Brain Delivery of Thyrotropin-Releasing Hormone via a Novel Prodrug Approach

 ,
,  ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Instruments
2.3. Animals
2.4. Synthesis
2.5. Membrane Affinity Studies
2.6. In Vitro Metabolic Stability Studies
2.7. Neuropharmacodynamic Assesment: PST
2.8. Neurochemical Assestment: ACh Release Using In Vivo Microdialysis
2.9. Statistical Analysis
3. Results and Discussion
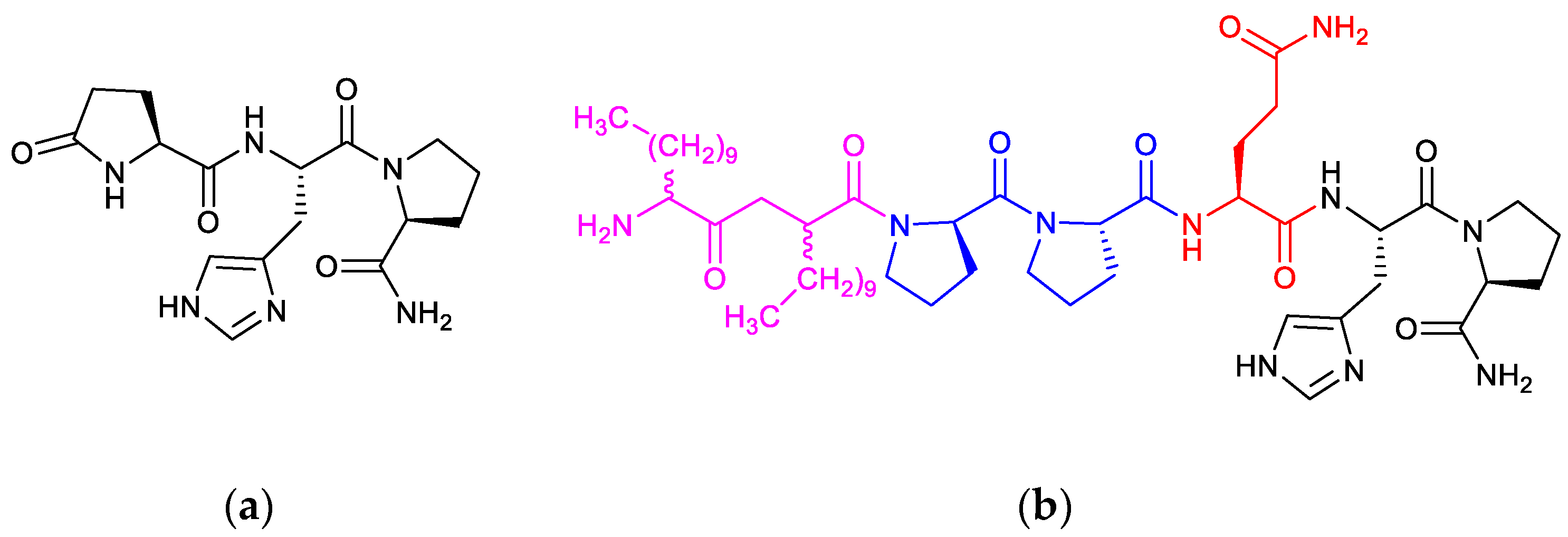
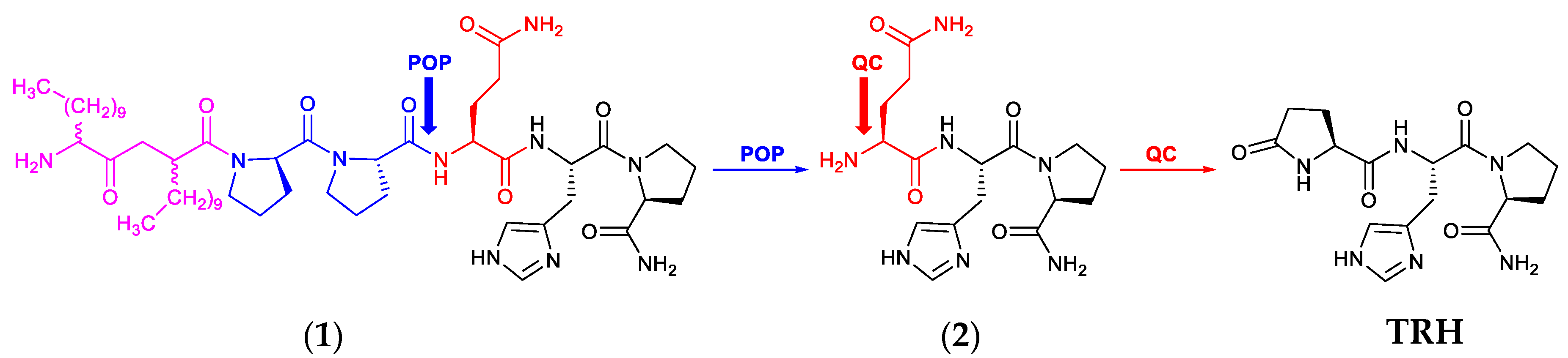
3.1. Synthesis and IAMC Studies
3.2. In Vitro Metabolic Stability Studies
3.3. In Vivo Studies
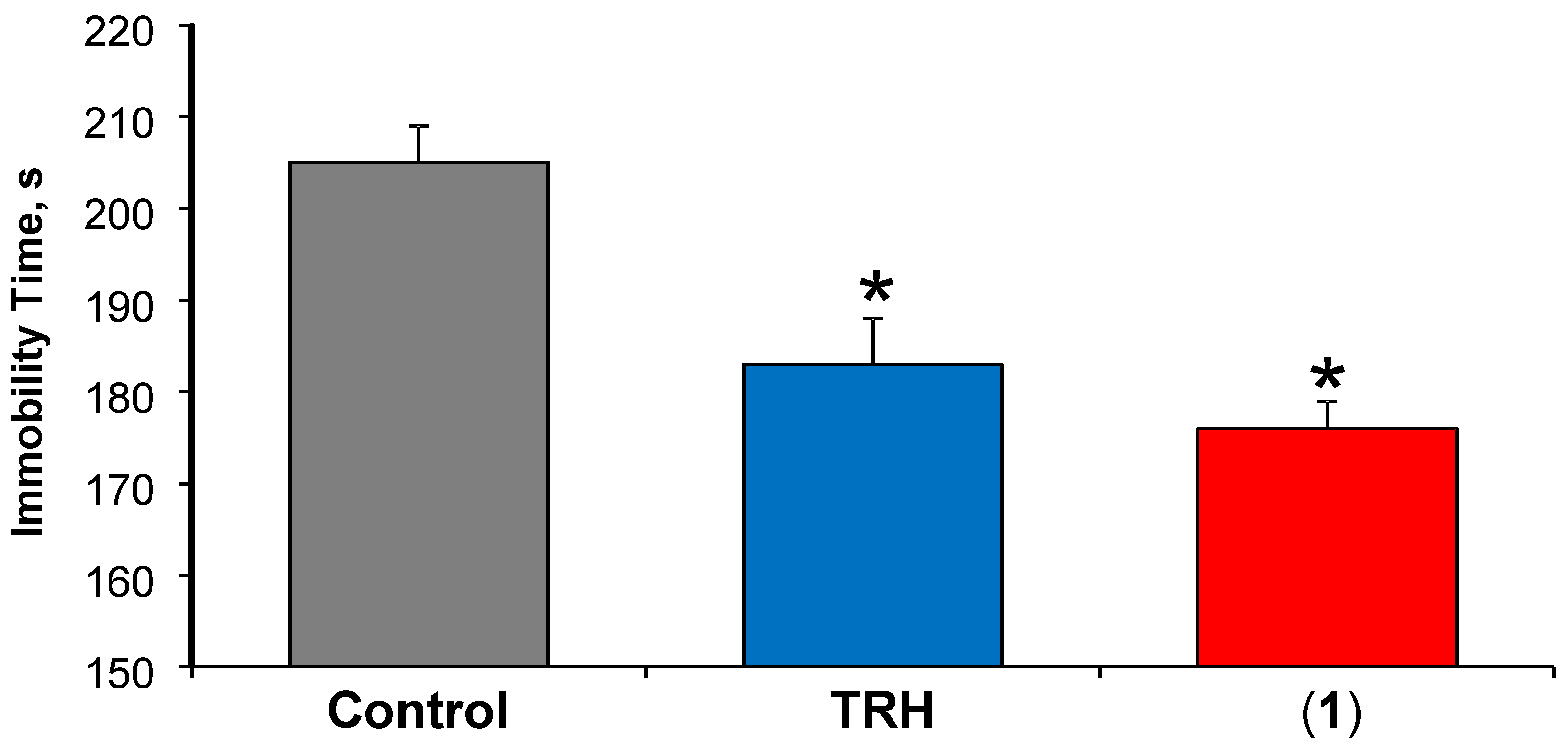
3.3.1. Antidepressant-Like Effect
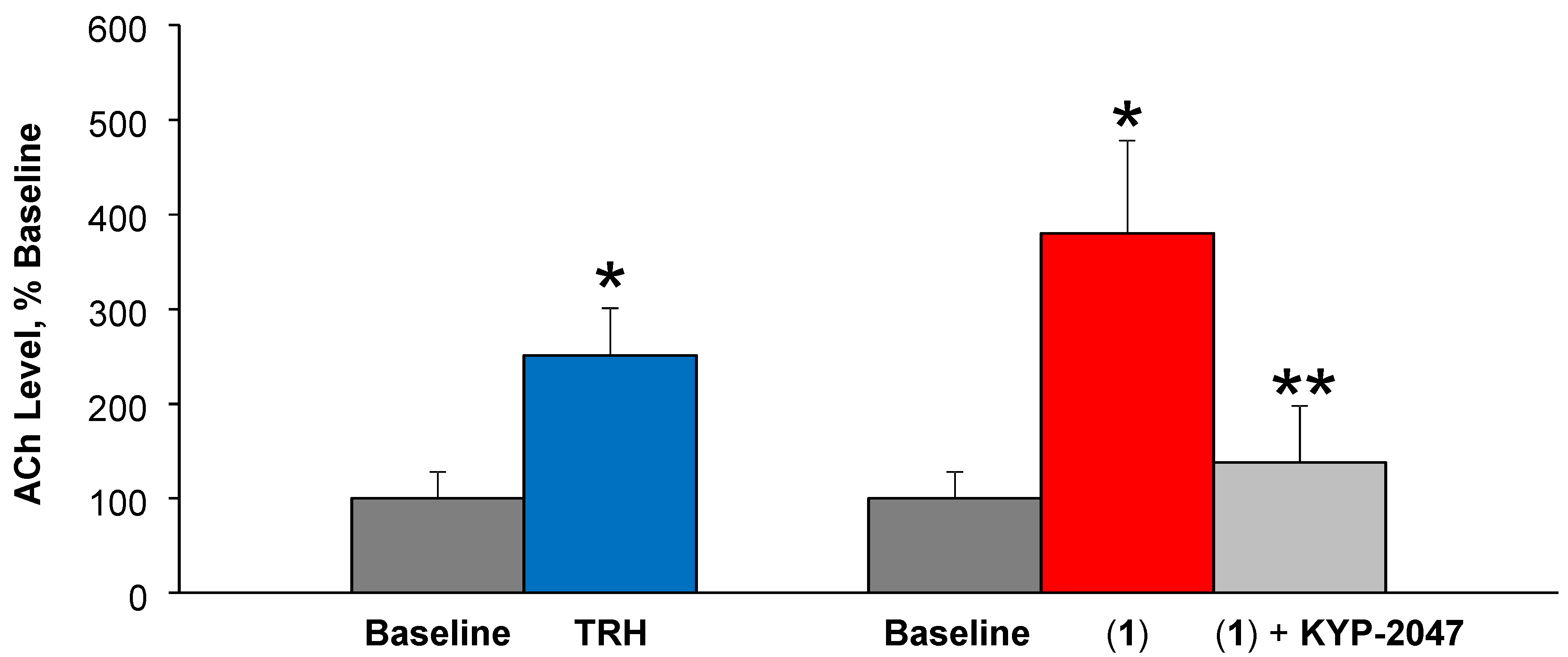
3.3.2. ACh Release
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Monga, V.; Meena, C.L.; Kaur, N.; Jain, R. Chemistry and biology of thyrotropin-releasing hormone (TRH) and its analogs. Curr. Med. Chem. 2008, 15, 2718–2733. [Google Scholar] [CrossRef]
- Duval, F. Thyroid hormone treatment of mood disorders. Curr. Treat. Options Psych. 2018. [Google Scholar] [CrossRef]
- Daimon, C.M.; Chirdon, P.; Maudsley, S.; Martin, B. The role of thyrotropin releasing hormone in aging and neurodegenerative diseases. Am. J. Alzheimer’s Dis. (Columbia) 2013, 1. [Google Scholar] [CrossRef] [PubMed]
- Gary, K.A.; Sevarino, K.A.; Yarbrough, G.G.; Prange, A.J.; Winokur, A. The thyrotropin-releasing hormone (TRH) hypothesis of homeostatic regulation: Implications for TRH-based therapeutics. J. Pharm. Exp. 2003, 305, 410–416. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez-Mariscal, M.; de Gortari, P.; López-Rubalcava, C.; Martínez, A.; Joseph-Bravo, P. Analysis of the anxiolytic-like effect of TRH and the response of amygdalar TRHergic neurons in anxiety. Psychoneuroendocrinology 2008, 33, 198–213. [Google Scholar] [CrossRef] [PubMed]
- Prokai, L. Central nervous system effects of thyrotropin-releasing hormone and its analogues: Opportunities and perspectives for drug discovery and development. In Progress of Drug Research; Jucker, E., Ed.; Birkhauser: Basel, Switzerland, 2002; Volume 59, pp. 133–170. [Google Scholar]
- Prokai-Tatrai, K.; Prokai, L. Prodrugs of thyrotropin-releasing hormone and related peptides as central nervous system agents. Molecules 2009, 6, 633–654. [Google Scholar] [CrossRef]
- Khomane, K.S.; Meena, C.L.; Jain, R.; Bansal, A.K. Novel thyrotropin-releasing hormone analogs: A patent review. Expert Opin. Ther. Pat. 2011, 21, 1673–1691. [Google Scholar] [CrossRef]
- Kelly, J.A.; Boyle, N.T.; Cole, N.; Slator, G.R.; Colivicchi, M.A.; Stefanini, C.; Gobbo, O.L.; Scalabrino, G.A.; Ryan, S.M.; Elamin, M.; et al. First-in-class thyrotropin-releasing hormone (TRH)-based compound binds to a pharmacologically distinct TRH receptor subtype in human brain and is effective in neurodegenerative models. Neuropharmacology 2015, 89, 193–203. [Google Scholar] [CrossRef]
- Kobayashi, N.; Sato, N.; Fujimura, Y.; Kihara, T.; Sugita, K.; Takahashi, K.; Koike, K.; Sugawara, T.; Tada, Y.; Nakai, H.; et al. Discovery of the orally effective thyrotropin-releasing hormone mimetic: 1-{N-[(4S,5S)-(5-Methyl-2-oxooxazolidine-4-yl)carbonyl]-3-(thiazol-4-yl)-l-alanyl}-(2R)-2-methyl- pyrrolidine trihydrate (rovatirelin hydrate). Acs Omega 2018, 3, 13647–13666. [Google Scholar] [CrossRef]
- Bundgaard, H.; Møss, J. Prodrugs of peptides. 6. Bioreversible derivatives of thyrotropin-releasing hormone (TRH) with increased lipophilicity and resistance to cleavage by the TRH-specific serum enzyme. Pharm. Res. 1990, 7, 885–892. [Google Scholar] [CrossRef]
- Prokai-Tatrai, K.; Prokai, L. Prodrug design for brain delivery of small- and medium-sized neuropeptides. Methods Mol. Biol. 2011, 789, 313–336. [Google Scholar]
- Prokai-Tatrai, K.; Perjesi, P.; Zharikova, A.D.; Li, X.; Prokai, L. Design, synthesis, and biological evaluation of novel, centrally-acting thyrotropin-releasing hormone analogues. Bioorg. Med. Chem. Lett. 2002, 12, 2171–2174. [Google Scholar] [CrossRef]
- Prokai, L.; Nguyen, V.; Szarka, S.; Garg, P.; Sabnis, G.; Bimonte-Nelson, H.B.; McLaughlin, K.J.; Talboom, J.S.; Conrad, C.D.; Shughrue, P.J.; et al. The prodrug DHED selectively delivers 17β-estradiol to the brain for treating estrogen-responsive disorders. Sci. Transl. Med. 2015, 7, 297ra113. [Google Scholar] [CrossRef]
- Prokai-Tatrai, K.; Prokai, L. Modifying peptide properties by prodrug design for enhanced transport into the CNS Peptide transport and delivery into the central nervous system. In Progress in Drug Research; Prokai, L., Prokai-Tatrai, K., Eds.; Birkhäuser: Basel, Switzerland, 2003; Volume 61, pp. 155–188. [Google Scholar]
- Prokai-Tatrai, K.; Teixido, M.; Nguyen, V.; Zharikova, A.D.; Prokai, L. A pyridinium-substituted analog of the TRH-like tripeptide pGlu-Glu-Pro-NH2 and its prodrugs as central nervous system agents. Med. Chem. 2005, 1, 141–152. [Google Scholar] [CrossRef] [PubMed]
- Prokai, L.; Prokai-Tatrai, K.; Bodor, N. Targeting drugs to the brain by redox chemical delivery systems. Med. Res. Rev. 2000, 20, 367–416. [Google Scholar] [CrossRef]
- Prokai-Tatrai, K.; Szarka, S.; Nguyen, V.; Sahyouni, F.; Walker, C.; White, S.; Talamantes, T.; Prokai, L. “All in the mind”? Brain-targeting chemical delivery system of 17β-estradiol (estredox) produces significant uterotrophic side effect. Pharm. Anal. Acta 2012, S7, 002. [Google Scholar] [CrossRef] [PubMed]
- Baranda, A.B.; Alonso, R.M.; Jiménez, R.M.; Weinmann, J.M. Instability of calcium channel antagonists during sample preparation for LC–MS–MS analysis of serum samples. Forensic Sci. Internat. 2006, 156, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Herraiz, T. N-methyltetrahydropyridines and pyridinium cations as toxins and comparison with naturally-occurring alkaloids. Food Chem. Toxicol. 2016, 97, 23–39. [Google Scholar] [CrossRef]
- Sirker, A.; Missouris, C.G.; MacGregor, G.A. Dihydropyridine calcium channel blockers and peripheral side effects. J. Hum. Hypertens. 2001, 5, 745–746. [Google Scholar] [CrossRef] [PubMed]
- Prokai, L.; Prokai-Tatrai, K.; Ouyang, X.; Kim, H.S.; Wu, W.M.; Zharikova, A.; Bodor, N. Metabolism-based brain-targeting system for a thyrotropin-releasing hormone analogue. J. Med. Chem. 1999, 42, 4563–4571. [Google Scholar] [CrossRef] [PubMed]
- Prokai-Tatrai, K.; Kim, H.S.; Prokai, L. The utility of oligopeptidase in brain-targeting delivery of an enkephalin analogue by prodrug design. Open Med. Chem. J. 2008, 2, 97–100. [Google Scholar] [CrossRef] [PubMed]
- Polgar, L.; Szeltner, Z. Structure, function and biological relevance of prolyl oligopeptidase. Curr. Prot. Pept. Sci. 2008, 9, 96–107. [Google Scholar] [CrossRef]
- Myöhänen, T.T.; Venäläinen, J.I.; García-Horsman, J.A.; Piltonen, M.; Männistö, P.T. Distribution of prolyl oligopeptidase in the mouse whole-body sections and peripheral tissues. Histochem. Cell. Biol. 2008, 130, 993–1003. [Google Scholar] [CrossRef]
- Pajouhesh, H.; Lenz, G.R. Medicinal chemical properties of successful central nervous system drugs. NeuroRx 2005, 2, 541–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tóth, I.; Malkinson, J.P.; Flinn, N.S.; Drouillat, B.; Horváth, A.; Erchegyi, J.; Idei, M.; Venetianer, A.; Artursson, P.; Lazorova, L.; et al. Novel lipoamino acid- and liposaccharide-based system for peptide delivery: Application for oral administration of tumor-selective somatostatin analogues. J. Med. Chem. 1999, 42, 4010–4013. [Google Scholar] [CrossRef] [PubMed]
- Blanchfield, J.T.; Toth, I. Lipids, sugars and liposaccharides in drug delivery 2: An update. Curr. Med. Chem. 2004, 11, 2375–2382. [Google Scholar] [CrossRef] [PubMed]
- Valko, K.; Du, C.M.; Bevan, C.D.; Reynolds, D.P.; Abraham, M.H. Rapid-gradient HPLC method for measuring drug interactions with immobilized artificial membrane: Comparison with other lipophilicity measures. J. Pharm. Sci. 2000, 89, 1085–1096. [Google Scholar] [CrossRef]
- Jalkanen, A.J.; Leikas, J.V.; Forsberg, M.M. KYP-2047 penetrates mouse brain and effectively inhibits mouse prolyl oligopeptidase. Basic Clin. Pharm. Toxicol. 2014, 114, 460–463. [Google Scholar] [CrossRef]
- Ramli, S.; Gentle, I.R.; Ross, B.P. Efficient manual Fmoc solid-phase synthesis of the N-terminal segment of surfactant protein B (SP-B1-25). Protein Peptide Lett. 2009, 16, 810–814. [Google Scholar] [CrossRef]
- Ross, B.P.; Falconer, R.A.; Toth, I. N-1-(4,4-dimethyl-2,6-dioxocyclohex-1-ylidene)ethyl (N-Dde) lipoamino acids. Molbank 2008, 2, M566. [Google Scholar] [CrossRef]
- Prokai, L.; Zharikova, A.D.; Janáky, T.; Prokai-Tatrai, K. Exploratory pharmacokinetics and brain distribution study of a neuropeptide FF antagonist by liquid chromatography/atmospheric pressure ionization tandem mass spectrometry. Rapid Comm. Mass Spectrom. 2000, 14, 2412–2418. [Google Scholar] [CrossRef]
- Prokai-Tatrai, K.; Nguyen, V.; Szarka, S.; Konya, K.; Prokai, L. Design and exploratory neuropharmacological evaluation of novel thyrotropin-releasing hormone analogs and their brain-targeting bioprecursor prodrugs. Pharmaceutics 2013, 5, 318–328. [Google Scholar] [CrossRef]
- Nguyen, V.; Zharikova, A.D.; Prokai, L. Evidence for interplay between thyrotropin-releasing hormone (TRH) and its structural analogue pGlu-Glu-Pro-NH2 ([Glu2]TRH) in the brain: An in vivo microdialysis study. Neurosci. Lett. 2007, 415, 64–67. [Google Scholar] [CrossRef] [PubMed]
- Prokai, L.; Fryčák, P.; Stevens, S.M.; Nguyen, V. Measurement of acetylcholine in rat brain microdialysates by LC–isotope dilution tandem MS. Chromatographia 2008, 68, S101–S105. [Google Scholar] [CrossRef] [PubMed]
- Ghose, A.K.; Pritchett, A.; Crippen, G.M. Atomic physicochemical parameters for three dimensional structure directed quantitative structure-activity relationships III: Modeling hydrophobic interactions. J. Comput. Chem. 1988, 9, 80–90. [Google Scholar] [CrossRef]
- Braddy, A.C.; Janáky, T.; Prokai, L. Immobilized artificial membrane chromatography coupled with atmospheric pressure ionization mass spectrometry. J. Chromatogr. A 2002, 966, 81–87. [Google Scholar] [CrossRef]
- Ross, B.P.; Braddy, A.C.; McGeary, R.P.; Blanchfield, J.T.; Prokai, L.; Toth, I. Membrane partitioning and micellar aggregation of bile salts, fatty acids, SDS, and sugar conjugated fatty acids: Correlation with hemolytic activity. Mol. Pharm. 2004, 1, 233–245. [Google Scholar] [CrossRef]
- Lloyd, R.L.; Pekary, A.E.; Sattin, A.; Amundson, T. Antidepressant effects of thyrotropin-releasing hormone analogues using a rodent model of depression. Pharmacol. Biochem. Behav. 2001, 70, 15–22. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | CHIIAM 1 | clogP 2 |
|---|---|---|
| TRH | −11.2 ± 3.2 | −3.50 |
| (1) | 66.0 ± 10.3 | 1.85 |
| Test Compound | t1/2 in Plasma | t1/2 in 20% (w/v) Brain Homogenate |
|---|---|---|
| TRH | 7 ± 4 min | 4 ± 1 min |
| (1) | 100 ± 7 min | 47 ± 6 min |
| (1) + KYP-2047 | 115 ± 13 min | >24 h |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Prokai-Tatrai, K.; De La Cruz, D.L.; Nguyen, V.; Ross, B.P.; Toth, I.; Prokai, L. Brain Delivery of Thyrotropin-Releasing Hormone via a Novel Prodrug Approach. Pharmaceutics 2019, 11, 349. https://doi.org/10.3390/pharmaceutics11070349
Prokai-Tatrai K, De La Cruz DL, Nguyen V, Ross BP, Toth I, Prokai L. Brain Delivery of Thyrotropin-Releasing Hormone via a Novel Prodrug Approach. Pharmaceutics. 2019; 11(7):349. https://doi.org/10.3390/pharmaceutics11070349
Chicago/Turabian StyleProkai-Tatrai, Katalin, Daniel L. De La Cruz, Vien Nguyen, Benjamin P. Ross, Istvan Toth, and Laszlo Prokai. 2019. "Brain Delivery of Thyrotropin-Releasing Hormone via a Novel Prodrug Approach" Pharmaceutics 11, no. 7: 349. https://doi.org/10.3390/pharmaceutics11070349