Anti-Angiogenic Effect of Orally Available Pemetrexed for Metronomic Chemotherapy

 ,
,
Abstract
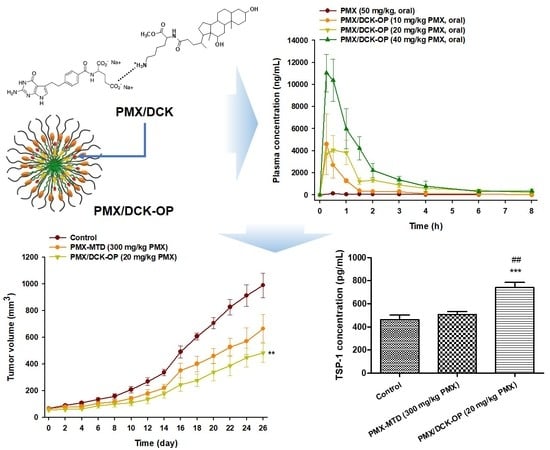
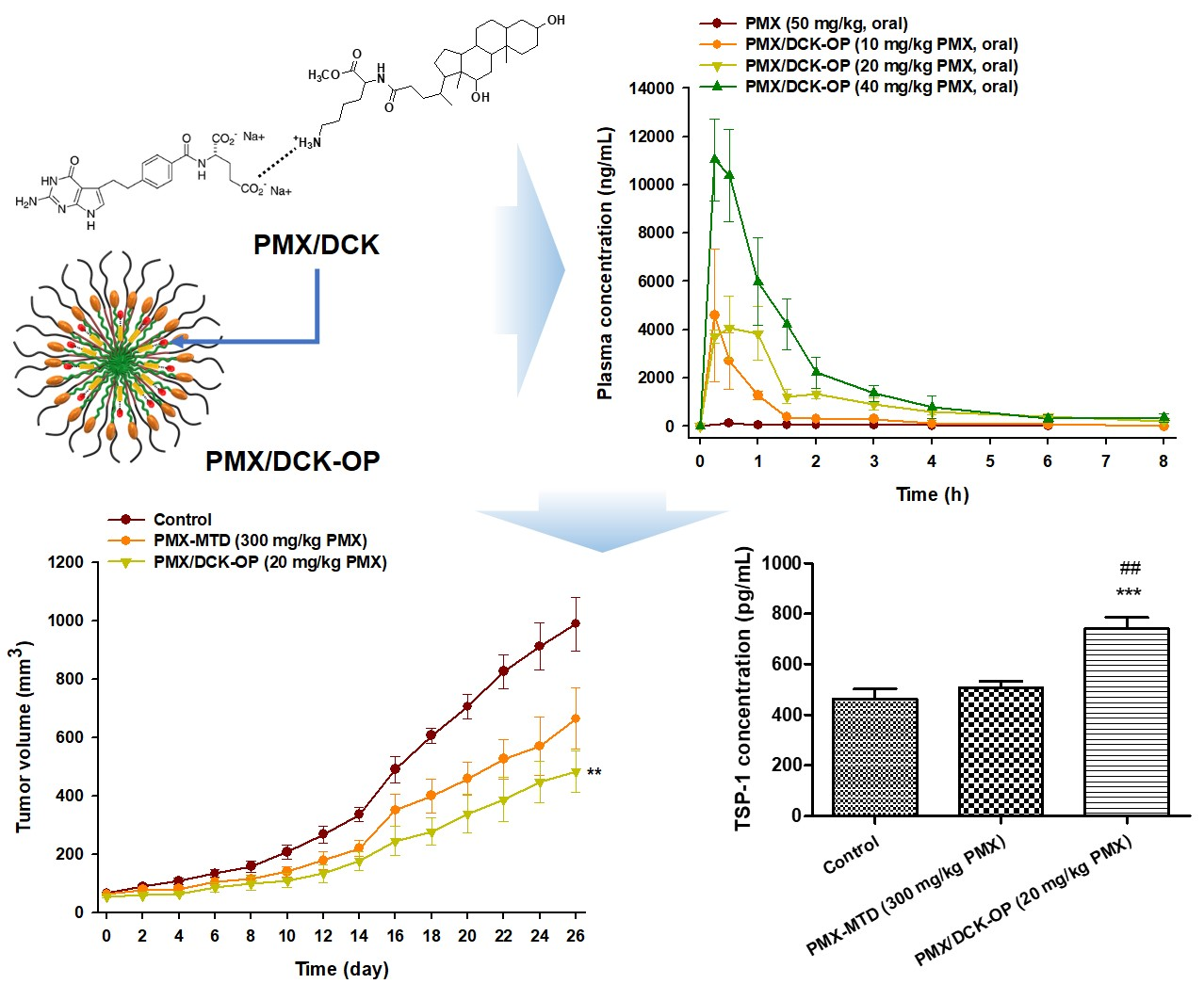
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Animals
2.3. In Vitro Anti-Angiogenic Effect of PMX
2.3.1. Cytotoxicity Assay
2.3.2. Tube Formation Assay
2.3.3. Scratch Wound Recovery Assay
2.4. Preparation and Characterization of PMX/DCK-OP
2.5. In Vitro Artificial Intestinal Membrane and Caco-2 Cell Monolayer Permeability
2.6. Oral Bioavailability of PMX/DCK-OP in Mice
2.7. In Vivo Matrigel Plug Assay of PMX/DCK-OP
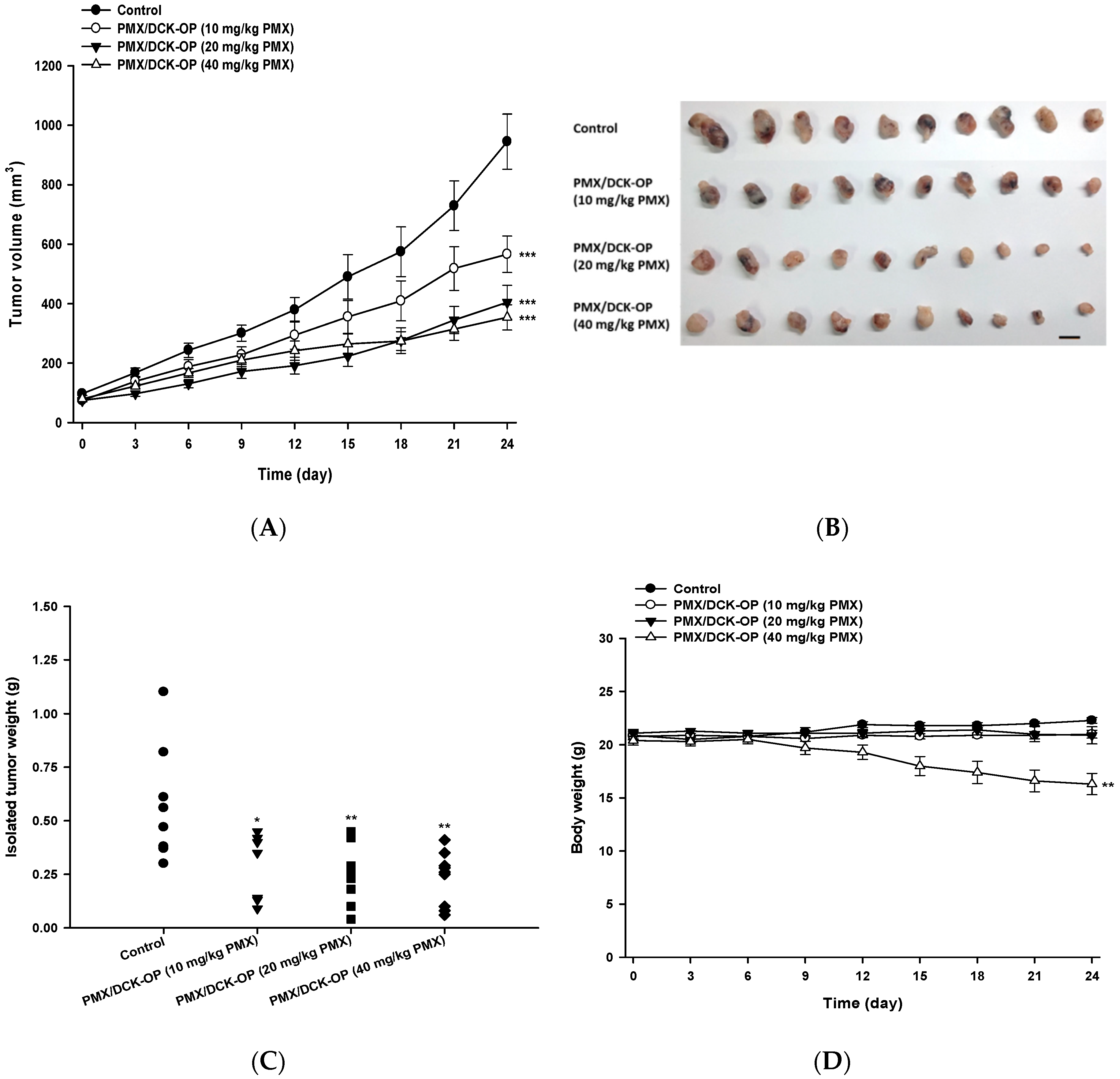
2.8. In Vivo Antitumor Efficacy of PMX/DCK-OP in Mice
2.9. In Vivo Anti-Angiogenic Effect of Oral Metronomic Chemotherapy of PMX-DCK-OP
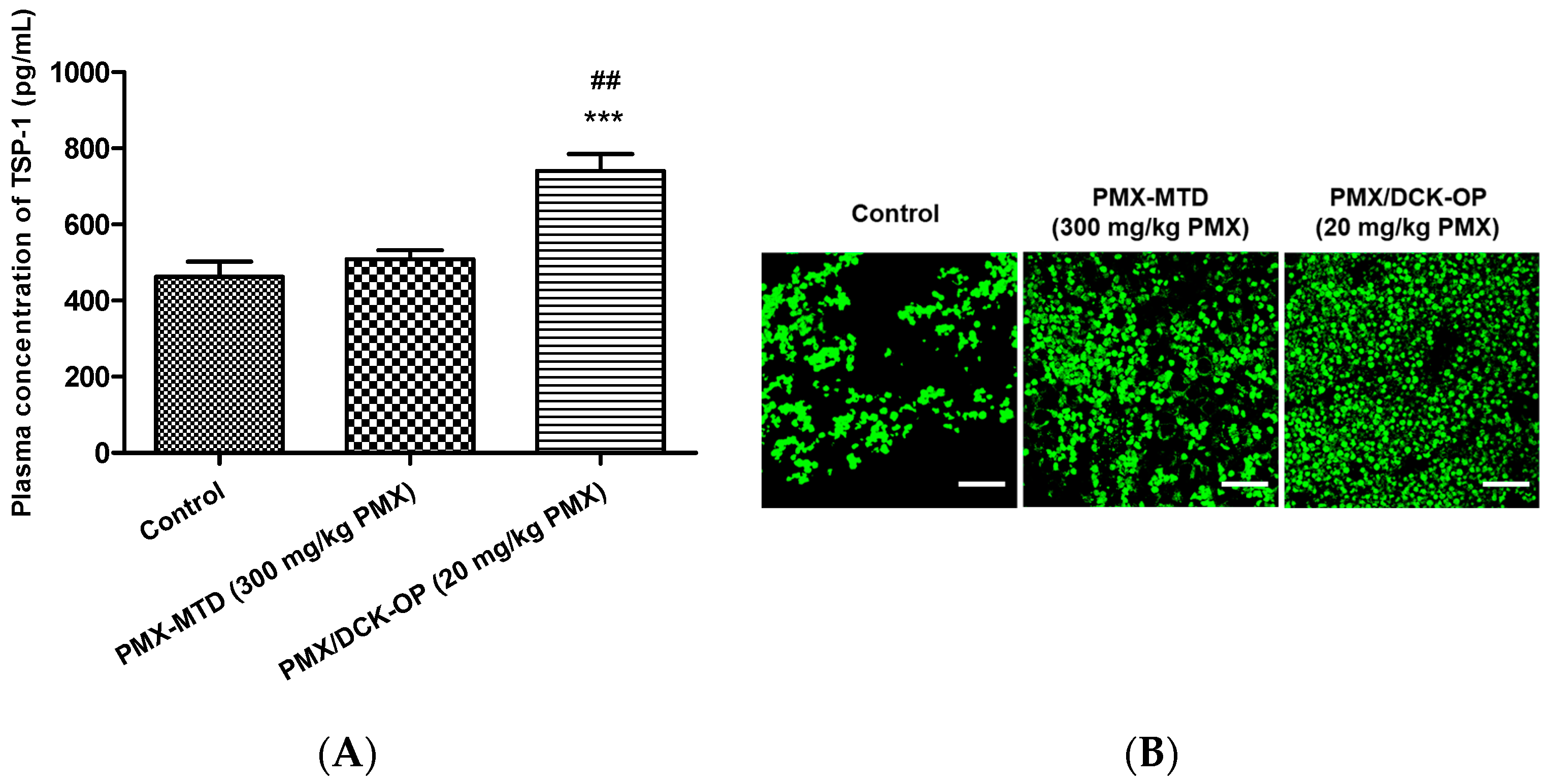
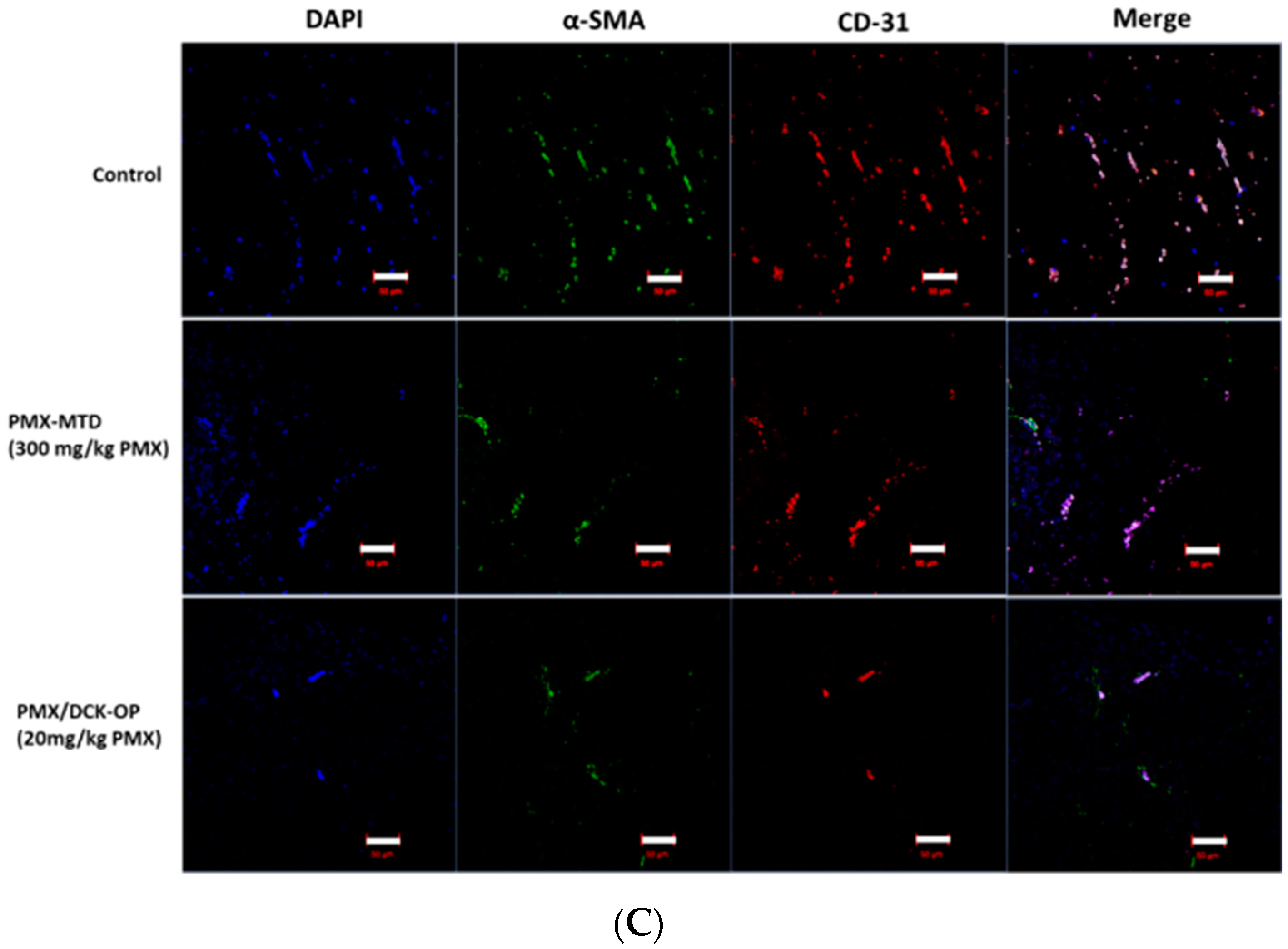
2.9.1. In Vivo Anti-Angiogenic Effect
2.9.2. In Vivo Antitumor Efficacy
2.9.3. Circulating TSP-1 by Immunoassay
2.9.4. Immunofluorescence Study
2.10. Pharmacokinetic and Statistical Analyses
3. Results and Discussion
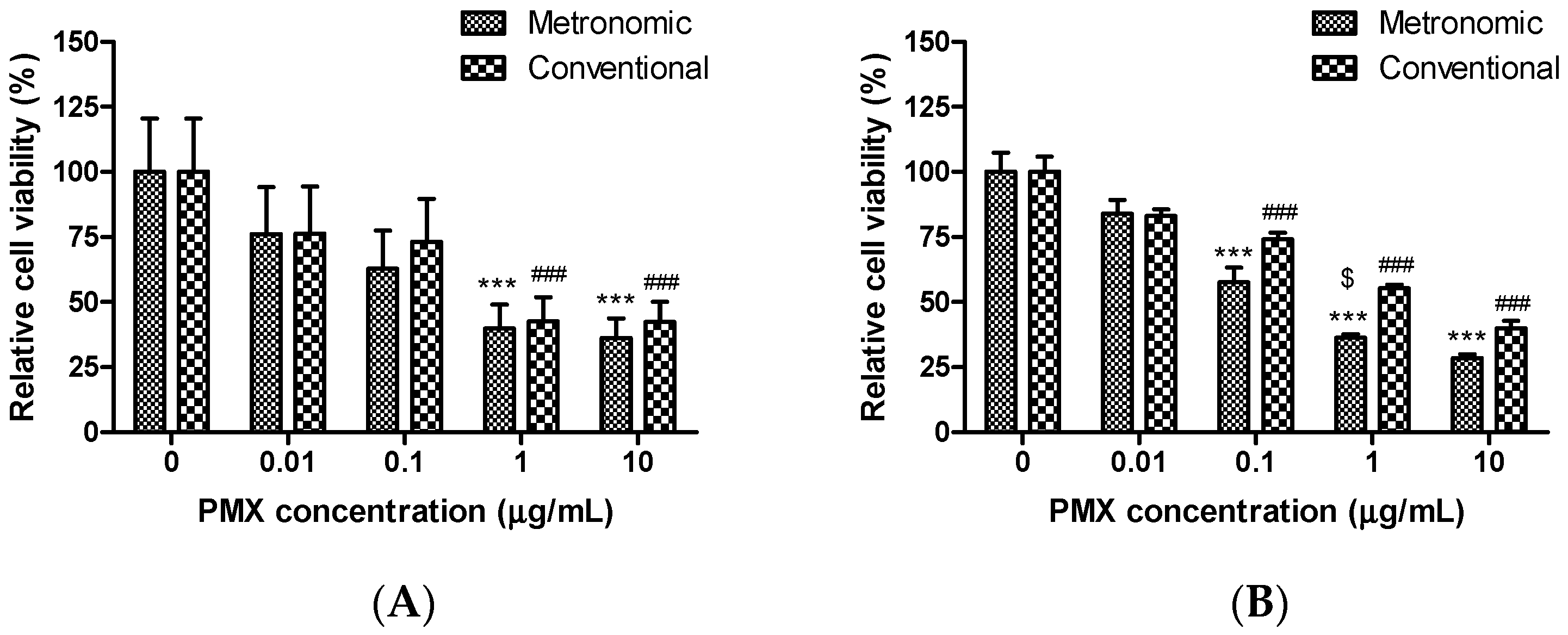
3.1. Cytotoxic Effects of Conventional and Metronomic Doses of PMX
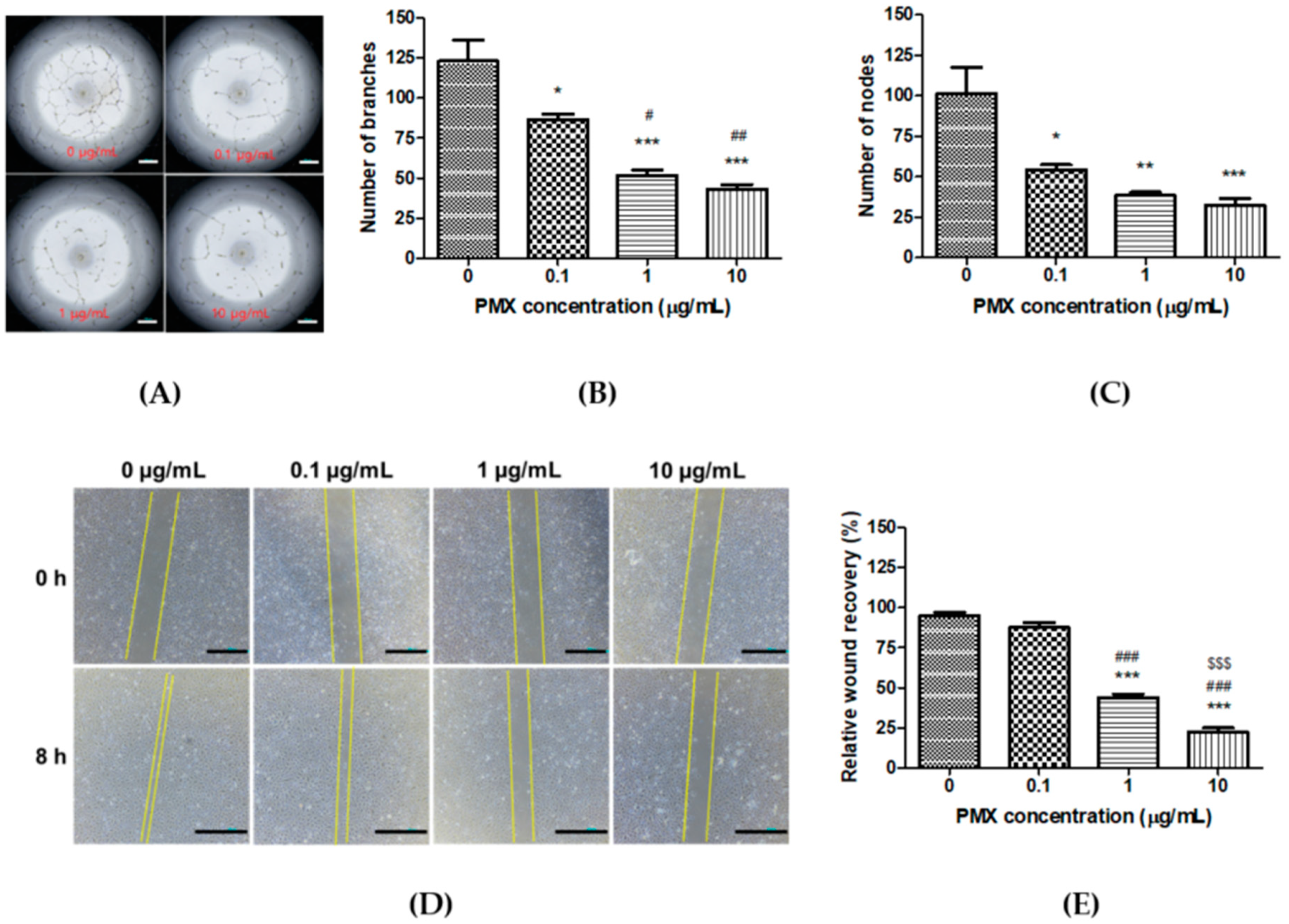
3.2. Anti-Angiogenic Effect of PMX
3.3. Characterization of PMX/DCK-OP
3.4. In Vitro Artificial Intestinal Membrane and Caco-2 Cell Permeability
3.5. Pharmacokinetic Property of PMX/DCK-OP in Mice
3.6. In Vivo Anti-Angiogenic Effect of PMX/DCK-OP
3.7. In Vivo Tumor Growth Inhibition Effect of PMX/DCK-OP
3.8. In Vivo Anti-Angiogenic Effect of Oral Metronomic Chemotherapy of PMX/DCK-OP
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Hanahan, D.; Bergers, G.; Bergsland, E. Less is more, regularly: Metronomic dosing of cytotoxic drugs can target tumor angiogenesis in mice. J. Clin. Investig. 2000, 105, 1045–1047. [Google Scholar] [CrossRef] [PubMed]
- Andre, N.; Carre, M.; Pasquier, E. Metronomics: Towards personalized chemotherapy? Nat. Rev. Clin. Oncol. 2014, 11, 413–431. [Google Scholar] [CrossRef] [PubMed]
- Gasparini, G. Metronomic scheduling: The future of chemotherapy? Lancet Oncol. 2001, 2, 733–740. [Google Scholar] [CrossRef]
- Kerbel, R.S.; Kamen, B.A. The anti-angiogenic basis of metronomic chemotherapy. Nat. Rev. Cancer 2004, 4, 423–436. [Google Scholar] [CrossRef] [PubMed]
- Pasquier, E.; Kavallaris, M.; Andre, N. Metronomic chemotherapy: New rationale for new directions. Nat. Rev. Clin. Oncol. 2010, 7, 455–465. [Google Scholar] [CrossRef]
- Gnoni, A.; Silvestris, N.; Licchetta, A.; Santini, D.; Scartozzi, M.; Ria, R.; Pisconti, S.; Petrelli, F.; Vacca, A.; Lorusso, V. Metronomic chemotherapy from rationale to clinical studies: A dream or reality? Crit. Rev. Oncol./Hematol. 2015, 95, 46–61. [Google Scholar] [CrossRef]
- Chen, Y.L.; Chang, M.C.; Cheng, W.F. Metronomic chemotherapy and immunotherapy in cancer treatment. Cancer Lett. 2017, 400, 282–292. [Google Scholar] [CrossRef]
- Hashimoto, K.; Man, S.; Xu, P.; Cruz-Munoz, W.; Tang, T.; Kumar, R.; Kerbel, R.S. Potent preclinical impact of metronomic low-dose oral topotecan combined with the antiangiogenic drug pazopanib for the treatment of ovarian cancer. Mol. Cancer Ther. 2010, 9, 996–1006. [Google Scholar] [CrossRef]
- Bocci, G.; Francia, G.; Man, S.; Lawler, J.; Kerbel, R.S. Thrombospondin 1, a mediator of the antiangiogenic effects of low-dose metronomic chemotherapy. Proc. Natl. Acad. Sci. USA 2003, 100, 12917–12922. [Google Scholar] [CrossRef] [Green Version]
- Hamano, Y.; Sugimoto, H.; Soubasakos, M.A.; Kieran, M.; Olsen, B.R.; Lawler, J.; Sudhakar, A.; Kalluri, R. Thrombospondin-1 associated with tumor microenvironment contributes to low-dose cyclophosphamide-mediated endothelial cell apoptosis and tumor growth suppression. Cancer Res. 2004, 64, 1570–1574. [Google Scholar] [CrossRef]
- Shi, H.; Jiang, J.; Ji, J.; Shi, M.; Cai, Q.; Chen, X.; Yu, Y.; Liu, B.; Zhu, Z.; Zhang, J. Anti-angiogenesis participates in antitumor effects of metronomic capecitabine on colon cancer. Cancer Lett. 2014, 349, 128–135. [Google Scholar] [CrossRef] [PubMed]
- Morgan, C.; Jeremiah, S.; Wagstaff, J. Metronomic administration of ibandronate and its anti-angiogenic effects In Vitro. Microvasc. Res. 2009, 78, 453–458. [Google Scholar] [CrossRef] [PubMed]
- Gyawali, B.; Prasad, V. Pemetrexed in nonsquamous non-small-cell lung cancer: The billion dollar subgroup analysis. JAMA Oncol. 2018, 4, 17–18. [Google Scholar] [CrossRef] [PubMed]
- Browder, T.; Butterfield, C.E.; Kraling, B.M.; Shi, B.; Marshall, B.; O’Reilly, M.S.; Folkman, J. Antiangiogenic scheduling of chemotherapy improves efficacy against experimental drug-resistant cancer. Cancer Res. 2000, 60, 1878–1886. [Google Scholar] [PubMed]
- Colleoni, M.; Rocca, A.; Sandri, M.T.; Zorzino, L.; Masci, G.; Nole, F.; Peruzzotti, G.; Robertson, C.; Orlando, L.; Cinieri, S.; et al. Low-dose oral methotrexate and cyclophosphamide in metastatic breast cancer: Antitumor activity and correlation with vascular endothelial growth factor levels. Ann. Oncol. 2002, 13, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Yuan, F.; Shi, H.; Ji, J.; Cai, Q.; Chen, X.; Yu, Y.; Liu, B.; Zhu, Z.; Zhang, J. Capecitabine metronomic chemotherapy inhibits the proliferation of gastric cancer cells through anti-angiogenesis. Oncol. Rep. 2015, 33, 1753–1762. [Google Scholar] [CrossRef] [PubMed]
- Bottini, A.; Generali, D.; Brizzi, M.P.; Fox, S.B.; Bersiga, A.; Bonardi, S.; Allevi, G.; Aguggini, S.; Bodini, G.; Milani, M.; et al. Randomized phase ii trial of letrozole and letrozole plus low-dose metronomic oral cyclophosphamide as primary systemic treatment in elderly breast cancer patients. J. Clin. Oncol. 2006, 24, 3623–3628. [Google Scholar] [CrossRef] [PubMed]
- Laquente, B.; Lacasa, C.; Ginesta, M.M.; Casanovas, O.; Figueras, A.; Galan, M.; Ribas, I.G.; Germa, J.R.; Capella, G.; Vinals, F. Antiangiogenic effect of gemcitabine following metronomic administration in a pancreas cancer model. Mol. Cancer Ther. 2008, 7, 638–647. [Google Scholar] [CrossRef] [Green Version]
- Sigmond, J.; Backus, H.H.; Wouters, D.; Temmink, O.H.; Jansen, G.; Peters, G.J. Induction of resistance to the multitargeted antifolate Pemetrexed (ALIMTA) in WiDr human colon cancer cells is associated with thymidylate synthase overexpression. Biochem. Pharmacol. 2003, 66, 431–438. [Google Scholar] [CrossRef]
- Paz-Ares, L.; Bezares, S.; Tabernero, J.M.; Castellanos, D.; Cortes-Funes, H. Review of a promising new agent—Pemetrexed disodium. Cancer 2003, 97, 2056–2063. [Google Scholar] [CrossRef]
- Walling, J. From methotrexate to pemetrexed and beyond. A review of the pharmacodynamic and clinical properties of antifolates. Investig. New Drugs 2006, 24, 37–77. [Google Scholar] [CrossRef] [PubMed]
- Langer, C.J.; Gadgeel, S.M.; Borghaei, H.; Papadimitrakopoulou, V.A.; Patnaik, A.; Powell, S.F.; Gentzler, R.D.; Martins, R.G.; Stevenson, J.P.; Jalal, S.I.; et al. Carboplatin and pemetrexed with or without pembrolizumab for advanced, non-squamous non-small-cell lung cancer: A randomised, phase 2 cohort of the open-label keynote-021 study. Lancet Oncol. 2016, 17, 1497–1508. [Google Scholar] [CrossRef]
- Siew, A.; Le, H.; Thiovolet, M.; Gellert, P.; Schatzlein, A.; Uchegbu, I. Enhanced oral absorption of hydrophobic and hydrophilic drugs using quaternary ammonium palmitoyl glycol chitosan nanoparticles. Mol. Pharm. 2012, 9, 14–28. [Google Scholar] [CrossRef] [PubMed]
- Ling, S.S.; Yuen, K.H.; Magosso, E.; Barker, S.A. Oral bioavailability enhancement of a hydrophilic drug delivered via folic acid-coupled liposomes in rats. J. Pharm. Pharmacol. 2009, 61, 445–449. [Google Scholar] [CrossRef] [PubMed]
- Pavlovic, N.; Golocorbin-Kon, S.; Ethanic, M.; Stanimirov, B.; Al-Salami, H.; Stankov, K.; Mikov, M. Bile acids and their derivatives as potential modifiers of drug release and pharmacokinetic profiles. Front. Pharmacol. 2018, 9. [Google Scholar] [CrossRef]
- Hanauske, A.R.; Chen, V.; Paoletti, P.; Niyikiza, C. Pemetrexed disodium: A novel antifolate clinically active against multiple solid tumors. Oncologist 2001, 6, 363–373. [Google Scholar] [CrossRef] [PubMed]
- Soni, K.; Mujtaba, A.; Kohli, K. Lipid drug conjugate nanoparticle as a potential nanocarrier for the oral delivery of pemetrexed diacid: Formulation design, characterization, Ex Vivo, and In Vivo assessment. Int. J. Biol. Macromol. 2017, 103, 139–151. [Google Scholar] [CrossRef]
- Ghasemiyeh, P.; Mohammadi-Samani, S. Solid lipid nanoparticles and nanostructured lipid carriers as novel drug delivery systems: Applications, advantages and disadvantages. Res. Pharm. Sci. 2018, 13, 288–303. [Google Scholar]
- Pangeni, R.; Choi, J.U.; Panthi, V.K.; Byun, Y.; Park, J.W. Enhanced oral absorption of pemetrexed by ion-pairing complex formation with deoxycholic acid derivative and multiple nanoemulsion formulations: Preparation, characterization, and In Vivo oral bioavailability and anticancer effect. Int. J. Nanomed. 2018, 13, 3329–3351. [Google Scholar] [CrossRef]
- Aljuffali, I.A.; Mock, J.N.; Costyn, L.J.; Nguyen, H.; Nagy, T.; Cummings, B.S.; Arnold, R.D. Enhanced antitumor activity of low-dose continuous administration schedules of topotecan in prostate cancer. Cancer Biol. Ther. 2011, 12, 407–420. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.Y.; Al-Hilal, T.A.; Chung, S.W.; Kim, S.Y.; Ryu, G.H.; Son, W.C.; Byun, Y. Antiangiogenic and anticancer effect of an orally active low molecular weight heparin conjugate and its application to lung cancer chemoprevention. J. Control. Release 2015, 199, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.W.; Bae, S.M.; Lee, M.; Al-Hilal, T.A.; Lee, C.K.; Kim, J.K.; Kim, I.S.; Kim, S.Y.; Byun, Y. Lht7, a chemically modified heparin, inhibits multiple stages of angiogenesis by blocking vegf, fgf2 and pdgf-b signaling pathways. Biomaterials 2015, 37, 27–28. [Google Scholar] [CrossRef] [PubMed]
- Mavroeidis, L.; Sheldon, H.; Briasoulis, E.; Marselos, M.; Pappas, P.; Harris, A.L. Metronomic vinorelbine: Anti-angiogenic activity In Vitro in normoxic and severe hypoxic conditions, and severe hypoxia-induced resistance to its anti-proliferative effect with reversal by akt inhibition. Int. J. Oncol. 2015, 47, 455–464. [Google Scholar] [CrossRef] [PubMed]
- Pangeni, R.; Kang, S.-W.; Oak, M.; Park, E.Y.; Park, J.W. Oral delivery of quercetin in oil-in-water nanoemulsion: In Vitro characterization and In Vivo anti-obesity efficacy in mice. J. Funct. Foods 2017, 38, 571–581. [Google Scholar] [CrossRef]
- Bocci, G.; Falcone, A.; Fioravanti, A.; Orlandi, P.; Di Paolo, A.; Fanelli, G.; Viacava, P.; Naccarato, A.G.; Kerbel, R.S.; Danesi, R.; et al. Antiangiogenic and anticolorectal cancer effects of metronomic irinotecan chemotherapy alone and in combination with semaxinib. Br. J. Cancer 2008, 98, 1619–1629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamat, A.A.; Kim, T.J.; Landen, C.N., Jr.; Lu, C.; Han, L.Y.; Lin, Y.G.; Merritt, W.M.; Thaker, P.H.; Gershenson, D.M.; Bischoff, F.Z.; et al. Metronomic chemotherapy enhances the efficacy of antivascular therapy in ovarian cancer. Cancer Res. 2007, 67, 281–288. [Google Scholar] [CrossRef]
- Sharif Makhmal Zadeh, B.; Esfahani, G.; Salimi, A. Permeability of ciprofloxacin-loaded polymeric micelles including ginsenoside as p-glycoprotein inhibitor through a caco-2 cells monolayer as an intestinal absorption model. Molecules 2018, 23, 1904. [Google Scholar] [CrossRef]
- Fischer, S.M.; Parmentier, J.; Buckley, S.T.; Reimold, I.; Brandl, M.; Fricker, G. Oral bioavailability of ketoprofen in suspension and solution formulations in rats: The influence of poloxamer 188. J. Pharm. Pharmacol. 2012, 64, 1631–1637. [Google Scholar] [CrossRef]
- Newa, M.; Bhandari, K.H.; Li, D.X.; Kwon, T.H.; Kim, J.A.; Yoo, B.K.; Woo, J.S.; Lyoo, W.S.; Yong, C.S.; Choi, H.G. Preparation, characterization and in vivo evaluation of ibuprofen binary solid dispersions with poloxamer 188. Int. J. Pharm. 2007, 343, 228–237. [Google Scholar] [CrossRef]
- Loureiro, A.; Nogueira, E.; Azoia, N.G.; Sarria, M.P.; Abreu, A.S.; Shimanovich, U.; Rollett, A.; Harmark, J.; Hebert, H.; Guebitz, G.; et al. Size controlled protein nanoemulsions for active targeting of folate receptor positive cells. Colloids Surf. B Biointerfaces 2015, 135, 90–98. [Google Scholar] [CrossRef] [Green Version]
- Prasad, Y.V.; Puthli, S.P.; Eaimtrakarn, S.; Ishida, M.; Yoshikawa, Y.; Shibata, N.; Takada, K. Enhanced intestinal absorption of vancomycin with labrasol and d-alpha-tocopheryl peg 1000 succinate in rats. Int. J. Pharm. 2003, 250, 181–190. [Google Scholar] [CrossRef]
- Kommuru, T.R.; Gurley, B.; Khan, M.A.; Reddy, I.K. Self-emulsifying drug delivery systems (sedds) of coenzyme q10: Formulation development and bioavailability assessment. Int. J. Pharm. 2001, 212, 233–246. [Google Scholar] [CrossRef]
- Gupta, S.; Kesarla, R.; Omri, A. Formulation strategies to improve the bioavailability of poorly absorbed drugs with special emphasis on self-emulsifying systems. ISRN Pharm. 2013, 2013. [Google Scholar] [CrossRef] [PubMed]
- Raimondi, F.; Santoro, P.; Barone, M.V.; Pappacoda, S.; Barretta, M.L.; Nanayakkara, M.; Apicella, C.; Capasso, L.; Paludetto, R. Bile acids modulate tight junction structure and barrier function of caco-2 monolayers via egfr activation. Am. J. Physiol.-Gastrointest. Liver Physiol. 2008, 294, G906–G913. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.; Mundada, V.; Sawant, K. Enhanced intestinal absorption of asenapine maleate by fabricating solid lipid nanoparticles using tpgs: Elucidation of transport mechanism, permeability across caco-2 cell line and in vivo pharmacokinetic studies. Artif. Cells Nanomed. Biotechnol. 2019, 47, 144–153. [Google Scholar] [CrossRef]
- Sahay, G.; Batrakova, E.V.; Kabanov, A.V. Different internalization pathways of polymeric micelles and unimers and their effects on vesicular transport. Bioconjug. Chem. 2008, 19, 2023–2029. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Lu, Y.; Jv, M.; Hu, J.; Li, Q.; Tu, J. Enhancing effect of labrasol on the intestinal absorption of ganciclovir in rats. Drug Dev. Ind. Pharm. 2011, 37, 1415–1421. [Google Scholar] [CrossRef]
- Sha, X.; Yan, G.; Wu, Y.; Li, J.; Fang, X. Effect of self-microemulsifying drug delivery systems containing labrasol on tight junctions in caco-2 cells. Eur. J. Pharm. Sci. 2005, 24, 477–486. [Google Scholar] [CrossRef]
- Zhou, J.; Zhou, M.; Yang, F.F.; Liu, C.Y.; Pan, R.L.; Chang, Q.; Liu, X.M.; Liao, Y.H. Involvement of the inhibition of intestinal glucuronidation in enhancing the oral bioavailability of resveratrol by labrasol containing nanoemulsions. Mol. Pharm. 2015, 12, 1084–1095. [Google Scholar] [CrossRef]
- Lin, Y.; Shen, Q.; Katsumi, H.; Okada, N.; Fujita, T.; Jiang, X.; Yamamoto, A. Effects of labrasol and other pharmaceutical excipients on the intestinal transport and absorption of rhodamine123, a p-glycoprotein substrate, in rats. Biol. Pharm. Bull. 2007, 30, 1301–1307. [Google Scholar] [CrossRef]
- Hatakeyama, Y.; Kobayashi, K.; Nagano, T.; Tamura, D.; Yamamoto, M.; Tachihara, M.; Kotani, Y.; Nishimura, Y. Synergistic effects of pemetrexed and amrubicin in non-small cell lung cancer cell lines: Potential for combination therapy. Cancer Lett. 2014, 343, 74–79. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Test Materials | Effective Permeability (Pe, × 10−6, cm/s) | Apparent Permeability (Papp, × 10−6, cm/s) |
|---|---|---|
| PMX | 1.52 ± 0.25 | 1.78 ± 0.12 |
| PMX/DCK | 8.88 ± 0.54 ***,### | 5.97 ± 1.38 ***,### |
| PMX/DCK-OP | 31.6 ± 2.22 ***,### | 18.9 ± 3.60 ***,### |
| Test Materials | PMX (20 mg/kg, IV) | PMX (50 mg/kg, Oral) | PMX/DCK-OP (10 mg/kg PMX, Oral) | PMX/DCK-OP (20 mg/kg PMX, Oral) | PMX/DCK-OP (40 mg/kg PMX, Oral) |
|---|---|---|---|---|---|
| Administration | Intravenous | Oral | Oral | Oral | Oral |
| Dose of PMX (mg/kg) | 20 | 50 | 10 | 20 | 40 |
| Tmax (h) | 0.17 ± 0.00 | 0.44 ± 0.05 | 0.33 ± 0.08 | 0.41 ± 0.08 | 0.25 ± 0.00 |
| T1/2 (h) | 2.57 ± 0.57 | 2.62 ± 0.67 | 3.08 ± 2.18 | 2.48 ± 0.27 | 1.57 ± 0.42 |
| Cmax (ng/mL) | 58,000 ± 2669 | 151 ± 5.77 | 4653 ± 2728 | 4558 ± 913 | 11,032 ± 1693 |
| AUClast (ng·h/mL) | 37,294 ± 928 | 316 ± 31.6 | 3622 ± 1001 | 8874 ± 1871 | 16,973 ± 3407 |
| AUCinf (ng·h/mL) | 38,521 ± 1.129 | 443 ± 62.2 | 4755 ± 674 | 9604 ± 1868 | 17,944 ± 3346 |
| Bioavailability (%) | 100 | 0.34 ± 0.01 | 19.4 ± 5.37 | 23.8 ± 5.02 | 22.8 ± 4.57 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maharjan, R.; Pangeni, R.; Jha, S.K.; Choi, J.U.; Chang, K.-Y.; Choi, Y.K.; Park, J.W.; Byun, Y. Anti-Angiogenic Effect of Orally Available Pemetrexed for Metronomic Chemotherapy. Pharmaceutics 2019, 11, 332. https://doi.org/10.3390/pharmaceutics11070332
Maharjan R, Pangeni R, Jha SK, Choi JU, Chang K-Y, Choi YK, Park JW, Byun Y. Anti-Angiogenic Effect of Orally Available Pemetrexed for Metronomic Chemotherapy. Pharmaceutics. 2019; 11(7):332. https://doi.org/10.3390/pharmaceutics11070332
Chicago/Turabian StyleMaharjan, Ruby, Rudra Pangeni, Saurav Kumar Jha, Jeong Uk Choi, Kwan-Young Chang, Young Kweon Choi, Jin Woo Park, and Youngro Byun. 2019. "Anti-Angiogenic Effect of Orally Available Pemetrexed for Metronomic Chemotherapy" Pharmaceutics 11, no. 7: 332. https://doi.org/10.3390/pharmaceutics11070332