Physiologically Based Pharmacokinetic Modeling of Transdermal Selegiline and Its Metabolites for the Evaluation of Disposition Differences between Healthy and Special Populations


Abstract
:1. Introduction
2. Methods
2.1. Pharmacokinetic Data
2.2. Simcyp Simulator Setup
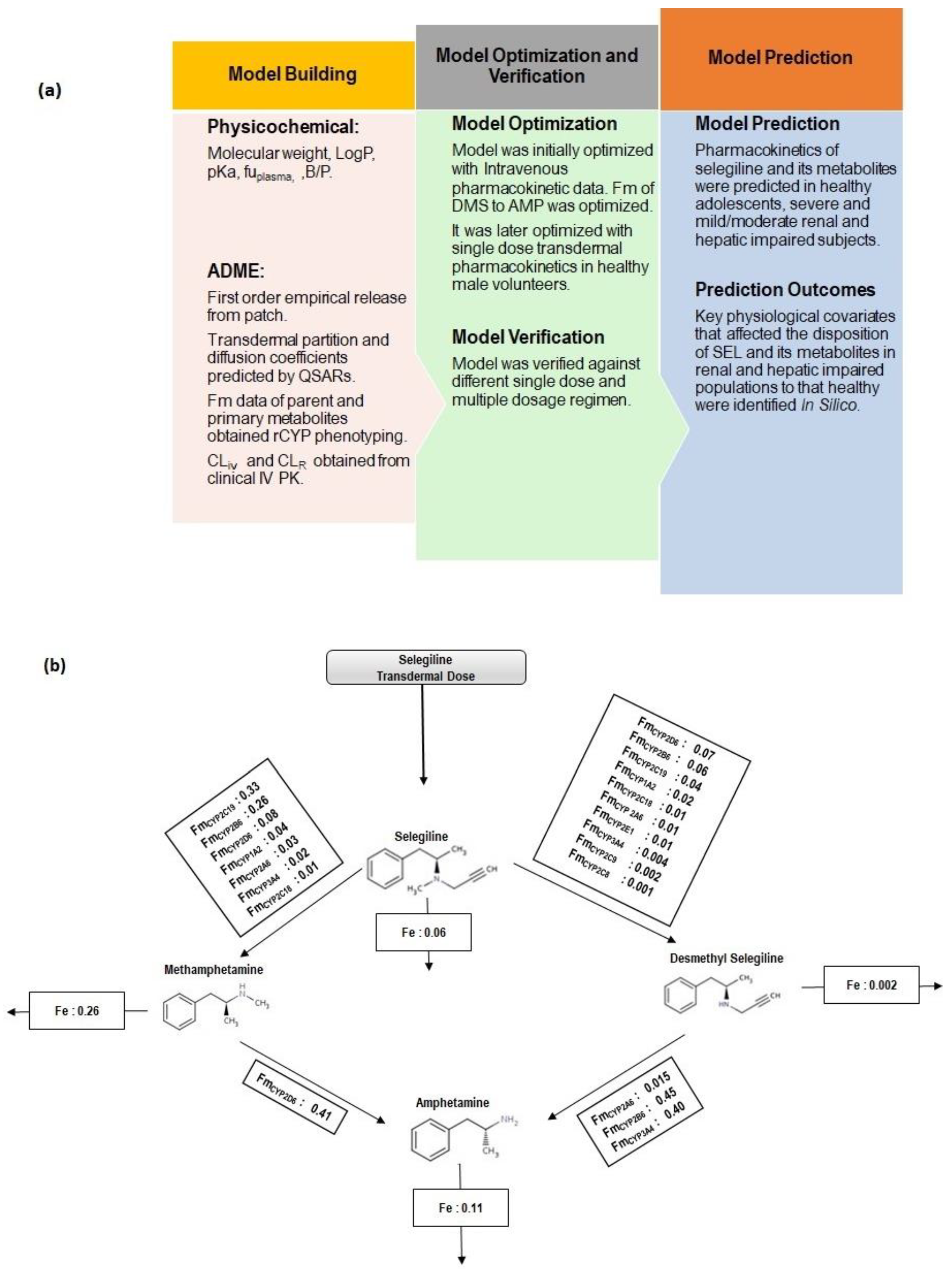
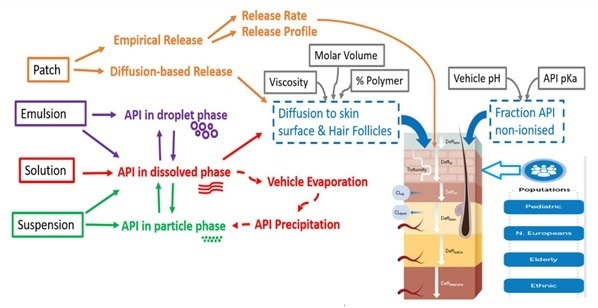
2.3. PBPK Model Workflow
2.4. Intravenous Pharmacokinetics
2.5. Transdermal Pharmacokinetics
2.6. Prediction of Pharmacokinetics in Special Populations
2.7. Model Evaluation
3. Results
3.1. Model Development and Optimization
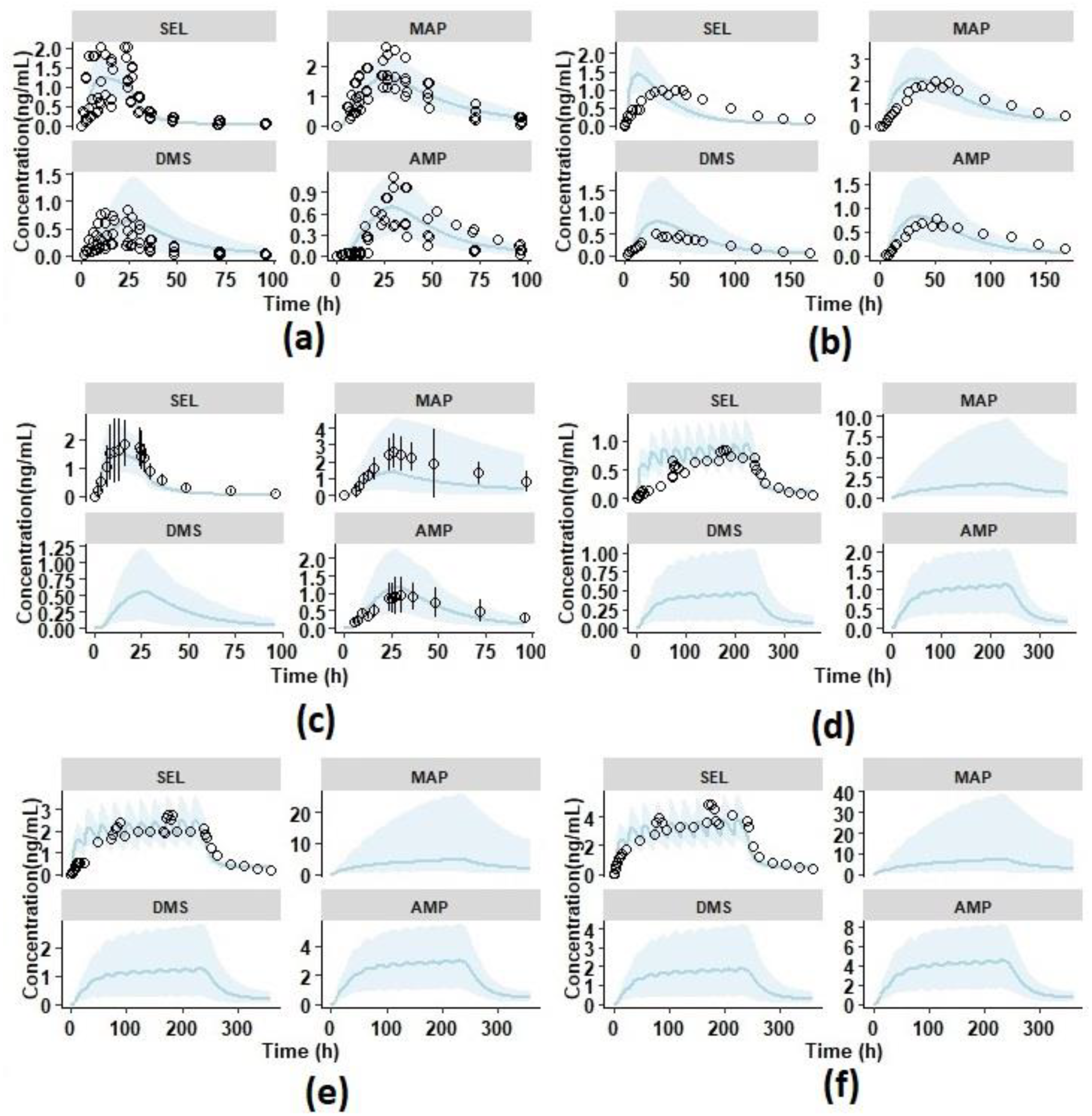
3.1.1. Intravenous Pharmacokinetics
3.1.2. Single Dose Transdermal Pharmacokinetics in Healthy Male Subjects
3.2. Model Verification
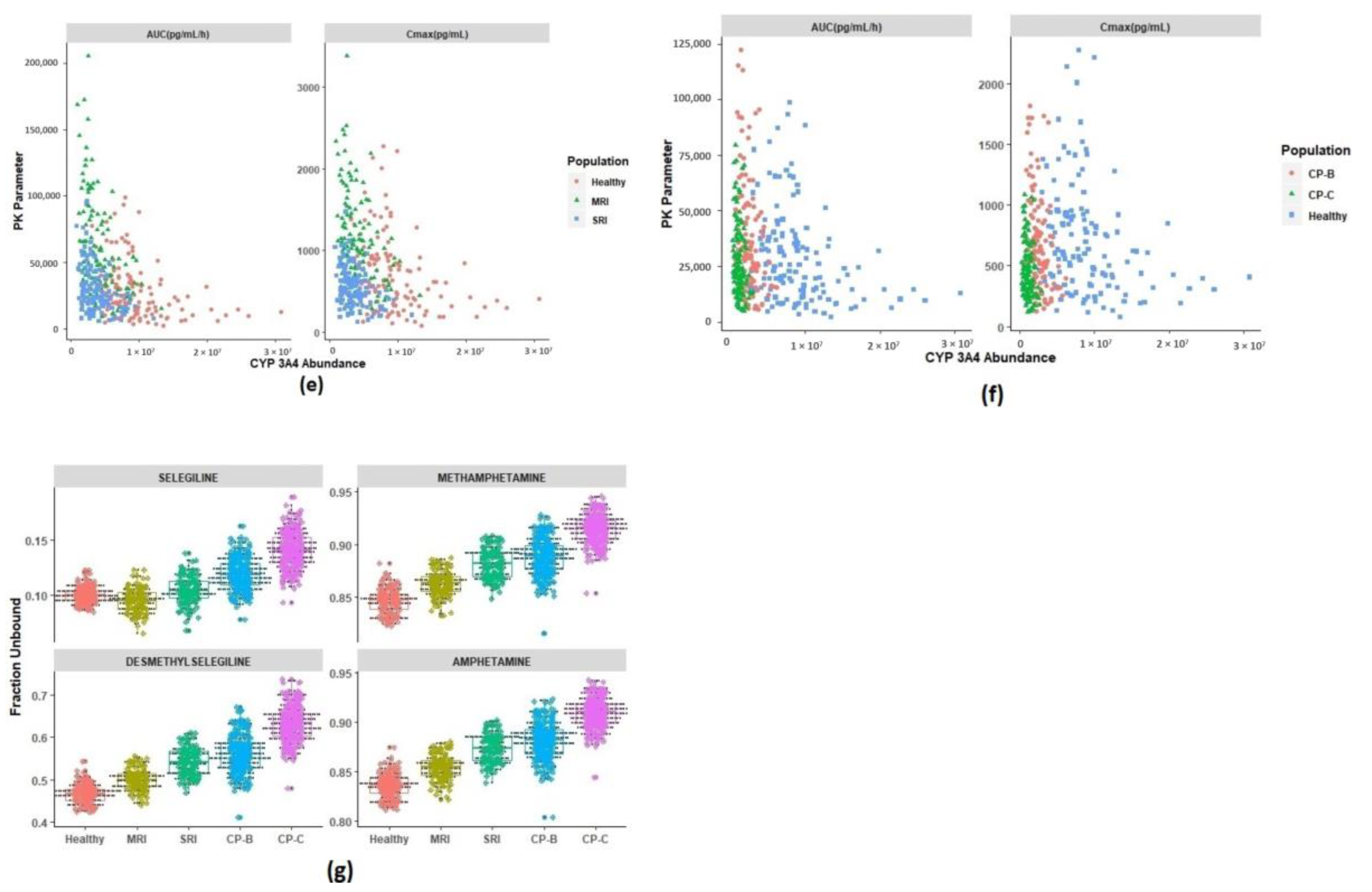
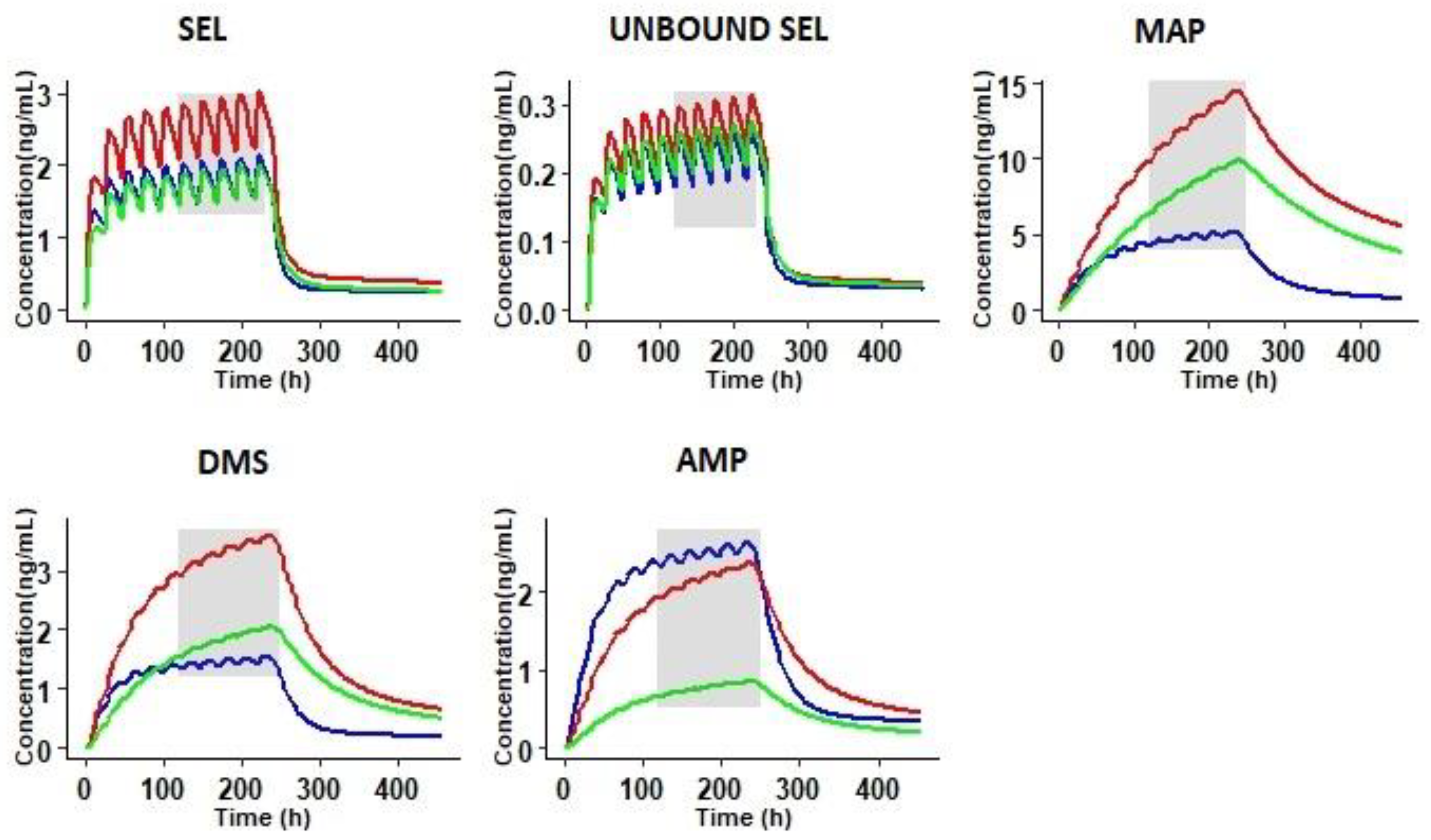
3.3. Prediction of Pharmacokinetics in Special Populations
3.3.1. Pharmacokinetics in Adolescents
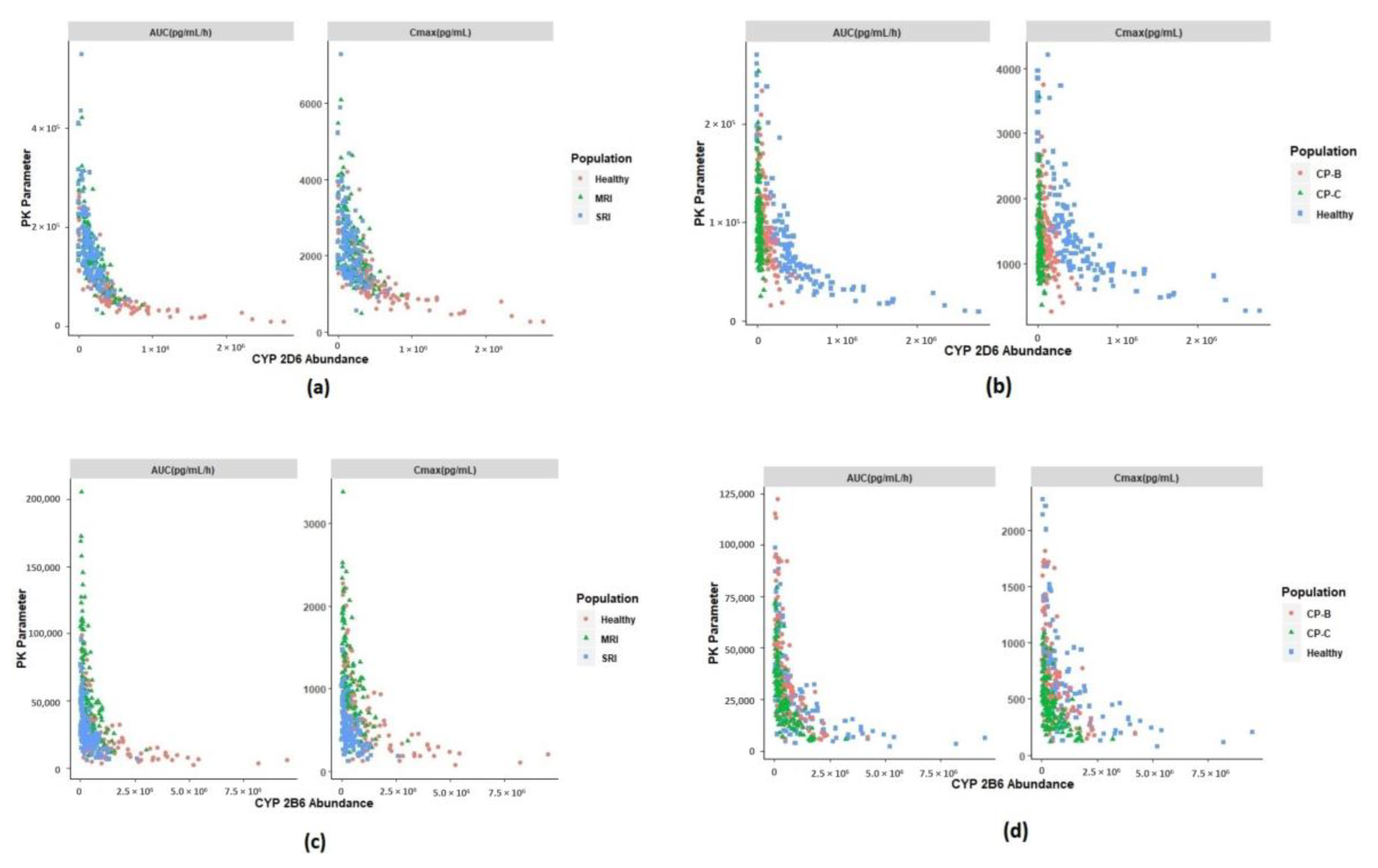
3.3.2. Prediction of Pharmacokinetics in Renally Impaired Subjects
3.3.3. Prediction of Pharmacokinetics in Hepatic Cirrhosis Subjects
3.4. Identification of Physiological Covariates
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chrisp, P.; Mammen, G.J.; Sorkin, E.M. Selegiline. Drugs Aging 1991, 1, 228–248. [Google Scholar] [CrossRef]
- Amsterdam, J.D. A double-blind, placebo-controlled trial of the safety and efficacy of selegiline transdermal system without dietary restrictions in patients with major depressive disorder. J. Clin. Psychiatry 2003, 64, 208–214. [Google Scholar] [CrossRef]
- Patel, N.K.; Clarke, J.; Arora, S.; Martins, F.; Salem, F.; Abdulla, T.; Tsakalozou, E.; Hodgkinson, A.; Tash, O.A.; Cristea, S.; et al. Multi-Phase Multi-LAYER Mechanistic Dermal Absorption (MPML MechDermA) Model to Predict Local and Systemic Exposure of Dermally Applied Drug Products 1: Model Structure and Parameterization (Manuscript under Preparation); 2020; Unpublished Work. [Google Scholar]
- Jessen, L.; Kovalick, L.J.; Azzaro, A.J. The Selegiline Transdermal System (EMSAM): A therapeutic option for the treatment of major depressive disorder. Pharm. Ther. 2008, 33, 212–246. [Google Scholar]
- Drug Label: EMSAM® (Selegiline Transdermal System) Continuous Delivery for Once-Daily Application; United States Food and Drug Administration: Silver Spring, MD, USA, 2006.
- Robinson, D.S.; Gilmor, M.L.; Yang, Y.; Moonsammy, G.; Azzaro, A.J.; A Oren, D.; Campbell, B.J. Treatment effects of selegiline transdermal system on symptoms of major depressive disorder: A meta-analysis of short-term, placebo-controlled, efficacy trials. Psychopharmacol. Bull. 2007, 40, 15–28. [Google Scholar]
- Heinonen, E.H.; I Anttila, M.; Lammintausta, R.A.S. Pharmacokinetic aspects of l-deprenyl (selegiline) and its metabolites *. Clin. Pharmacol. Ther. 1994, 56, 742–749. [Google Scholar] [CrossRef]
- Mahmood, I. Clinical Pharmacokinetics and Pharmacodynamics of Selegiline. Clin. Pharmacokinet. 1997, 33, 91–102. [Google Scholar] [CrossRef]
- Barrett, J.S.; Hochadel, T.J.; Morales, R.J.; Rohatagi, S.; DeWitt, K.E.; Watson, S.K.; Disanto, A.R. Pharmacokinetics and Safety of a Selegiline Transdermal System Relative to Single-Dose Oral Administration in the Elderly. Am. J. Ther. 1996, 3, 688–698. [Google Scholar] [CrossRef] [PubMed]
- Rohatagi, S.; Barrett, J.S.; DeWitt, K.E.; Morales, R.J. Integrated pharmacokinetic and metabolic modeling of selegiline and metabolites after transdermal administration. Biopharm. Drug Dispos. 1997, 18, 567–584. [Google Scholar] [CrossRef]
- New Drug Application Number: 21-336/21-708 Clinical Pharmacology and Biopharmaceutics Review; 21-336/21-708; United States Food and Drug Administration: Silver Spring, MD, USA, 2005.
- Shin, H.S. Metabolism of selegiline in humans. Identification, excretion, and stereochemistry of urine metabolites. Drug Metab. Dispos. 1997, 25, 657–662. [Google Scholar] [PubMed]
- Salonen, J.S.; Nyman, L.; Boobis, A.R.; Edwards, R.J.; Watts, P.; Lake, B.G.; Price, R.J.; Renwick, A.B.; Gómez-Lechón, M.-J.; Castell, J.; et al. Comparative Studies on the Cytochrome P450-Associated Metabolism and Interaction Potential of Selegiline between Human Liver-Derived In Vitro Systems. Drug Metab. Dispos. 2003, 31, 1093–1102. [Google Scholar] [CrossRef] [Green Version]
- Benetton, S.A.; Fang, C.; Yang, Y.-O.; Alok, R.; Year, M.; Lin, C.-C.; Yeh, L.-T. P450 phenotyping of the metabolism of selegiline to desmethylselegiline and methamphetamine. Drug Metab. Pharmacokinet. 2007, 22, 78–87. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.Y.; Di Stefano, E.W.; A Schmitz, D.; Hsu, L.; Ellis, S.W.; Lennard, M.S.; Tucker, G.T.; Cho, A.K. Oxidation of methamphetamine and methylenedioxymethamphetamine by CYP2D6. Drug Metab. Dispos. 1997, 25, 1059–1064. [Google Scholar] [PubMed]
- Heinonen, E.H.; Anttila, M.I.; Lammintausta, R.A.S. Pharmacokinetics and Clinical Pharmacology of Selegiline. In Milestones in Drug Therapy; Springer Science and Business Media LLC: Berlin/Heidelberg, Germany, 1993; pp. 201–213. [Google Scholar]
- Yanagisawa, Y.; Nakazato, K.; Nagai, T. Binding of Methamphetamine to SERUM Albumin in Various Speciesin Vitro. Pharmacol. Res. 1997, 35, 99–102. [Google Scholar] [CrossRef] [PubMed]
- Szökö, E.; Kalasz, H.; Kerecsen, L.; Magyar, K. Binding of (−) deprenyl to serum proteins. Pol. J. Pharmacol. Pharm. 1984, 36, 413–421. [Google Scholar] [PubMed]
- Kalász, H.; Kerecsen, L.; Knoll, J.; Pucsok, J. Chromatographic studies on the binding, action and metabolism of (−)-deprenyl. J. Chromatogr. A 1990, 499, 589–599. [Google Scholar] [CrossRef]
- Jeong, H.-C.; Bae, S.H.; Bae, J.-W.; Lee, S.; Kim, A.; Jang, Y.; Shin, K.-H. Evaluation of the Effect of CYP2D6 Genotypes on Tramadol and O-Desmethyltramadol Pharmacokinetic Profiles in a Korean Population Using Physiologically-Based Pharmacokinetic Modeling. Pharmaceutics 2019, 11, 618. [Google Scholar] [CrossRef] [Green Version]
- Balant, L.; Francis, R.J.; Tozer, T.N.; Marmy, A.; Tschopp, J.-M.; Fabre, J. Influence of renal failure on the hepatic clearance of bufuralol in man. J. Pharmacokinet. Biopharm. 1980, 8, 421–438. [Google Scholar] [CrossRef]
- Zanger, U.M.; Klein, K.; Saussele, T.; Blievernicht, J.; Hofmann, M.H.; Schwab, M. PolymorphicCYP2B6: Molecular mechanisms and emerging clinical significance. Pharmacogenomics 2007, 8, 743–759. [Google Scholar] [CrossRef]
- Burger, D.; Van Der Heiden, I.; La Porte, C.; Van Der Ende, M.; Groeneveld, P.; Richter, C.; Koopmans, P.; Kroon, F.; Sprenger, H.; Lindemans, J.; et al. Interpatient variability in the pharmacokinetics of the HIV non-nucleoside reverse transcriptase inhibitor efavirenz: The effect of gender, race, and CYP2B6 polymorphism. Br. J. Clin. Pharmacol. 2005, 61, 148–154. [Google Scholar] [CrossRef]
- Yeung, C.K.; Shen, D.D.; Thummel, K.E.; Himmelfarb, J. Effects of chronic kidney disease and uremia on hepatic drug metabolism and transport. Kidney Int. 2014, 85, 522–528. [Google Scholar] [CrossRef] [Green Version]
- Prasad, B.; Bhatt, D.K.; Johnson, K.; Chapa, R.; Chu, X.; Salphati, L.; Xiao, G.; Lee, C.; Hop, C.E.C.A.; Mathias, A.; et al. Abundance of Phase 1 and 2 Drug-Metabolizing Enzymes in Alcoholic and Hepatitis C Cirrhotic Livers: A Quantitative Targeted Proteomics Study. Drug Metab. Dispos. 2018, 46, 943–952. [Google Scholar] [CrossRef] [PubMed]
- New Drug Application Number: 21-479 Clinical Pharmacology and Biopharmaceutics Review; 21–479; United States Food and Drug Administration: Silver Spring, MD, USA, 2006.
- Jamei, M.; Dickinson, G.L.; Rostami-Hodjegan, A. A Framework for Assessing Inter-individual Variability in Pharmacokinetics Using Virtual Human Populations and Integrating General Knowledge of Physical Chemistry, Biology, Anatomy, Physiology and Genetics: A Tale of ‘Bottom-Up’ vs ‘Top-Down’ Recognition of Covariates. Drug Metab. Pharmacokinet. 2009, 24, 53–75. [Google Scholar] [CrossRef] [PubMed]
- Anttila, M.; Pelkonen, O.; Sotaniemi, E.A.; Rautio, A. Marked effect of liver and kidney function on the pharmacokinetics of selegiline. Clin. Pharmacol. Ther. 2005, 77, 54–62. [Google Scholar] [CrossRef]
- Zhou, L.; Sharma, P.; Yeo, K.R.; Higashimori, M.; Xu, H.; Al-Huniti, N.; Zhou, D. Assessing pharmacokinetic differences in Caucasian and East Asian (Japanese, Chinese and Korean) populations driven by CYP2C19 polymorphism using physiologically-based pharmacokinetic modelling. Eur. J. Pharm. Sci. 2019, 139, 105061. [Google Scholar] [CrossRef] [PubMed]
- Vogt-Ferrier, N. Older patients, multiple comorbidities, polymedication… should we treat everything? Eur. Geriatr. Med. 2011, 2, 48–51. [Google Scholar] [CrossRef]
- Small, B.G.; Wendt, B.; Jamei, M.; Johnson, T.N. Prediction of liver volume-A population-based approach to meta-analysis of paediatric, adult and geriatric populations-An update. Biopharm. Drug Dispos. 2017, 38, 290–300. [Google Scholar] [CrossRef] [PubMed]
- Yeo, K.R.; Aarabi, M.; Jamei, M.; Rostami-Hodjegan, A. Modeling and predicting drug pharmacokinetics in patients with renal impairment. Expert Rev. Clin. Pharmacol. 2011, 4, 261–274. [Google Scholar] [CrossRef]
- Johnson, T.N.; Boussery, K.; Rowland-Yeo, K.; Tucker, G.T.; Rostami-Hodjegan, A. A Semi-Mechanistic Model to Predict the Effects of Liver Cirrhosis on Drug Clearance. Clin. Pharmacokinet. 2010, 49, 189–206. [Google Scholar] [CrossRef]
- Tunstall, A.M.; Merriman, J.M.; Milne, I.; James, K. Normal and pathological serum levels of alpha2-macroglobulins in men and mice. J. Clin. Pathol. 1975, 28, 133–139. [Google Scholar] [CrossRef]
- Housley, J. Alpha2-macroglobulin levels in disease in man. J. Clin. Pathol. 1968, 21, 27–31. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Everhart, T.; Iii, P.J.; Jones, R.; Mendelson, J.; Jacob, P. Stereoselectivity in the human metabolism of methamphetamine. Br. J. Clin. Pharmacol. 2010, 69, 187–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gunne, L.-M.; Niklasson, F. Pharmacokinetic and clinical studies on amphetamine dependent subjects. Eur. J. Clin. Pharmacol. 1970, 3, 3–11. [Google Scholar] [CrossRef]
- Chen, Y.; Jin, J.Y.; Mukadam, S.; Malhi, V.; Kenny, J.R. Application of IVIVE and PBPK modeling in prospective prediction of clinical pharmacokinetics: Strategy and approach during the drug discovery phase with four case studies. Biopharm. Drug Dispos. 2012, 33, 85–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodgers, T.; Leahy, D.; Rowland, M. Physiologically Based Pharmacokinetic Modeling 1: Predicting the Tissue Distribution of Moderate-to-Strong Bases. J. Pharm. Sci. 2005, 94, 1259–1276. [Google Scholar] [CrossRef]
- Rodgers, T.; Rowland, M. Physiologically based pharmacokinetic modelling 2: Predicting the tissue distribution of acids, very weak bases, neutrals and zwitterions. J. Pharm. Sci. 2006, 95, 1238–1257. [Google Scholar] [CrossRef]
- Salem, F.; Johnson, T.N.; Abduljalil, K.; Tucker, G.T.; Rostami-Hodjegan, A. A Re-evaluation and Validation of Ontogeny Functions for Cytochrome P450 1A2 and 3A4 Based on In Vivo Data. Clin. Pharmacokinet. 2014, 53, 625–636. [Google Scholar] [CrossRef]
- Proctor, N.J.; Tucker, G.T.; Rostami-Hodjegan, A. Predicting drug clearance from recombinantly expressed CYPs: Intersystem extrapolation factors. Xenobiotica 2004, 34, 151–178. [Google Scholar] [CrossRef]
- Patel, N.K.; Clarke, J.; Arora, S.; Martins, F.; Salem, F.; Abdulla, T.; Tsakalozou, E.; Hodgkinson, A.; Tash, O.A.; Cristea, S.; et al. Multi-phase Multi-Layer Mechanistic Dermal Absorption (MPML MechDermA) Model to Predict Local and Systemic Exposure of Dermally Applied Drug Products 2: Complex Formulations, Applications and Verification (Manuscript under Preparation); 2020; Unpublished Work. [Google Scholar]
- Abdulla, T.; Patel, N.K.; Polak, S.; Martins, F.S.; Jamei, M.; Rostomi, A.H. Quantitative Prediction of Dermal Drug Absorption: Relative Effects of Application Site. In Proceedings of the Simcyp Consortium 2017, Sheffield, UK, 19–21 September 2017. [Google Scholar]
- Arrora, S.; Patel, N.K.; Polak, S. Modeling in vitro skin permeation experiments to mechanistically understand in vivo dermal absorption: Application of in vitro-in vivo extrapolation (IVIVE) and physiological based pharmacokinetic (PBPK) modeling using testosterone as model drug. Eur. Fed. Pharm. Sci. In Proceedings of the Eur. Federation for Pharmaceutical Sciences; Available online: https://www.certara.com/app/uploads/2019/09/Arora_2019_EUFEPS_Skin.pdf (accessed on 20 September 2020).
- Patel, N.K.; Cristea, S.; Rose, R.; Salem, F.; Johnson, T.N.; Jamei, M.; Raney, S.G.; Zhang, X.; Lin, H.-P.; Newman, B.; et al. Mechanistic modelling of dermal drug absorption using the Simcyp Multi-phase Multi-layer MechDermA model: Case study of a transdermal patch formulation of weak base drug timolol. In Proceedings of the 14th Barrier Function of Mammalian Skin Gordon Research Conference, Waterville Valley, NH, USA, 16–21 August 2015. [Google Scholar]
- Puttrevu, S.K.; Arora, S.; Polak, S.; Patel, N.K. Evaluation of Methamphetamine and Amphetamine Disposition Discrepancy upon Selegiline Transdermal Patch Administration in Healthy Volunteers versus Special Populations Using PBPK Modelling. In Proceedings of the American Association of Pharmaceutical Scientists, AAPS Pharmsci 360, San Antanio, TX, USA, 3–6 November 2019. [Google Scholar]
- Salem, F.; Patel, N.K.; Polak, S. Development of a PBPK model for topical lidocaine in order to predict systemic absorption in healthy volunteers, geriatrics and paediatrics. In Proceedings of the American Society of Clinical Pharmacology and Therapeutics, Washington, DC, USA, 13–16 March 2019. [Google Scholar]
- Hansen, S.; Lehr, C.; Schaefer, U.F. Improved input parameters for diffusion models of skin absorption. Adv. Drug Deliv. Rev. 2013, 65, 251–264. [Google Scholar] [CrossRef]
- Valiveti, S.; Wesley, J.; Lu, G.W. Investigation of drug partition property in artificial sebum. Int. J. Pharm. 2008, 346, 10–16. [Google Scholar] [CrossRef]
- Chen, L.; Han, L.; Saib, O.; Lian, G. In Silico Prediction of Percutaneous Absorption and Disposition Kinetics of Chemicals. Pharm. Res. 2014, 32, 1779–1793. [Google Scholar] [CrossRef]
- Shatkin, J.A.; Brown, H.S. Pharmacokinetics of the dermal route of exposure to volatile organic chemicals in water: A computer simulation model. Environ. Res. 1991, 56, 90–108. [Google Scholar] [CrossRef]
- Mitragotri, S. Modeling skin permeability to hydrophilic and hydrophobic solutes based on four permeation pathways. J. Control. Release 2003, 86, 69–92. [Google Scholar] [CrossRef]
- Johnson, M.E.; Blankschtein, D.; Langer, R. Evaluation of Solute Permeation through the Stratum Corneum: Lateral Bilayer Diffusion as the Primary Transport Mechanism. J. Pharm. Sci. 1997, 86, 1162–1172. [Google Scholar] [CrossRef] [PubMed]
- Seif, S.; Hansen, S. Measuring the stratum corneum reservoir: Desorption kinetics from keratin. J. Pharm. Sci. 2012, 101, 3718–3728. [Google Scholar] [CrossRef]
- Johnson, T.N.; Zhou, D.; Bui, K.H. Development of physiologically based pharmacokinetic model to evaluate the relative systemic exposure to quetiapine after administration of IR and XR formulations to adults, children and adolescents. Biopharm. Drug Dispos. 2014, 35, 341–352. [Google Scholar] [CrossRef]
- Abduljalil, K. Predicting the Developmental PK/PD of Cyclosporine (CsA) in Paediatrics K; Abduljalil, T.N., Johnson, M., Jamei1, D., Edwards, A., Eds.; Rostami-Hodjegan Simcyp Limited: Sheffield, UK, 2012. [Google Scholar]
- Abduljalil, K.; Cain, T.; Humphries, H.; Rostami-Hodjegan, A. Deciding on Success Criteria for Predictability of Pharmacokinetic Parameters from In Vitro Studies: An Analysis Based on In Vivo Observations. Drug Metab. Dispos. 2014, 42, 1478–1484. [Google Scholar] [CrossRef]
- Shebley, M.; Sandhu, P.; Riedmaier, A.E.; Jamei, M.; Narayanan, R.; Patel, A.; Peters, S.A.; Reddy, V.P.; Zheng, M.; De Zwart, L.; et al. Physiologically Based Pharmacokinetic Model Qualification and Reporting Procedures for Regulatory Submissions: A Consortium Perspective. Clin. Pharmacol. Ther. 2018, 104, 88–110. [Google Scholar] [CrossRef]
- Rostami-Hodjegan, A. Reverse Translation in PBPK and QSP: Going Backwards in Order to Go Forward With Confidence. Clin. Pharmacol. Ther. 2017, 103, 224–232. [Google Scholar] [CrossRef] [Green Version]
- Zhou, W.; Johnson, T.; Xu, H.; Cheung, S.; Bui, K.; Li, J.; Al-Huniti, N.; Zhou, D. Predictive Performance of Physiologically Based Pharmacokinetic and Population Pharmacokinetic Modeling of Renally Cleared Drugs in Children. CPT: Pharmacometrics Syst. Pharmacol. 2016, 5, 475–483. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, H.; Yoshida, K.; Nakada, T.; Kojima, K.; Ogasawara, A.; Nakamaru, Y.; Yamazaki, H. Prediction of Human Distribution Volumes of Compounds in Various Elimination Phases Using Physiologically Based Pharmacokinetic Modeling and Experimental Pharmacokinetics in Animals. Drug Metab. Dispos. 2018, 47, 114–123. [Google Scholar] [CrossRef] [Green Version]
- Maharaj, A.R.; Wu, H.; Hornik, C.P.; Arrieta, A.; James, L.; Bhatt-Mehta, V.; Bradley, J.; Muller, W.J.; Al-Uzri, A.; Downes, K.J.; et al. Use of normalized prediction distribution errors for assessing population physiologically-based pharmacokinetic model adequacy. J. Pharmacokinet. Pharmacodyn. 2020, 47, 199–218. [Google Scholar] [CrossRef] [PubMed]
- Azzaro, A.J.; Ziemniak, J.; Kemper, E.; Campbell, B.J.; Vandenberg, C. Pharmacokinetics and Absolute Bioavailability of Selegiline Following Treatment of Healthy Subjects With the Selegiline Transdermal System (6 mg/24 h): A Comparison With Oral Selegiline Capsules. J. Clin. Pharmacol. 2007, 47, 1256–1267. [Google Scholar] [CrossRef] [PubMed]
- Logan, B.K. Methamphetamine-Effects on Human Performance and Behavior. Forensic Sci. Rev. 2002, 14, 133–151. [Google Scholar] [PubMed]
- Korashy, H.M.; Elbekai, R.H.; El-Kadi, A.O.S. Effects of renal diseases on the regulation and expression of renal and hepatic drug-metabolizing enzymes: A review. Xenobiotica 2004, 34, 1–29. [Google Scholar] [CrossRef]
- Dreisbach, A.W.; Lertora, J.J.L. The effect of chronic renal failure on hepatic drug metabolism and drug disposition. Semin. Dial. 2003, 16, 45–50. [Google Scholar] [CrossRef]
- Pichette, V. Drug Metabolism in Chronic Renal Failure. Curr. Drug Metab. 2003, 4, 91–103. [Google Scholar] [CrossRef]
- Nolin, T.D.; Frye, R.F.; Matzke, G.R. Hepatic drug metabolism and transport in patients with kidney disease. Am. J. Kidney Dis. 2003, 42, 906–925. [Google Scholar] [CrossRef]
- Houston, J.B. Utility of in vitro drug metabolism data in predicting in vivo metabolic clearance. Biochem. Pharmacol. 1994, 47, 1469–1479. [Google Scholar] [CrossRef]
- Gabrielsson, J.; Weiner, D. Pharmacokinetic and Pharmacodynamic Data Analysis. Trends Pharmacol. Sci. 1995, 16, 143. [Google Scholar]
- Heinonen, E.H.; Anttila, M.I.; Karnani, H.L.; Nyman, L.M.; Vuorinen, J.A.; Pyykkö, K.A.; Lammintausta, R.A.S. Desmethylselegiline, a metabolite of selegiline, is an irreversible inhibitor of monoamine oxidase type B in humans. J. Clin. Pharmacol. 1997, 37, 602–609. [Google Scholar] [CrossRef]
- Cruickshank, C.C.; Dyer, K.R. A review of the clinical pharmacology of methamphetamine. Addiction 2009, 104, 1085–1099. [Google Scholar] [CrossRef] [PubMed]
- Patrick, K.S.; Markowitz, J.S. Pharmacology of methylphenidate, amphetamine enantiomers and pemoline in attention-deficit hyperactivity disorder. Hum. Psychopharmacol. Clin. Exp. 1997, 12, 527–546. [Google Scholar] [CrossRef]
- Albertson, T.E.; Derlet, R.W.; E Van Hoozen, B. Methamphetamine and the expanding complications of amphetamines. West. J. Med. 1999, 170, 214–219. [Google Scholar] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Selegiline | Methamphetamine | Desmethyl Selegiline | Amphetamine | |||
|---|---|---|---|---|---|---|---|
| Molecular weight (g/mol) # | 187.28 | 149.23 | 173.25 | 135.2 | |||
| Log P | 2.7 a | 2.07 b | 2.46 c | 1.8 b | |||
| Compound type | Monoprotic base | Monoprotic base | Monoprotic base | Monoprotic base | |||
| pKa | 7.44 d | 9.9 e | 9.8 c | 9.9 e | |||
| fu | 0.1 f | 0.85 e | 0.47 * | 0.84 e | |||
| B/P | 1.34 f | 1.487 $ | 1.70 $ | 1.35 $ | |||
| Distribution | |||||||
| Distribution model | Full PBPK | Full PBPK | Minimal PBPK | Minimal PBPK | |||
| Vss (L/Kg) | 9.26 | 7.14 | 5.37 | 5.29 | |||
| Elimination | |||||||
| CLiv | 84.56g | 20 h | 35.8 * | 20 * | |||
| CLRg | 0.46 | 8.74 | 0.96 | 8.04 | |||
| % Metabolized | 1A2 | MAP: 4.21 | 2D6 | AMP:40 | |||
| % Metabolized | 1A2 2A6 | DMS: 1.64 | 2D6 | OHAMP:60 | 2A6 | AMP: 15 | |
| MAP: 3.13 | 3A4 | 2B6 | AMP: 45 | ||||
| 2A6 2B6 | DMS: 0.77 | AMP: 40 | |||||
| MAP: 26.27 | |||||||
| 2B6 2C8 | DMS: 6.48 | ||||||
| DMS:0.10 | |||||||
| 2C9 | DMS:0.23 | ||||||
| 2C18 | MAP: 1.22 | ||||||
| 2C18 2C19 | DMS: 1.05 | ||||||
| MAP: 32.70 | |||||||
| 2C19 2D6 | DMS: 3.58 | ||||||
| MAP: 8.37 | |||||||
| 2D6 2E1 | DMS: 7.20 | ||||||
| DMS: 0.69 | |||||||
| 3A4 | MAP:1.89 | ||||||
| 3A4 | DMS:0.45 | ||||||
| Absorption Parameters | Parameter | Estimate | QSAR/Reference | Equation |
|---|---|---|---|---|
| Partition coefficients | Stratum corneum Lipid:vehicle | 85.03 | [49] | |
| Sebum:vehicle | 915.9 | [50] | ||
| Viable epidermis:Stratum corneum | 24.8 | [51] | ||
| Viable epidermis:Sebum | 0.09 | Calculated | ||
| Dermis:Viable epidermis | 1 | Assumed | ||
| Subcutis:Dermis | 1 × 10−6 | Assumed | ||
| Dermis:blood | 2.44 | [52] | ||
| Subcutis:blood | 1 | Assumed | ||
| Diffusion coefficients | Stratum corneum lipid | 3.22 × 10−5 | [53] | . rc: Molecular radius |
| Viable epidermis | 0.002 | [51] | ||
| Dermis | 0.002 | [51] | ||
| Sebum | 0.0008 | [54] | A = 0.000145; B = 1.32; Kbolt = 1.38 × 10−16 T [°K] = [°C] + 273.15 ( | |
| Keratin Binding | Kon/koff78.8/0.93 | [55] |
| Population | Moiety | Hepatic Intrinsic Clearance (L/h) | Cardiac Output (L/h) | Hepatic Blood Flow (L/h) | Fraction Unbound (fu) | Renal Function a | Hepatic Clearance (L/h) | Renal Clearance (L/h) |
|---|---|---|---|---|---|---|---|---|
| Healthy | SEL | 24217.92 | 350.19 | 89.30 | 0.10 | 0.99 | 86.11 | 0.34 |
| MAP | 27.70 | 350.19 | 1.00 | 21.14 | 8.6526 | |||
| DMS | 125.44 | 350.19 | 0.47 | 35.36 | 0.9504 | |||
| AMP | 24.745 | 350.19 | 0.84 | 16.79 | 7.9596 | |||
| SRI | SEL | 8221.1 | 268.79 | 71.82 | 0.10 | 0.16 | 66.19 | 0.05 |
| MAP | 8.56 | 268.79 | 1.00 | 7.65 | 1.37 | |||
| DMS | 45.31 | 268.79 | 0.55 | 18.40 | 0.15 | |||
| AMP | 36.29 | 268.79 | 0.87 | 22.01 | 1.26 | |||
| CP-C | SEL | 2109.18 | 299.67 | 70.88 | 0.14 | 0.47 | 57.05 | 0.16 |
| MAP | 1.45 | 299.67 | 1.00 | 1.42 | 4.05 | |||
| DMS | 37.62 | 299.67 | 0.63 | 17.66 | 0.45 | |||
| AMP | 30.11 | 299.67 | 0.91 | 19.70 | 3.73 | |||
| Adolescents | SEL | 20147.64 | 333.70 | 89.59 | 0.10 | 1.05 | 85.88 | 0.36 |
| MAP | 20.89 | 333.70 | 1.00 | 16.94 | 9.11 | |||
| DMS | 95.42 | 333.70 | 0.48 | 30.37 | 1.00 | |||
| AMP | 17.05 | 333.70 | 0.84 | 12.40 | 8.38 |
| Compound | Healthy | SRI | CP-C | ||||||
|---|---|---|---|---|---|---|---|---|---|
| AUC 0–24 h | AUC Last Dose | AR | AUC 0–24 h | AUC Last Dose | AR | AUC 0–24 h | AUC Last Dose | AR | |
| SEL | 25.70 | 113.30 | 4.41 | 34.60 | 174.39 | 5.04 | 21.54 | 121.91 | 5.66 |
| MAP | 21.75 | 479.48 | 22.05 | 29.34 | 2202.52 | 75.07 | 15.40 | 1577.26 | 102.39 |
| DMS | 8.14 | 115.26 | 14.15 | 9.47 | 384.40 | 40.58 | 3.81 | 258.65 | 67.82 |
| AMP | 10.94 | 210.16 | 19.21 | 5.61 | 258.96 | 46.18 | 1.67 | 102.29 | 61.28 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Puttrevu, S.K.; Arora, S.; Polak, S.; Patel, N.K. Physiologically Based Pharmacokinetic Modeling of Transdermal Selegiline and Its Metabolites for the Evaluation of Disposition Differences between Healthy and Special Populations. Pharmaceutics 2020, 12, 942. https://doi.org/10.3390/pharmaceutics12100942
Puttrevu SK, Arora S, Polak S, Patel NK. Physiologically Based Pharmacokinetic Modeling of Transdermal Selegiline and Its Metabolites for the Evaluation of Disposition Differences between Healthy and Special Populations. Pharmaceutics. 2020; 12(10):942. https://doi.org/10.3390/pharmaceutics12100942
Chicago/Turabian StylePuttrevu, Santosh Kumar, Sumit Arora, Sebastian Polak, and Nikunj Kumar Patel. 2020. "Physiologically Based Pharmacokinetic Modeling of Transdermal Selegiline and Its Metabolites for the Evaluation of Disposition Differences between Healthy and Special Populations" Pharmaceutics 12, no. 10: 942. https://doi.org/10.3390/pharmaceutics12100942