Enhanced Intradermal Delivery of Nanosuspensions of Antifilariasis Drugs Using Dissolving Microneedles: A Proof of Concept Study



Abstract
:
1. Introduction
2. Materials and Methods
2.1. Chemicals and Materials
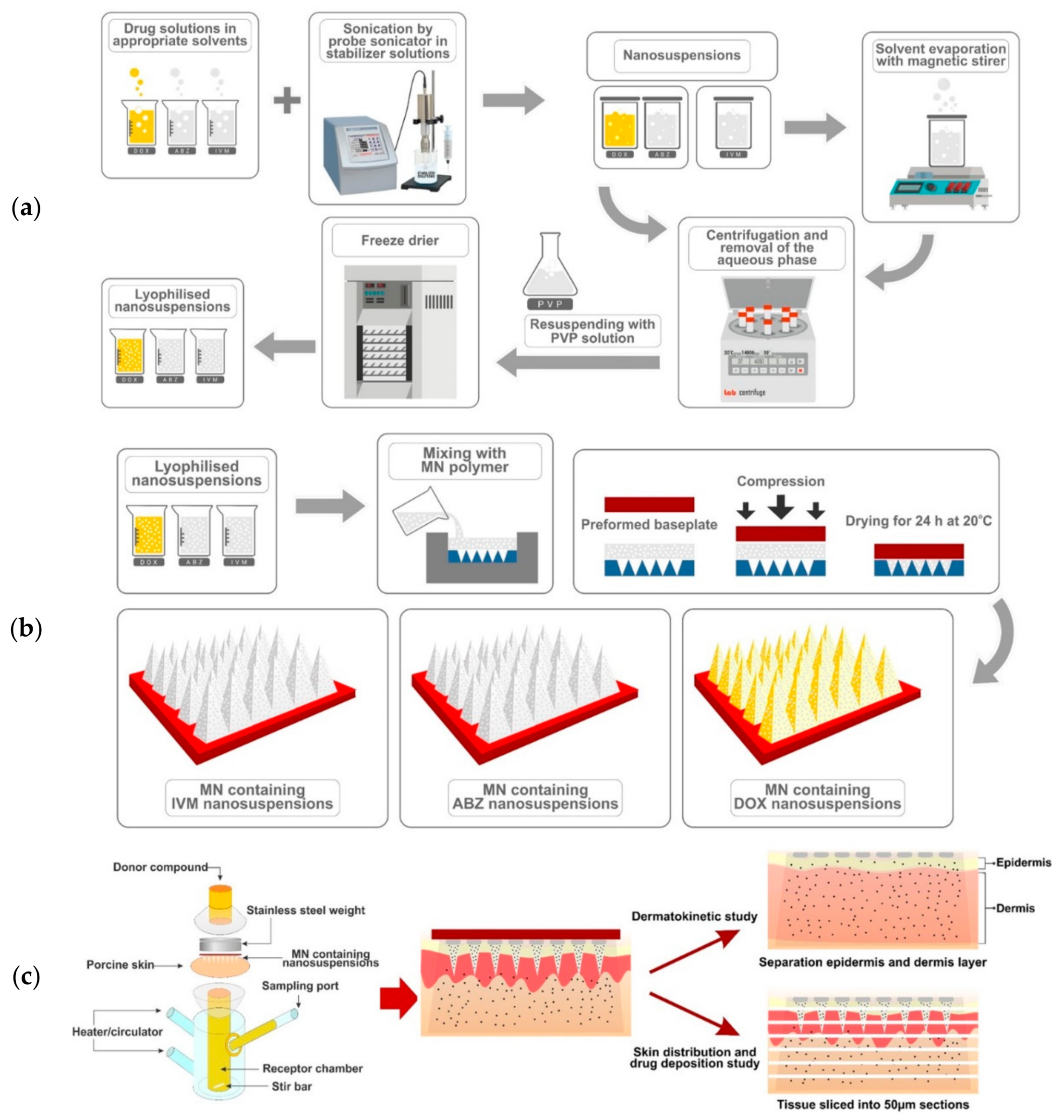
2.2. Preparation of NS
2.2.1. Doxycycline Nanosuspensions (DOX NS)
2.2.2. Albendazole Sulfoxide Nanosuspensions (ABZ-OX NS)
2.2.3. Ivermectin Nanosuspensions (IVM NS)
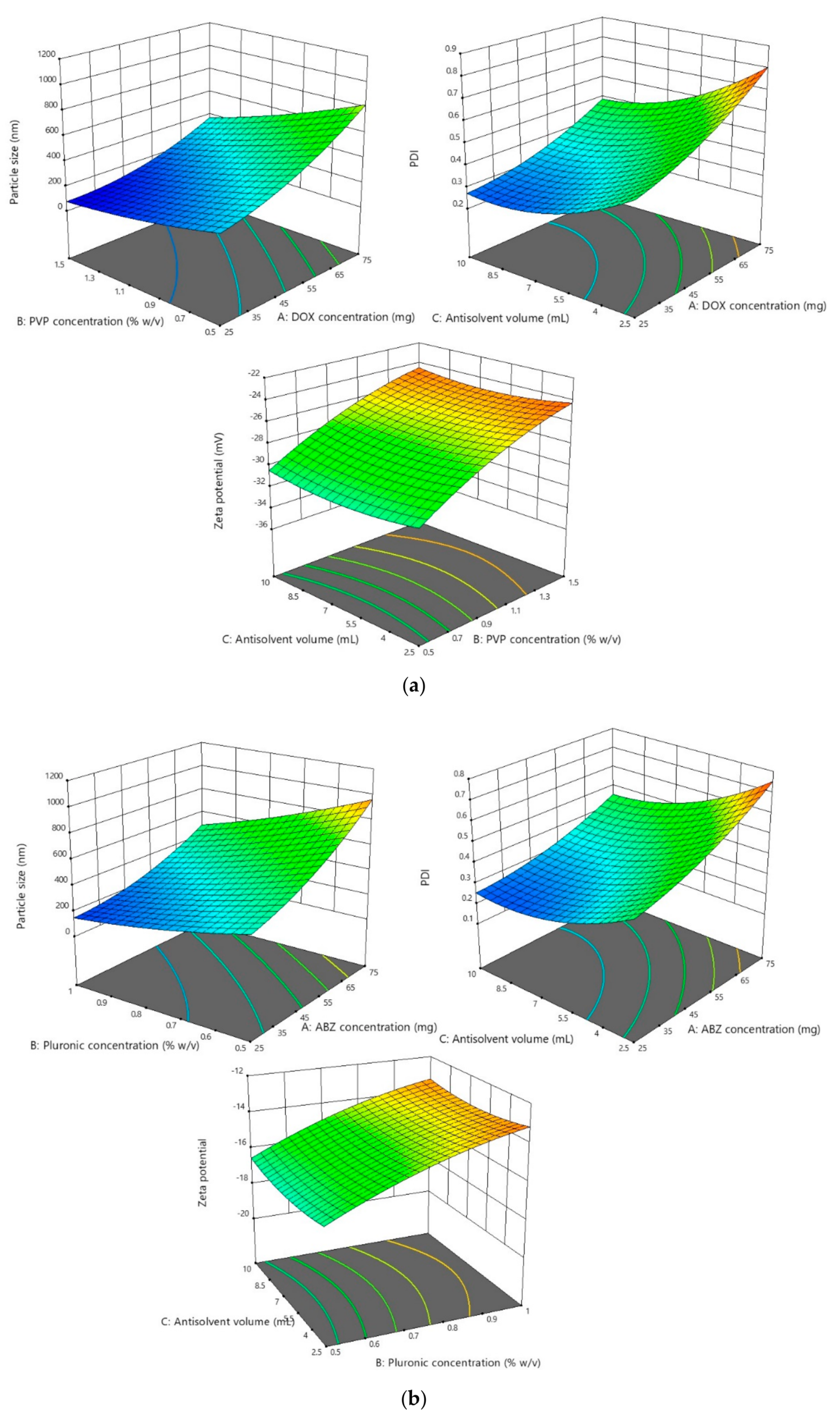
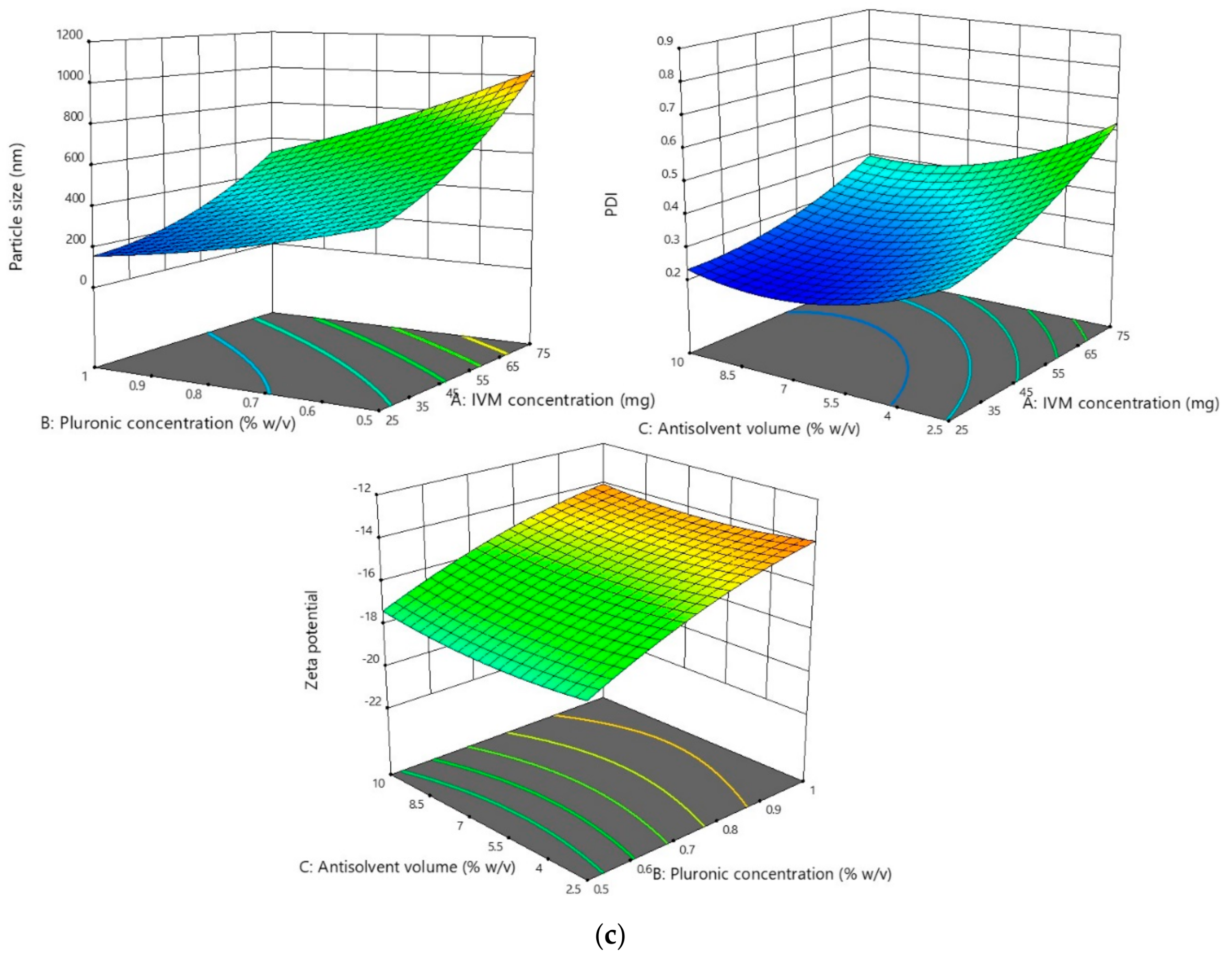
2.3. Optimization of NS Formulations
2.4. Characterization of Optimized NS
2.5. In Vitro Drug Release Study
2.6. Two-Step Casting of MN and Needle-Free Patch
2.7. Skin Sample Collection
2.8. Mechanical and Insertion Properties of MNs
2.9. Calculation of Theoretical Drug Content Localized to the Needles
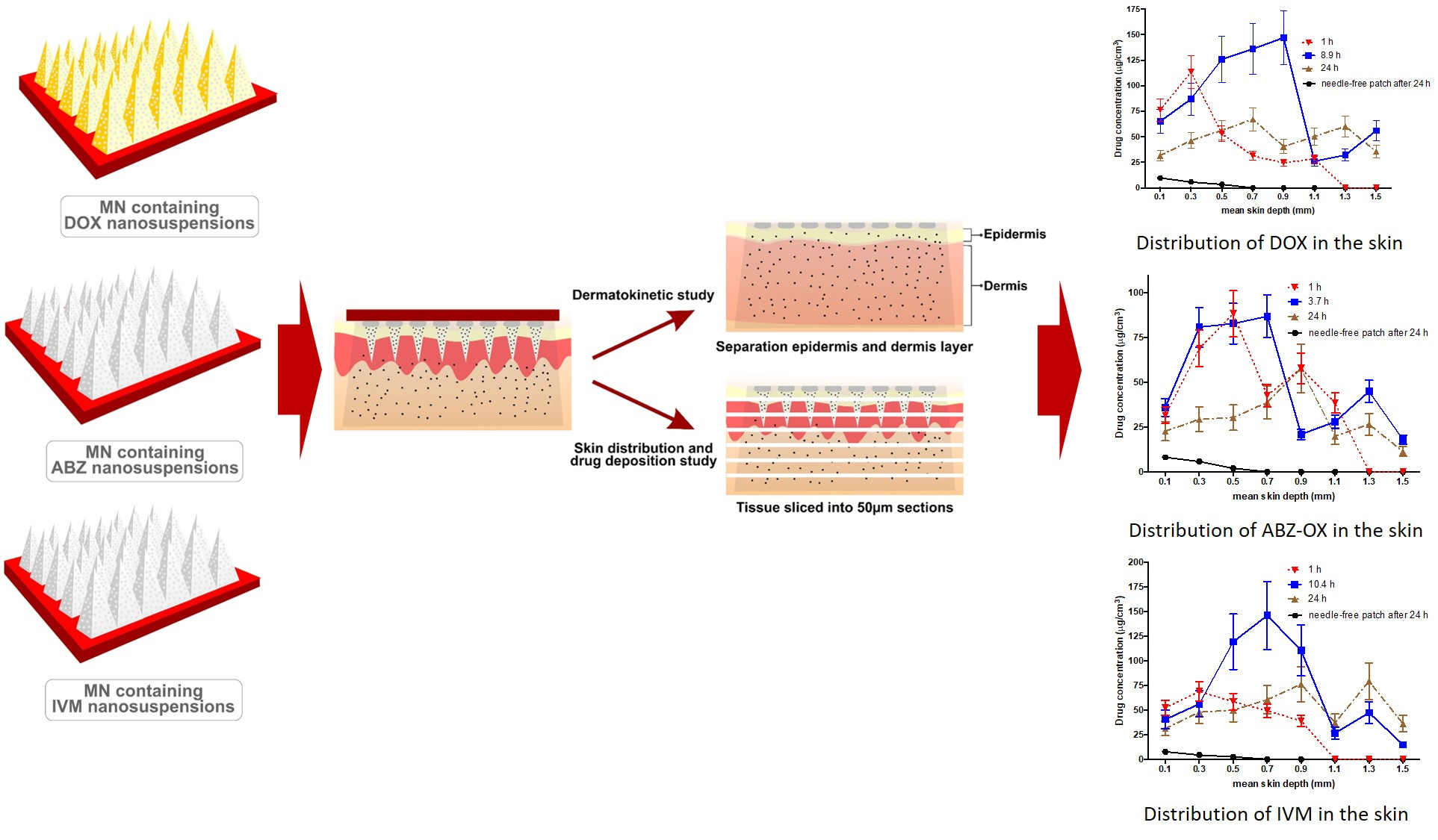
2.10. In Vitro Dermatokinetic, Skin Distribution and Deposition Studies
2.11. Instrumentation and Chromatographic Condition for Analytical Method
2.12. Statistical Analysis
3. Results and Discussion
3.1. Preparation of NS and Screening of Stabilizers
3.2. Optimization of NS Formulations
3.2.1. Effect of the Parameters on the Particle Size and PDI
3.2.2. Effect of the Parameters on Zeta Potential
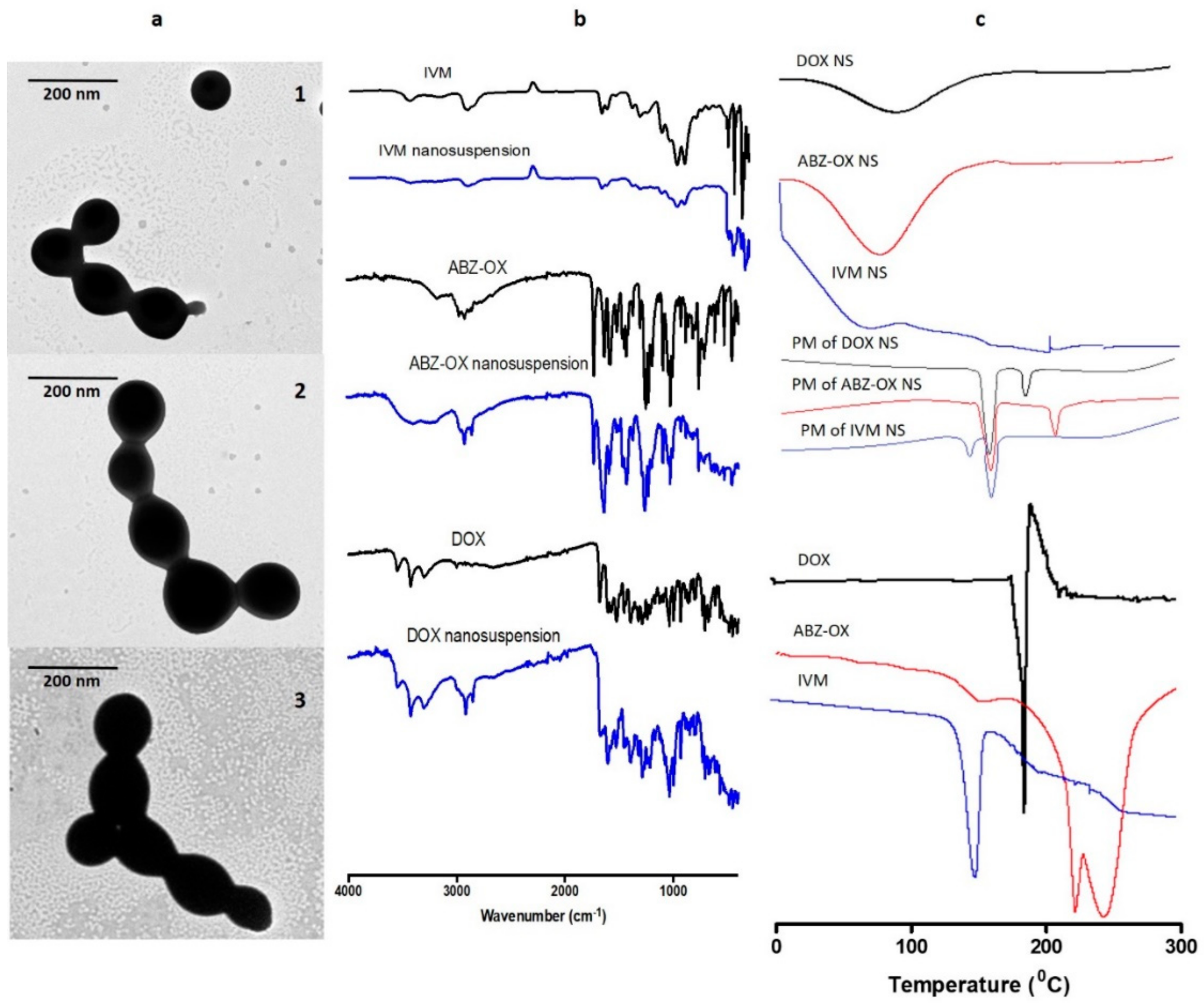
3.3. Characterization of Optimized NS
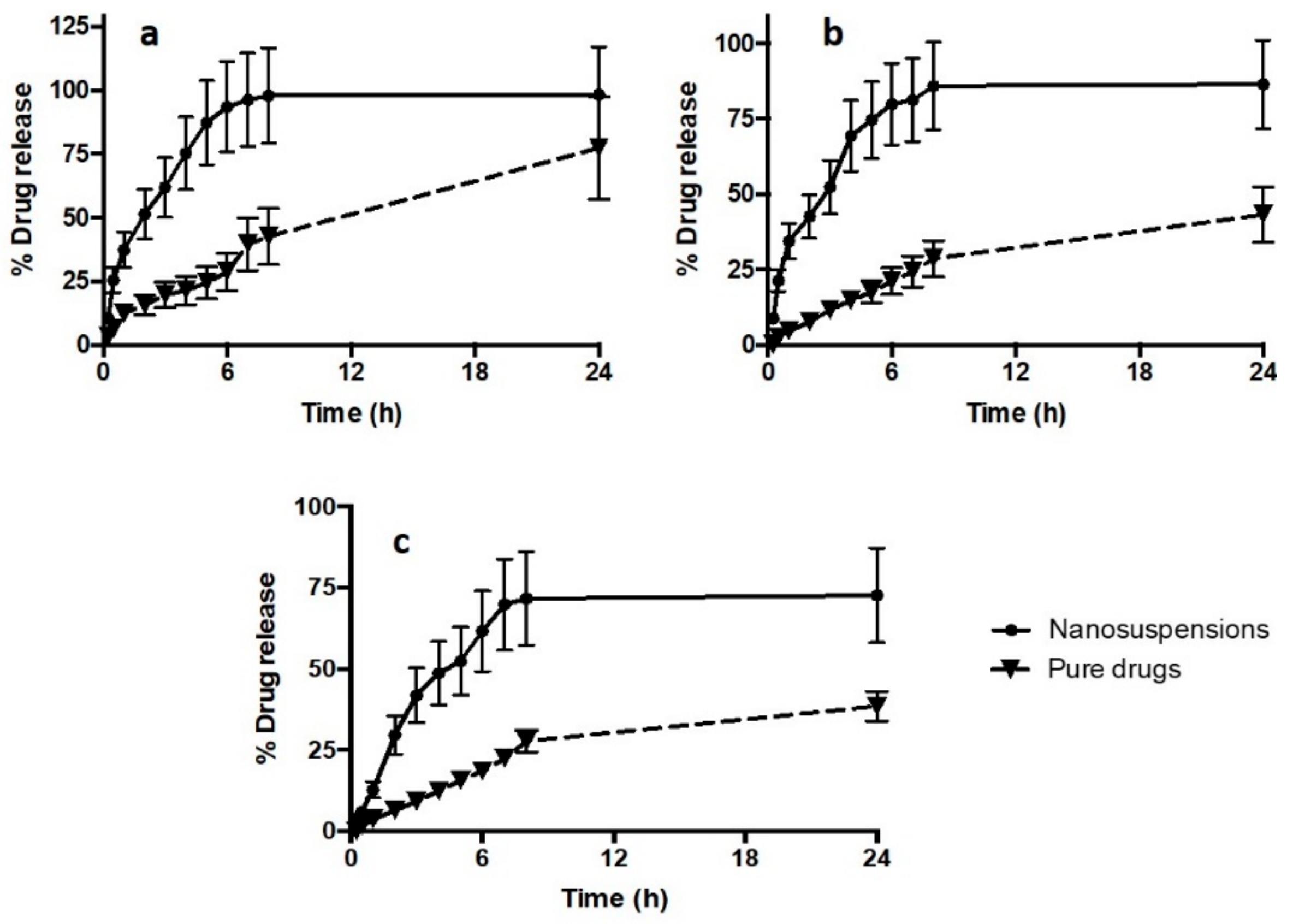
3.4. In Vitro Drug Release Study
3.5. MN Formulations
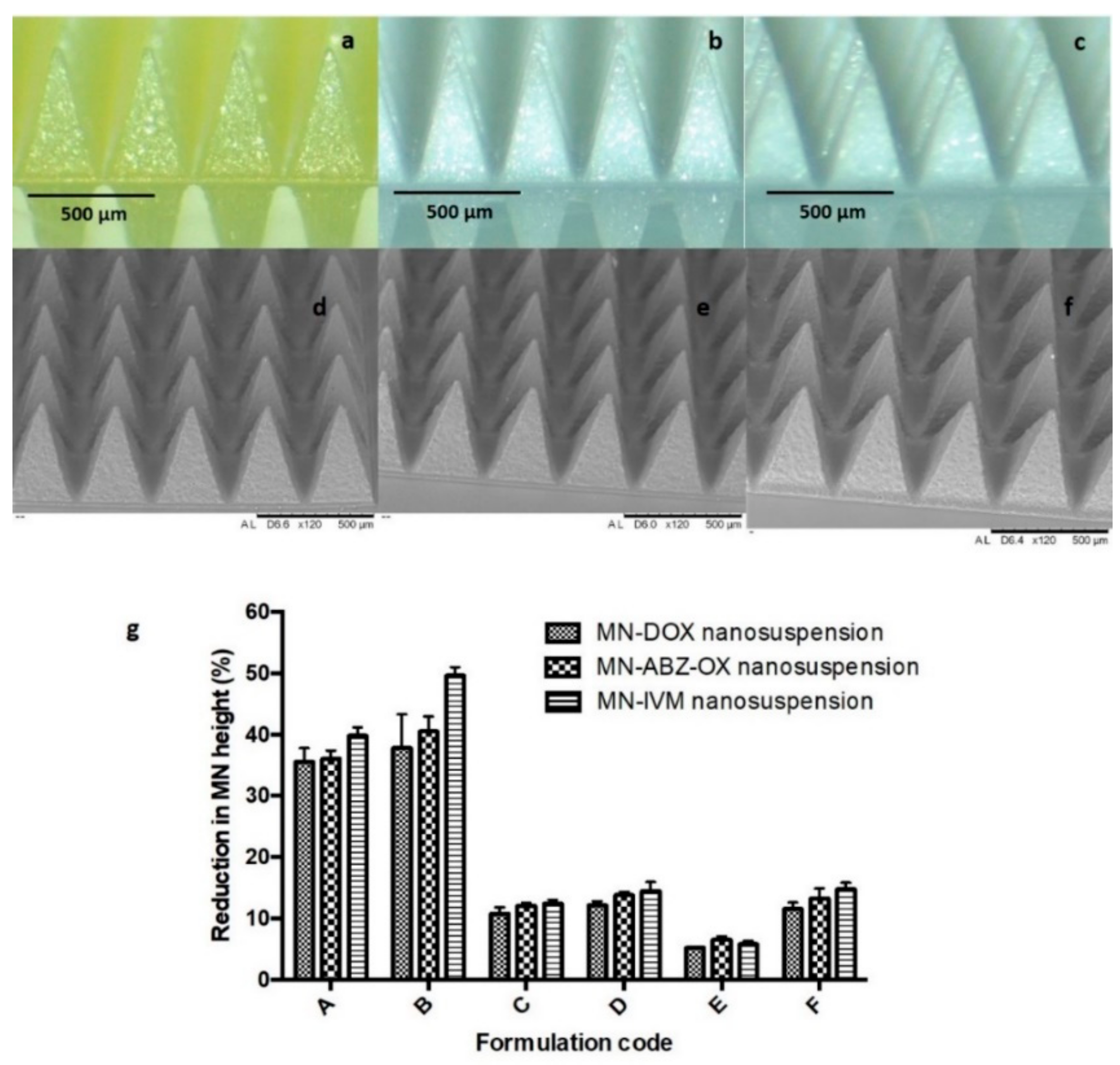
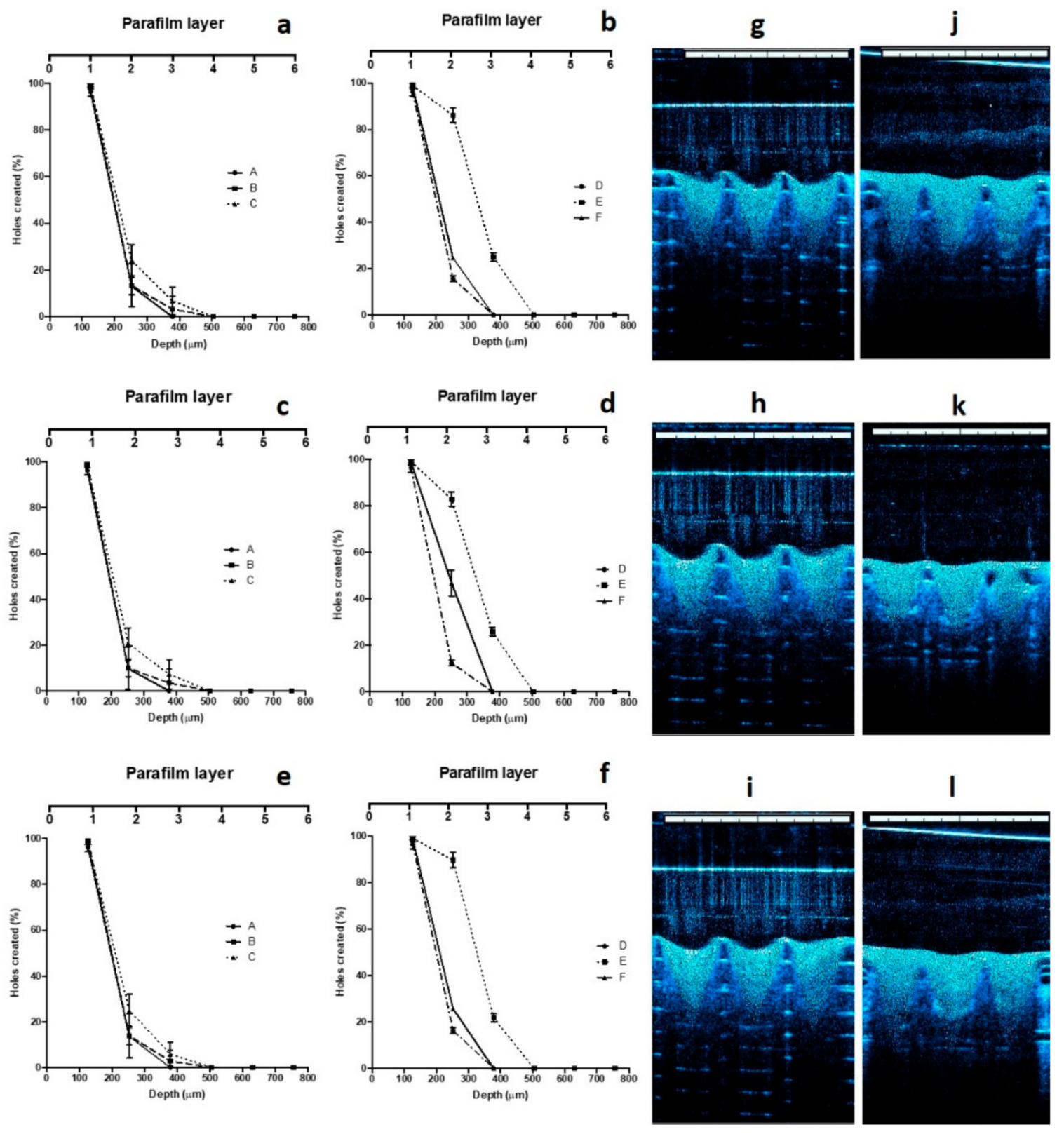
3.6. Mechanical and Insertion Properties of MNs
3.7. Drug Content Located in the Needles
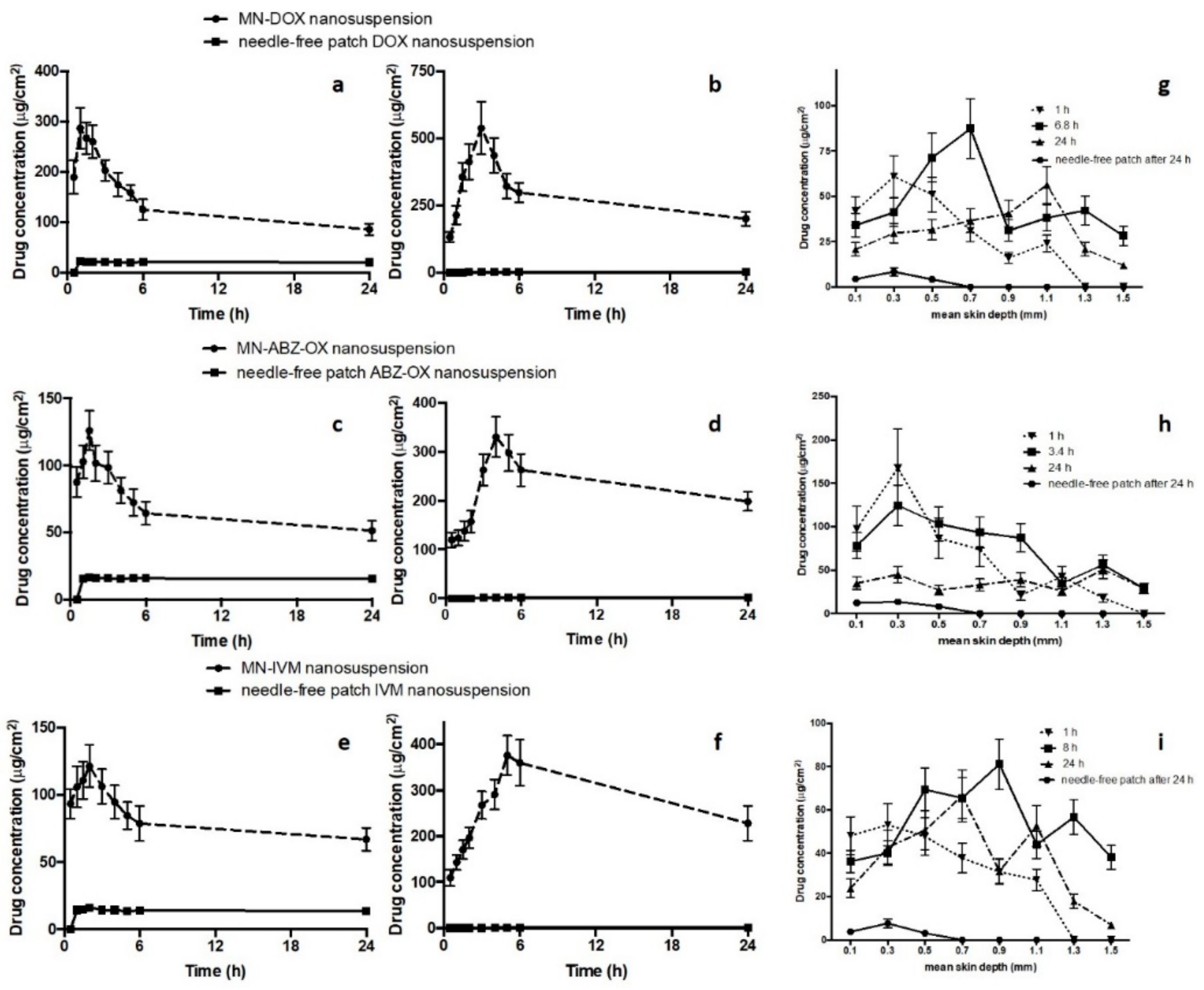
3.8. In Vitro Dermatokinetic, Skin Distribution and Deposition Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- WHO. Alternative Mass Drug Administration Regimens to Eliminate Lymphatic Filariasis; World Health Organization: Geneva, Switzerland, 2017; ISBN 9789241550161. [Google Scholar]
- Ottesen, E.A. Lymphatic filariasis: Treatment, control and elimination. Adv. Parasitol. 2006, 61, 395–441. [Google Scholar] [PubMed]
- Stoops, C.A.; Gionar, Y.R.; Rusmiarto, S.; Susapto, D.; Andris, H.; Elyazar, I.R.F.; Barbara, K.A.; Munif, A. Laboratory and field testing of bednet traps for mosquito (Diptera: Culicidae) sampling in West Java, Indonesia. J. Vector Ecol. 2010, 35, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Hoerauf, A. Filariasis: New drugs and new opportunities for lymphatic filariasis and onchocerciasis. Curr. Opin. Infect. Dis. 2008, 21, 673–681. [Google Scholar] [CrossRef] [PubMed]
- WHO. Global Programme to Eliminate Lymphatic Filariasis: Progress Report. World Health Organization: Geneva, Switzerland, 2018. [Google Scholar]
- Remme, J.H.F.; Feenstra, P.; Lever, P.R.; Medici, A.C.; Morel, C.M.; Noma, M.; Ramaiah, K.D.; Richards, F.; Seketeli, A.; Schmunis, G.; et al. Tropical diseases targeted for elimination: Chagas disease, lymphatic filariasis, onchocerciasis, and leprosy. In Disease Control Priorities in Developing Countries; Oxford University Press: New York, NY, USA, 2006; Volume 22, pp. 433–449. ISBN 0821361791. [Google Scholar]
- Hoerauf, A.; Mand, S.; Fischer, K.; Kruppa, T.; Marfo-Debrekyei, Y.; Debrah, A.Y.; Pfarr, K.M.; Adjei, O.; Büttner, D.W. Doxycycline as a novel strategy against bancroftian filariasis-depletion of Wolbachia endosymbionts from Wuchereria bancrofti and stop of microfilaria production. Med. Microbiol. Immunol. 2003, 192, 211–216. [Google Scholar] [CrossRef] [PubMed]
- WHO. Lymphatic Filariasis. Available online: https://www.who.int/news-room/fact-sheets/detail/lymphatic-filariasis (accessed on 22 March 2019).
- Trevaskis, N.L.; Kaminskas, L.M.; Porter, C.J.H. From sewer to saviour-targeting the lymphatic system to promote drug exposure and activity. Nat. Rev. Drug Discov. 2015, 14, 781–803. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.; Afzal, M.; Bhattacharya, S.M.; Ahmad, F.J.; Dinda, A.K. Nanopharmaceuticals to target antifilarials: A comprehensive review. Expert Opin. Drug Deliv. 2013, 10, 665–678. [Google Scholar] [CrossRef]
- Tiantian, Y.; Wenji, Z.; Mingshuang, S.; Rui, Y.; Shuangshuang, S.; Yuling, M.; Jianhua, Y.; Xinggang, Y.; Shujun, W.; Weisan, P. Study on intralymphatic-targeted hyaluronic acid-modified nanoliposome: Influence of formulation factors on the lymphatic targeting. Int. J. Pharm. 2014, 471, 245–257. [Google Scholar] [CrossRef]
- Singh, Y.; Srinivas, A.; Gangwar, M.; Meher, J.G.; Misra-Bhattacharya, S.; Chourasia, M.K. Subcutaneously Administered Ultrafine PLGA Nanoparticles Containing Doxycycline Hydrochloride Target Lymphatic Filarial Parasites. Mol. Pharm. 2016, 13, 2084–2094. [Google Scholar] [CrossRef]
- Siram, K.; Chellan, V.R.; Natarajan, T. Solid lipid nanoparticles of diethylcarbamazine citrate for enhanced delivery to the lymphatics: In vitro and in vivo evaluation. Expert Opin. Drug Deliv. 2014, 11, 1351–1365. [Google Scholar] [CrossRef]
- El-Laithy, H.M.; Basalious, E.B.; El-Hoseiny, B.M.; Adel, M.M. Novel self-nanoemulsifying self-nanosuspension (SNESNS) for enhancing oral bioavailability of diacerein: Simultaneous portal blood absorption and lymphatic delivery. Int. J. Pharm. 2015, 490, 146–154. [Google Scholar] [CrossRef]
- McConnachie, L.A.; Kinman, L.M.; Koehn, J.; Kraft, J.C.; Lane, S.; Lee, W.; Collier, A.C.; Ho, R.J.Y. Long-acting profile of 4 drugs in 1 anti-HIV nanosuspension in nonhuman primates for 5 weeks after a single subcutaneous injection. J. Pharm. Sci. 2018, 107, 1787–1790. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Wu, X.; Zhou, Z.; Chen, X.; Kong, M. Enhanced transdermal lymphatic delivery of doxorubicin via hyaluronic acid based transfersomes/microneedle complex for tumor metastasis therapy. Int. J. Biol. Macromol. 2019, 125, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Rabinow, B.E. Nanosuspensions in drug delivery. Drug Discov. 2004, 3, 785–796. [Google Scholar] [CrossRef] [PubMed]
- George, M.; Ghosh, I. Identifying the correlation between drug/stabilizer properties and critical quality attributes (CQAs) of nanosuspension formulation prepared by wet media milling technology. Eur. J. Pharm. Sci. 2013, 48, 142–152. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Li, X.; Lansakara-P, D.S.P.; Kumar, A.; Cui, Z. A nanoparticle depot formulation of 4-(N)-stearoyl gemcitabine shows a strong antitumor activity. J. Pharm. Pharmacol. 2013, 65, 236–242. [Google Scholar] [CrossRef] [PubMed]
- Indermun, S.; Luttge, R.; Choonara, Y.E.; Kumar, P.; Du Toit, L.C.; Modi, G.; Pillay, V. Current advances in the fabrication of microneedles for transdermal delivery. J. Control. Release 2014, 185, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Tarnow, K.; King, N. Intradermal injections: Traditional bevel up versus bevel down. Appl. Nurs. Res. 2004, 17, 275–282. [Google Scholar] [CrossRef]
- Donnelly, R.F.; Singh, T.R.R.; Morrow, D.I.J.; Woolfson, A.D. Microneedle-Mediated Transdermal and Intradermal Drug Delivery. In Wiley-Blackwell; Wiley Online Library: New York, NY, USA, 2012; ISBN 9780470654897. [Google Scholar]
- Donnelly, R.F.; Majithiya, R.; Raghu, T.; Singh, R.; Morrow, D.I.J.; Garland, M.J.; Demir, Y.K.; Ryan, E.; Gillen, D.; Scott, C.J.; et al. Design, optimization and characterisation of polymeric microneedle arrays prepared by a novel laser-based micromoulding technique. Pharm. Res. 2011, 28, 41–57. [Google Scholar] [CrossRef]
- Niu, L.; Chu, L.Y.; Burton, S.A.; Hansen, K.J.; Panyam, J. Intradermal delivery of vaccine nanoparticles using hollow microneedle array generates enhanced and balanced immune response. J. Control. Release 2018, 294, 268–278. [Google Scholar] [CrossRef]
- Vora, L.K.; Vavia, P.R.; Larrañeta, E.; Bell, S.E.J.; Donnelly, R.F. Novel nanosuspension-based dissolving microneedle arrays for transdermal delivery of a hydrophobic drug. J. Interdiscip. Nanomed. 2018, 3, 89–101. [Google Scholar] [CrossRef]
- Mc Crudden, M.T.C.; Larrañeta, E.; Clark, A.; Jarrahian, C.; Rein-Weston, A.; Lachau-Durand, S.; Niemeijer, N.; Williams, P.; Haeck, C.; McCarthy, H.O.; et al. Design, formulation and evaluation of novel dissolving microarray patches containing a long-acting rilpivirine nanosuspension. J. Control. Release 2018, 292, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Ng, W.K.; Shen, S.; Kim, S.; Tan, R.B.H. Preparation and characterization of spironolactone nanoparticles by antisolvent precipitation. Int. J. Pharm. 2009, 375, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Liu, X.; Lian, R.; Zheng, S.; Yin, Z.; Lu, Y.; Wu, W. Enhanced dissolution and oral bioavailability of aripiprazole nanosuspensions prepared by nanoprecipitation/homogenization based on acid-base neutralization. Int. J. Pharm. 2012, 438, 287–295. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Ng, W.K.; Tan, R.B.H. Are nanostructured lipid carriers (NLCs) better than solid lipid nanoparticles (SLNs): Development, characterizations and comparative evaluations of clotrimazole-loaded SLNs and NLCs? Eur. J. Pharm. Sci. 2012, 47, 139–151. [Google Scholar] [CrossRef] [PubMed]
- Costa, P.; Lobo, J.M.S. Modeling and comparison of dissolution profiles. Eur. J. Pharm. Sci. 2001, 13, 123–133. [Google Scholar] [CrossRef]
- Zhang, Y.; Huo, M.; Zhou, J.; Zou, A.; Li, W.; Yao, C.; Xie, S. DDSolver: An add-in program for modeling and comparison of drug dissolution profiles. AAPS J. 2010, 12, 263–271. [Google Scholar] [CrossRef]
- González-Vázquez, P.; Larrañeta, E.; McCrudden, M.T.C.; Jarrahian, C.; Rein-Weston, A.; Quintanar-Solares, M.; Zehrung, D.; McCarthy, H.; Courtenay, A.J.; Donnelly, R.F. Transdermal delivery of gentamicin using dissolving microneedle arrays for potential treatment of neonatal sepsis. J. Control. Release 2017, 265, 30–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larrañeta, E.; Moore, J.; Vicente-Pérez, E.M.; González-Vázquez, P.; Lutton, R.; Woolfson, A.D.; Donnelly, R.F. A proposed model membrane and test method for microneedle insertion studies. Int. J. Pharm. 2014, 472, 65–73. [Google Scholar] [CrossRef] [Green Version]
- Garg, N.K.; Sharma, D.; Singh, B.; Nirbhavane, P.; Tyagi, R.K.; Shukla, R.; Katare, O.P. Quality by Design (QbD)-enabled development of aceclofenac loaded-nano structured lipid carriers (NLCs): An improved dermatokinetic profile for inflammatory disorder(s). Int. J. Pharm. 2017, 517, 413–431. [Google Scholar] [CrossRef]
- Zhang, Y.; Huo, M.; Zhou, J.; Xie, S. PKSolver: An add-in program for pharmacokinetic and pharmacodynamic data analysis in Microsoft Excel. Comput. Methods Programs Biomed. 2010, 99, 306–314. [Google Scholar] [CrossRef]
- Mandal, B.; Moulik, S.P.; Ghosh, S. Physicochemistry of interaction of polyvinylpyrrolidone (PVP) with sodium dodecyl sulfate (SDS) in salt solution. J. Surf. Interfaces Mater. 2013, 1, 77–86. [Google Scholar] [CrossRef]
- Patel, J.; Dhingani, A.; Garala, K.; Raval, M.; Sheth, N. Design and development of solid nanoparticulate dosage forms of telmisartan for bioavailability enhancement by integration of experimental design and principal component analysis. Powder Technol. 2014, 258, 331–343. [Google Scholar] [CrossRef]
- Liu, P.; Rong, X.; Laru, J.; Van Veen, B.; Kiesvaara, J.; Hirvonen, J.; Laaksonen, T.; Peltonen, L. Nanosuspensions of poorly soluble drugs: Preparation and development by wet milling. Int. J. Pharm. 2011, 411, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Abdelbary, A.A.; Al-Mahallawi, A.M.; Abdelrahim, M.E.; Ali, A.M.A. Preparation, optimization, and in vitro simulated inhalation delivery of carvedilol nanoparticles loaded on a coarse carrier intended for pulmonary administration. Int. J. Nanomed. 2015, 10, 6339–6353. [Google Scholar] [CrossRef] [PubMed]
- Gang, S.; Ling, C.; Li-Qiang, W.; Li-Hon, Z.; Bao-De, S.; Wei-Bo, L.; Juan-Juan, L.; Juan, Z.; Rong, X.; Hai-Long, Y. Formulation of dried lignans nanosuspension with high redispersibility to enhance stability, dissolution, and oral bioavailability. Chin. J. Nat. Med. 2016, 14, 757–768. [Google Scholar]
- Zu, Y.; Sun, W.; Zhao, X.; Wang, W.; Li, Y.; Ge, Y.; Liu, Y.; Wang, K. Preparation and characterization of amorphous amphotericin B nanoparticles for oral administration through liquid antisolvent precipitation. Eur. J. Pharm. Sci. 2014, 53, 109–117. [Google Scholar] [CrossRef]
- Rao, Q.; Qiu, Z.; Huang, D.; Lu, T.; Zhang, Z.J.; Luo, D.; Pan, P.; Zhang, L.; Liu, Y.; Guan, S.; et al. Enhancement of the apparent solubility and bioavailability of tadalafil nanoparticles via antisolvent precipitation. Eur. J. Pharm. Sci. 2019, 128, 222–231. [Google Scholar] [CrossRef]
- Sengel, C.; Hasçiçek, C.; Gönül, N. Design of vitamin E d-α-tocopheryl polyethylene glycol 1000 succinate-emulsified poly (D,L-Lactide-co-glycolide) nanoparticles: Influence of duration of ultrasonication energy. J. Young Pharm. 2011, 3, 171–175. [Google Scholar] [CrossRef]
- Shelar, D.B.; Pawar, S.K.; Vavia, P.R. Fabrication of isradipine nanosuspension by anti-solvent microprecipitation-high-pressure homogenization method for enhancing dissolution rate and oral bioavailability. Drug Deliv. Transl. Res. 2013, 3, 384–391. [Google Scholar] [CrossRef]
- Dash, S.; Murthy, P.N.; Nath, L.; Chowdhury, P. Kinetic modelling on drug release from controlled drug delivery systems. Acta Pol. Pharm. Drug Res. 2010, 67, 217–223. [Google Scholar]
- Prausnitz, M.R.; Mikszta, J.A.; Cormier, M.; Andrianov, A.K. Microneedle-based vaccines. Curr. Top. Microbiol. Immunol. 2009, 333, 369–393. [Google Scholar] [PubMed]
- Chen, W.; Wang, C.; Yan, L.; Huang, L.; Zhu, X.; Chen, B.; Sant, H.J.; Niu, X.; Zhu, G.; Yu, K.N.; et al. Improved polyvinylpyrrolidone microneedle arrays with non-stoichiometric cyclodextrin. J. Mater. Chem. B 2014, 2, 1699–1705. [Google Scholar] [CrossRef]
- Hamdan, I.M.N.; Tekko, I.A.; Matchett, K.B.; Arnaut, L.G.; Silva, C.S.; McCarthy, H.O.; Donnelly, R.F. Intradermal delivery of a near-infrared photosensitizer using dissolving microneedle arrays. J. Pharm. Sci. 2018, 107, 2439–2450. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Allen, M.G.; Prausnitz, M.R. Polymer microneedles for controlled-release drug delivery. Pharm. Res. 2006, 23, 1008–1019. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, R.F.; Moffat, K.; Alkilani, A.Z.; Vicente-Pérez, E.M.; Barry, J.; McCrudden, M.T.C.; Woolfson, A.D. Hydrogel-forming microneedle arrays can be effectively inserted in skin by self-application: A pilot study centred on pharmacist intervention and a patient information leaflet. Pharm. Res. 2014, 31, 1989–1999. [Google Scholar] [CrossRef] [PubMed]
- Garg, N.K.; Tyagi, R.K.; Singh, B.; Sharma, G.; Nirbhavane, P.; Kushwah, V.; Jain, S.; Katare, O.P. Nanostructured lipid carrier mediates effective delivery of methotrexate to induce apoptosis of rheumatoid arthritis via NF-κB and FOXO1. Int. J. Pharm. 2016, 499, 301–320. [Google Scholar] [CrossRef] [PubMed]
- Suami, H.; Pan, W.R.; Taylor, G.I. The lymphatics of the skin filled by a dermal backflow: An obsevation in a scarred cadaver leg. Lymphology 2007, 40, 122–126. [Google Scholar] [PubMed]
- Dumont, N.; Merrigan, S.; Turpin, J.; Lavoie, C.; Papavasiliou, V.; Geretti, E.; Espelin, C.W.; Luus, L.; Kamoun, W.S.; Ghasemi, O.; et al. Nanoliposome targeting in breast cancer is influenced by the tumor microenvironment. Nanomed. Nanotechnol. Biol. Med. 2019, 17, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Khadke, S.; Roces, C.B.; Cameron, A.; Devit, A.; Perrie, Y. Formulation and manufacturing of lymphatic targeting liposomes using microfluidics. J. Control. Release 2019, 307, 211–220. [Google Scholar] [CrossRef]
- Abellan-Pose, R.; Teijeiro-Valiño, C.; Santander-Ortega, M.J.; Borrajo, E.; Vidal, A.; Garcia-Fuentes, M.; Csaba, N.; Alonso, M.J. Polyaminoacid nanocapsules for drug delivery to the lymphatic system: Effect of the particle size. Int. J. Pharm. 2016, 509, 107–117. [Google Scholar] [CrossRef]
- Kennedy, J.; Larrañeta, E.; McCrudden, M.T.C.; McCrudden, C.M.; Brady, A.J.; Fallows, S.J.; McCarthy, H.O.; Kissenpfennig, A.; Donnelly, R.F. In vivo studies investigating biodistribution of nanoparticle-encapsulated rhodamine B delivered via dissolving microneedles. J. Control. Release 2017, 265, 57–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wenkert, D.; Ramirez, B.; Shen, Y.; Kron, M.A. In vitro activity of geldanamycin derivatives against Schistosoma japonicum and Brugia malayi. J. Parasitol. Res. 2010, 2010. [Google Scholar] [CrossRef] [PubMed]
- Tompkins, J.B.; Stitt, L.E.; Ardelli, B.F. Brugia malayi: In vitro effects of ivermectin and moxidectin on adults and microfilariae. Exp. Parasitol. 2010, 124, 394–402. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulation | R2 Value of Kinetic Model | n | ||||
|---|---|---|---|---|---|---|
| Zero Order | First Order | Higuchi | Hixcon-Crowell | Korsmeyer-Peppas | ||
| DOX NS | 0.4360 | 0.9311 | 0.5900 | 0.7032 | 0.7983 | 0.311 |
| ABZ-OX NS | 0.4486 | 0.9461 | 0.6049 | 0.7611 | 0.8054 | 0.314 |
| IVM NS | 0.4963 | 0.9759 | 0.76 | 0.8076 | 0.7891 | 0.391 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Permana, A.D.; McCrudden, M.T.C.; Donnelly, R.F. Enhanced Intradermal Delivery of Nanosuspensions of Antifilariasis Drugs Using Dissolving Microneedles: A Proof of Concept Study. Pharmaceutics 2019, 11, 346. https://doi.org/10.3390/pharmaceutics11070346
Permana AD, McCrudden MTC, Donnelly RF. Enhanced Intradermal Delivery of Nanosuspensions of Antifilariasis Drugs Using Dissolving Microneedles: A Proof of Concept Study. Pharmaceutics. 2019; 11(7):346. https://doi.org/10.3390/pharmaceutics11070346
Chicago/Turabian StylePermana, Andi Dian, Maelíosa T. C. McCrudden, and Ryan F. Donnelly. 2019. "Enhanced Intradermal Delivery of Nanosuspensions of Antifilariasis Drugs Using Dissolving Microneedles: A Proof of Concept Study" Pharmaceutics 11, no. 7: 346. https://doi.org/10.3390/pharmaceutics11070346