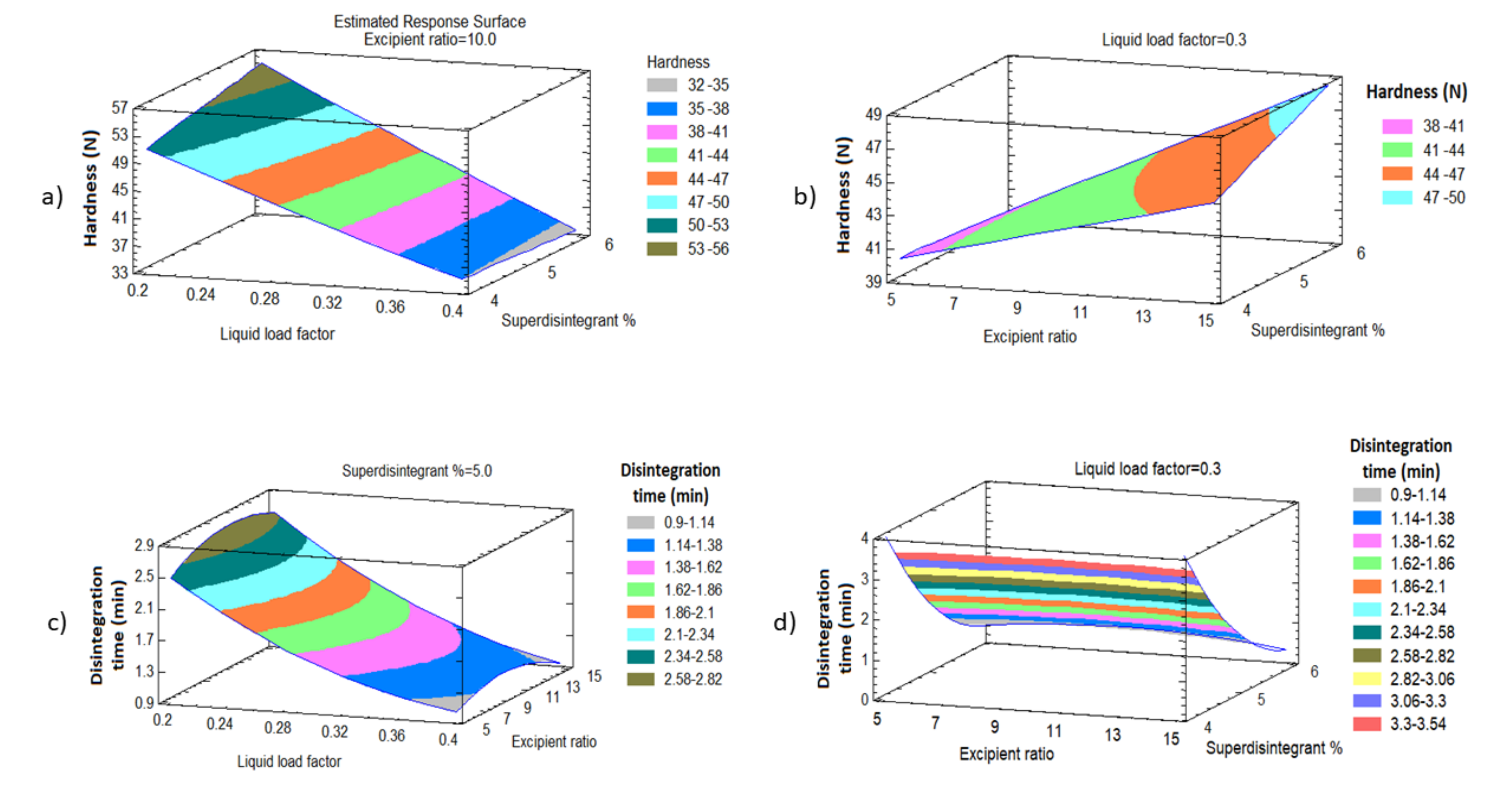
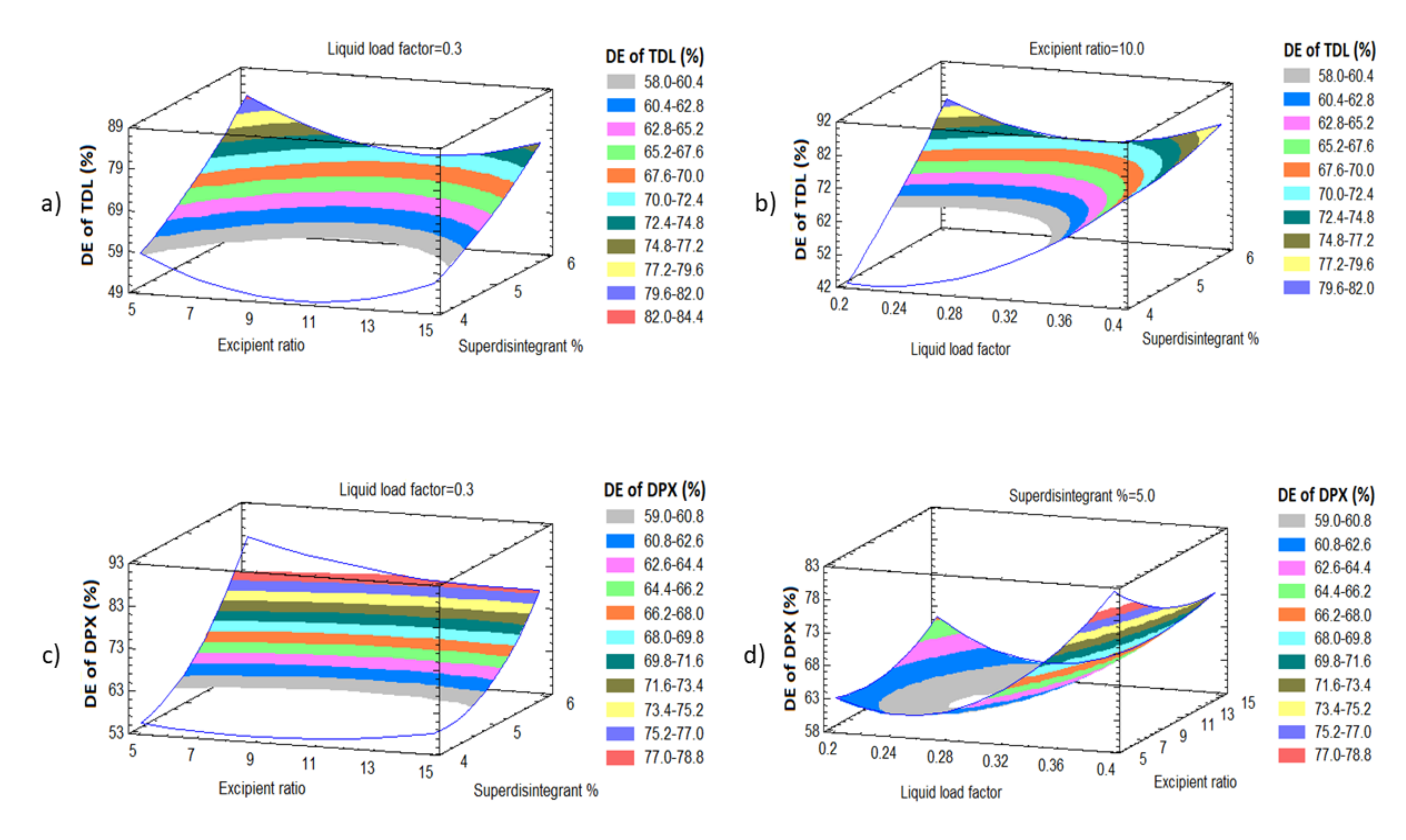
BBD was employed to evaluate the effect of the liquid load factor (Lf) as X1, the powder excipient ratio (R) as X2, and the superdisintegrant (Polyplasone XL-10) percentage as X3 on the quality attributes of the LS formulations. Fifteen experimental runs were suggested by the design. X1 was studied in the level from 0.2 to 0.4, X2 from 5 to 15, while X3 was studied from 4 to 6%. Statistical analysis was performed using Statgraphics Centurion 18 software, Statgraphics Technologies, Inc. (Virginia, VA, USA) to investigate the effect of these independent variables on the tablet hardness (Y1), the disintegration time (Y2), the dissolution efficiency percent of TDL after 60 min (Y3) and the dissolution efficiency percent of DPX after 60 min (Y4).
2.3. Preparation of TDL and DPX Liquisolid Tablets
Fifteen formulations of LSTs (LS-1 to LS-15) were prepared as shown in
Table 1. Briefly, 100 mg of TDL was dissolved in the first half of the calculated solvent mixture (PEG 200) and 600 mg of DPX was dissolved in the second half of the solvent mixture (Labrasol
®) and the mixture was mixed well. The calculated amounts of Avicel PH 101, fumed silica, magnesium trisilicate, polyplasdone XL-10, and Methocel
® ES were added to the mixture with continuous trituration for 10 min in a mortar and the dried mixture were passed through 20 mesh sieves. Finally, the mixture was mixed with 0.5% of talc and magnesium stearate. The obtained powder blend of the fifteen formulations was examined for the flowability parameters before their compression into LSTs. The powder excipients were de-lumped individually through a No. 40 mesh sieve. The de-lumped powders were mixed for 15 min. Talc powder and magnesium stearate were also de-lumped through the 40-mesh sieve and then added to the powder blend and mixed for 3 min. The LSTs were made at 10 KN compression force in a single punch tablet press (Erweka, GmbH, Heusenstamm, Germany) equipped with 9 mm flat round tooling sets.
2.4. Pre-Compression Evaluation of the Liquisolid Powder Formulations
Each LS powder blend was evaluated physically before compression into a tablet by determination of the angle of repose (direct method), bulk and tap density, calculation of Hausner ratio, and Car’s index (compressibility percent) from the Equations (1)–(3) [
35,
36]. The angle of repose was determined according to Equation (1). Where H is the height and D is the mean diameter of the cone.
The Hausner ratio and Carr’s index were calculated from Equations (2) and (3).
2.5. Post-Compression Evaluation of the Prepared Liquisolid Tablets
The LS tablets were visually inspected for any drawbacks during the compression and then examined for their quality attributes, such as weight and content uniformity, thickness, hardness, friability, and disintegration time, according to the requirements of the United States Pharmacopeia [
37].
2.5.1. In-Vitro Disintegration Study
The study was carried out on 6 tablets using a Pharma test disintegration tester according to the USP specifications. Distilled water at 37 ± 0.5 °C was used as a disintegration media and the time in seconds taken for complete disintegration of six tablets was recorded and the average of 6 determinations was reported.
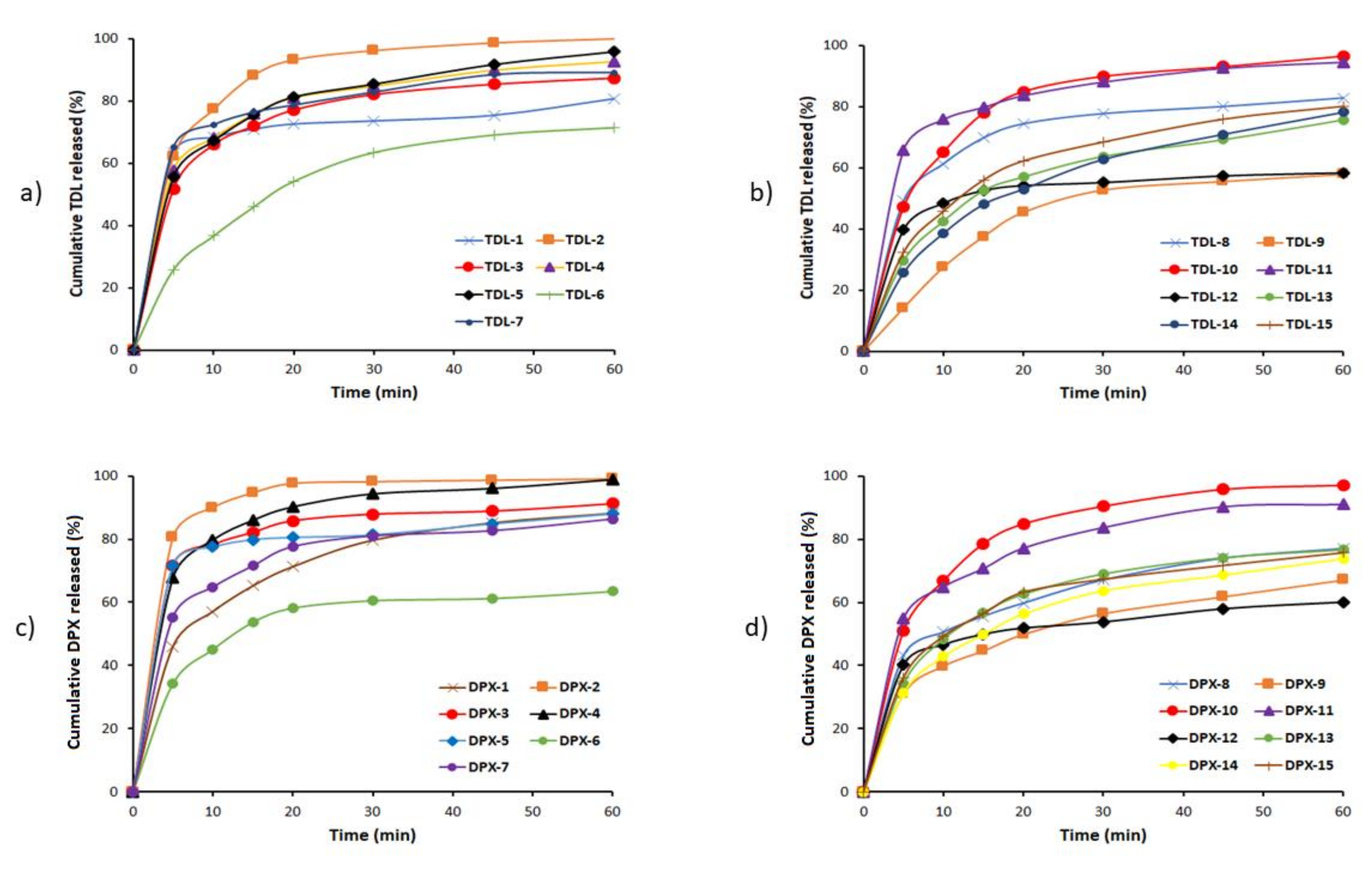
2.5.2. In-Vitro Dissolution Study
The study was performed with the dissolution apparatus II (paddle type). The dissolution medium of 900 mL distilled water at 37 ± 0.5 °C at a rotation of 100 rpm was used. Aliquots of 5 mL were withdrawn at predetermined time intervals 5, 10, 15, 20, 30, 45 and 60 min and filtered through a 0.45 µm Millipore filter (Millipore Corp., Bedford, MA, USA). The concentrations of TDL and DPX were determined spectrophotometrically at 284 and 291 nm, respectively, using a UV–Vis spectrophotometer (Jenway 7315, Bibby scientific Limited, Stone, Staffordshire, UK).
2.5.3. Mathematical Modeling of the Dissolution Data
The in vitro dissolution of TDL and DPX from the fifteen LST formulations and the optimized LSTs formulation were fitted to Zero [
38], First [
39], Weibull [
40], Hixson–Crowell [
41], Higuchi [
42], Korsmeyer–Peppas [
43] and Baker–Lonsdale [
44]. The highest value of the coefficient R
2 was used to identify the goodness of fit and the appropriate release model.
Dissolution Rate (DR10)
For comparative evaluation, TDL and DPX dissolution rates (D
R10) for the LS formulations were used [
45,
46]. The amount of TDL and DPX (in µg) dissolved per min during the first 10 min, was calculated from Equation (4).
where M is the total amount of TDL and DPX in each tablet (in this study it is 2500 and 15,000 µg for TDL and DPX, respectively) and D designates the percentage of drug dissolved during the first 10 min. In addition, other non-model parameters such as the mean dissolution time (MDT) and the dissolution efficiency at 60 min (DE
60) were calculated from the in vitro release data using DDSolver (an add-in program for modeling and comparison of drug dissolution profiles) [
47].
Mean Dissolution Time (MDT)
MDT is defined as the mean residence time of a drug in the formulation [
48]. It is a beneficial parameter for describing the drug release rate from a dosage form and is calculated using Equation (5).
where (j) is the sample number,
n is the number of dissolution sample times, (
) is the time at the midpoint between t and t − 1 (calculated with (t + t − 1)/2) and (
) is the additional amount of drug dissolved between t and t − 1.
Dissolution Efficiency after 60 Min (DE60)
DE
60% expresses the integrated area under the dissolution curve up to a certain time, t, as a percentage of rectangle area represents 100% dissolution at the same time [
49,
50]. The DE of the formulations was calculated using Equation (6).
where (Q) is the percent of drug released as a function of time, (t) is the total time of drug release, and (Q
100) is 100% drug release.
2.6. Prediction of the Optimized Formulation
Analysis of variance and multiple response optimization developed for responses showed the effect of each input variable and their interactions with other variables were utilized for predicting and obtaining the optimized TDL and DPX LSTs using the statistical package Statgraphics® Centurion 18 Software (StatPoint, Inc., Warrenton, VA, USA). The optimized formulation was prepared and fully characterized by the hardness, disintegration time, and dissolution efficiency after 60 min. This optimized formulation was scaled up to be evaluated in vivo for its pharmacokinetic parameters on human volunteers.
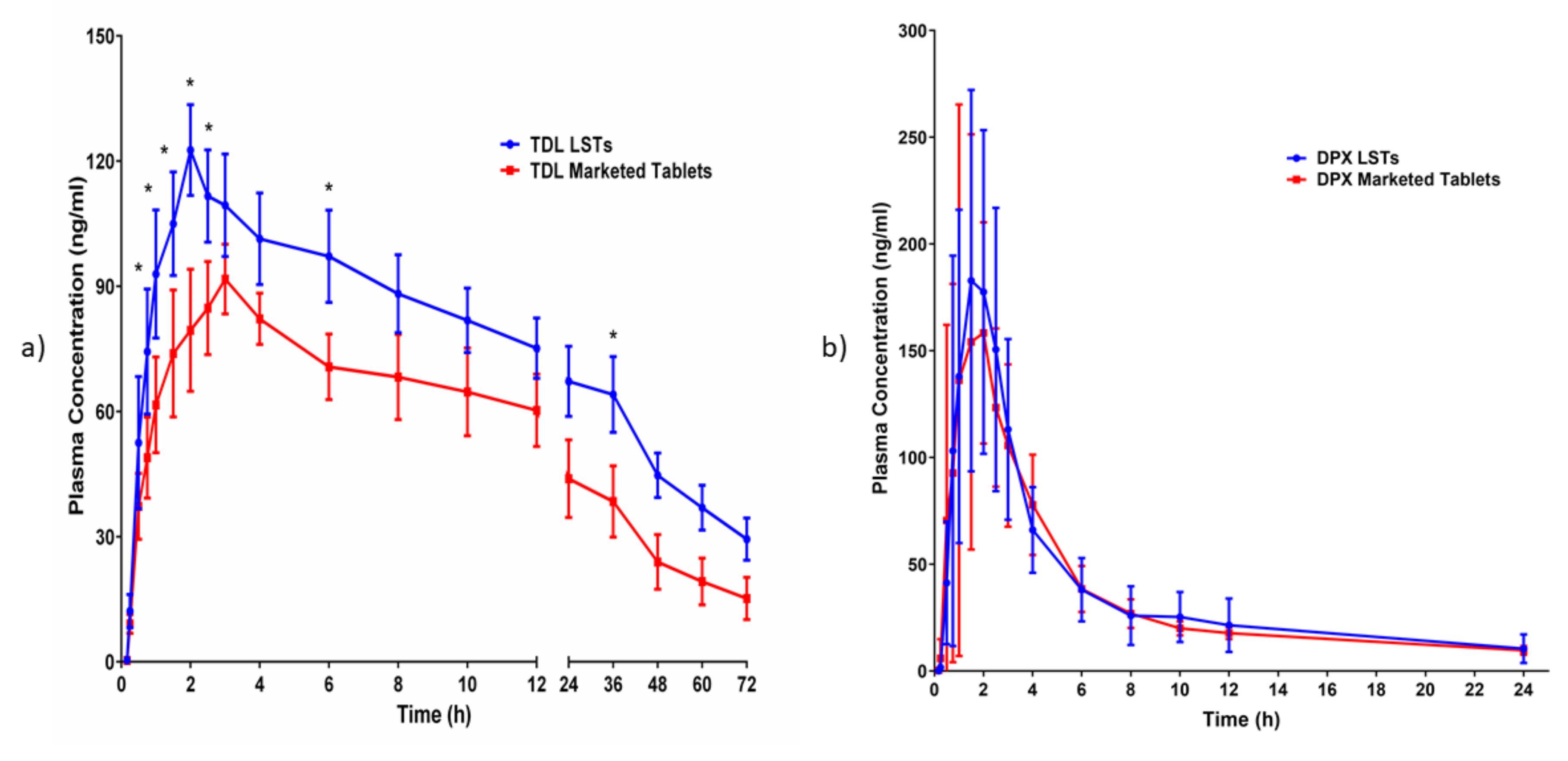
2.7. In-Vivo Pharmacokinetic Evaluation on Healthy Human Volunteers
A pharmacokinetic study was carried out for the optimized LST (test) in comparison with the marketed tablet (reference) on healthy human volunteers. The prepared LSTs and the marketed tablets were administered orally.
2.7.1. Study Design and Conduct
A single-dose one-period parallel design was used in the study. The study was performed following EMA (European Medicines Agency), ICH (International Conference on Harmonization), GCP (Good Clinical Practice), and FDA (Food and Drug Administration) guidelines. The Protocol was approved by the Egyptian Research and Development Company Research Ethics Committee (ERDC REC), Cairo, Egypt, on its expedited meeting on the 2nd of July 2018 with the Ethical Approval Code (0569/452). Six subjects per group gave written informed consent before participation in this study. The subjects who participated were in good health as determined by past medical history, physical examination, vital signs and laboratory tests (hematology, biochemistry, and urine analysis). They were also screened for viral infections and remained under close medical supervision until 24 h after the study period. Each subject fasted for at least 12 h before the administration of tablets. Subjects were kept in-house for 72 h before and after administration of the drug so that regular blood sampling could be withdrawn at a predetermined time (as described in the “Blood Sampling” section).
2.7.2. Subjects
Twelve healthy Egyptian male volunteers participated in the study. The age and body mass index (BMI) of the subjects ranged from 21 to 30 years and 20 to 30 kg/m2, respectively, with a median height of 172 ± 5.3 cm. Subjects were classified into two groups (6 per group); the first group was administered the optimized TDL and DPX LST, and the second group was given the marketed film-coated tablets. The marketed tablets used were TDL 5 mg (Cialis 5 mg, Lilly S.A., Madrid, Spain) and DPX 30 mg (Joypox 30 mg tablets that were produced by South Egypt Drug Industries Co. (SEDICO, Cairo, Egypt).
2.7.3. Blood Sampling
A sample of 5 mL of blood was drawn just before and at 0.25, 0.5, 0.75, 1, 1.5, 2, 2.5, 3, 4, 6, 8, 10, 12, 24, 36, 48, 60 and 72 h after the oral administration of both the test and reference and collected in heparinized tubes. Samples were centrifuged at 3000 rpm for 5 min and plasma samples were collected and stored at −20 °C until analysis.
2.7.4. Chromatographic Conditions
A high-performance liquid chromatographic method coupled with MS/MS detection (HPLC-MS/MS) was developed at ERDC laboratories for the determination of TDL and DPX in human plasma. Agilent series 1200, Agilent Technologies, Deutschland GmbH, (Waldbronn, Germany), equipped with G1311A quaternary pump, G1329A, autosampler, G1322A vacuum degasser, and mass hunter software were used. Chromatography was performed using 75% acetonitrile to 25% of 10 mmoles of ammonium acetate and 100 uL formic acid for each 100 mL water as the mobile phase at a flow rate of 0.3 mL/min and the reverse phase column Intersil ODS-3 (4.6 mm × 50 cm, dp 5µm) from (GL Sciences, Tokyo, Japan) temporized at 25 °C. Sildenafil was used as an internal standard (IS). TDL, DPX, and Sildenafil (IS) were monitored by applying positive multiple reaction monitoring (+MRM). The protonated precursor ions and the produced ions were m/z 390.1(268.0), m/z 306.1 (261.0), and m/z 475.3 (100.1), respectively. The retention time was 2.25, 2.32 and 2.17 min for TDL, DPX, and Sildenafil (IS), respectively. The linearity of the assay for TDL was verified within the concentration range of 1–200 ng/mL with a regression coefficient R2 = 0.998. All the results were within the acceptance criteria as stated in the recommended guidelines. The mean recovery of TDL was 100% at 1 ng/mL (lower limit of quantification; LLOQ) and 95.3% at 200 ng/mL (upper limit of quantification; ULOQ). The described method is proved to be sensitive, accurate, and reproducible with a lower limit of quantification of 1 ng/mL for TDL, while the linearity of the assay for DPX was verified within the concentration range of 2–500 ng/mL with a regression coefficient R2 = 0.994. All the results were within the acceptance criteria as stated in the recommended guidelines. The mean recovery of DPX was 99.8% at 2 ng/mL (LLOQ) and 92.2% at 500 ng/mL (ULOQ). The described method is proven to be sensitive, accurate, and reproducible with a lower limit of quantification of 5 ng/mL for DPX.
2.7.5. Pharmacokinetic Data Analysis
The plasma concentration of TDL and DPX versus time and the pharmacokinetic parameters were determined by the non-compartmental pharmacokinetic model using PKsolver (An add-in program for pharmacokinetic data). Maximum (peak) plasma concentration over the time specified (Cmax), and time point of maximum plasma concentration (Tmax), and the area under the plasma concentration–time curve from zero time to the last measurable concentration (AUC0–t) was calculated by the linear trapezoidal method. In addition, individual estimates were made of the terminal elimination rate constant (Lambda_z), the mean residence time (MRT0-inf) which was calculated by the ratio of AUMC to AUC, and the elimination half-life (t1/2) which was calculated as 0.693/Lambda_z. Moreover, the apparent total body clearance of the drug from plasma after oral administration (Cl/F) was calculated by dividing the dose by AUC and the apparent volume of distribution during the terminal phase after non-intravenous administration (Vz/F) was calculated by multiplying total body clearance by MRT. Finally, the relative bioavailability of the optimized LSTs (AUC test/AUC standard ×100) was determined.
2.8. Statistical Analysis
All statistical analyses were performed using GraphPad Prism 8 for Windows, Version 8.2.1 Software (San Diego, CA, USA). Regarding the plasma concentration–time curve, two-way ANOVA followed by Sidak’s multiple comparisons test was conducted to compare each mean with the others at all time points and assess the significance between groups, while a two-tailed unpaired t-test was used to assess the pharmacokinetic parameters of the formulations. Results with p < 0.05 were considered significant.
4. Conclusions
From the obtained results, we could conclude that BBD was successfully implemented in the optimization of the formulation factors to produce an optimized combined-dose of TDL and DPX LST with acceptable mechanical properties, short disintegration time, and good dissolution profile. The statistical analysis suggested the combination of the factor levels of 0.2 of the liquid load factor, 11.82 of the excipient ratio and 5.11% of polyplasdone XL-10 in the preparation of the optimized formulation. The pharmacokinetic evaluation revealed a significant improvement in the drug bioavailability after oral administration owing to the enhanced drug solubility and absorption. The maximum plasma concentration (Cmax) of TDL in LSTs was 122.61 ng/mL within 2 h (Tmax), compared to the marketed tablets which reach 91.72 ng/mL after 3 h. Amazingly, the optimized LST formulation achieved this Cmax of the marketed tablet after only 1 h, which revealed that LSTs’ formulation improved the rate and extent of TDL absorption. In addition, the optimized formulation showed a significant difference between the Cmax and AUC0–t of both groups with p-values of 0.0198 and 0.0280, respectively. The improved absorption of LSTs was probably due to the enhanced solubilization of the drugs and the high surface area available for absorption. The dissolved APIs in the mixture of solvents used can be directly absorbed, with a short time for the dissolution step, which is considered the rate-limiting step for drug absorption in BCS Class II compounds. Improvement of the dissolution rate leads to a significant increase in oral absorption and a subsequent enhancement of oral bioavailability. The relative bioavailability of TDL and DPX of LSTs was larger (170.6% and 117.05%) than that of the marketed tablet. Finally, these findings reveal that the onset was rapid enough, and the duration and the concentration achieved a level suitable to overcome male sexual dysfunction. So, the developed combined-dose LS formulation could be a promising approach in the treatment of male sexual dysfunction, particularly for diabetic patients.












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}