Boosting Drug Discovery for Parkinson’s: Enhancement of the Delivery of a Monoamine Oxidase-B Inhibitor by Brain-Targeted PEGylated Polycaprolactone-Based Nanoparticles

 , , , , , , and
, , , , , , and
Abstract
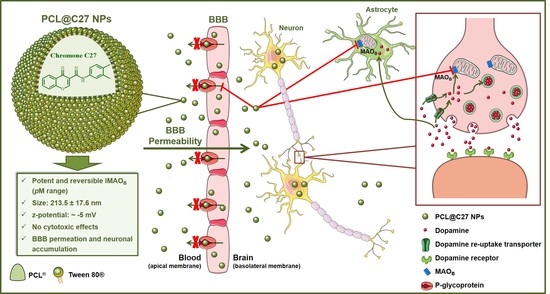
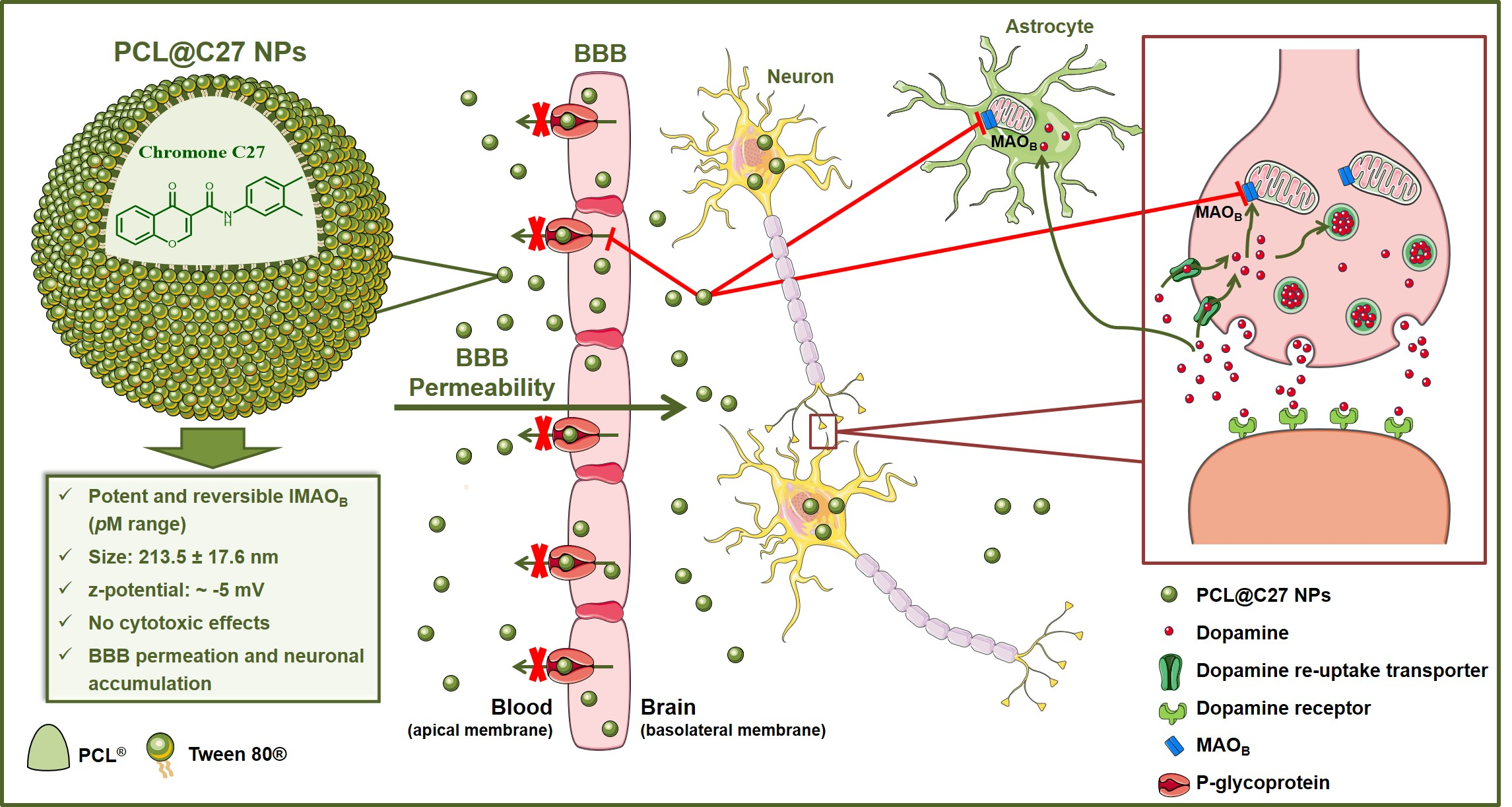
:
1. Introduction
2. Materials and Methods
2.1. Materials and Reagents
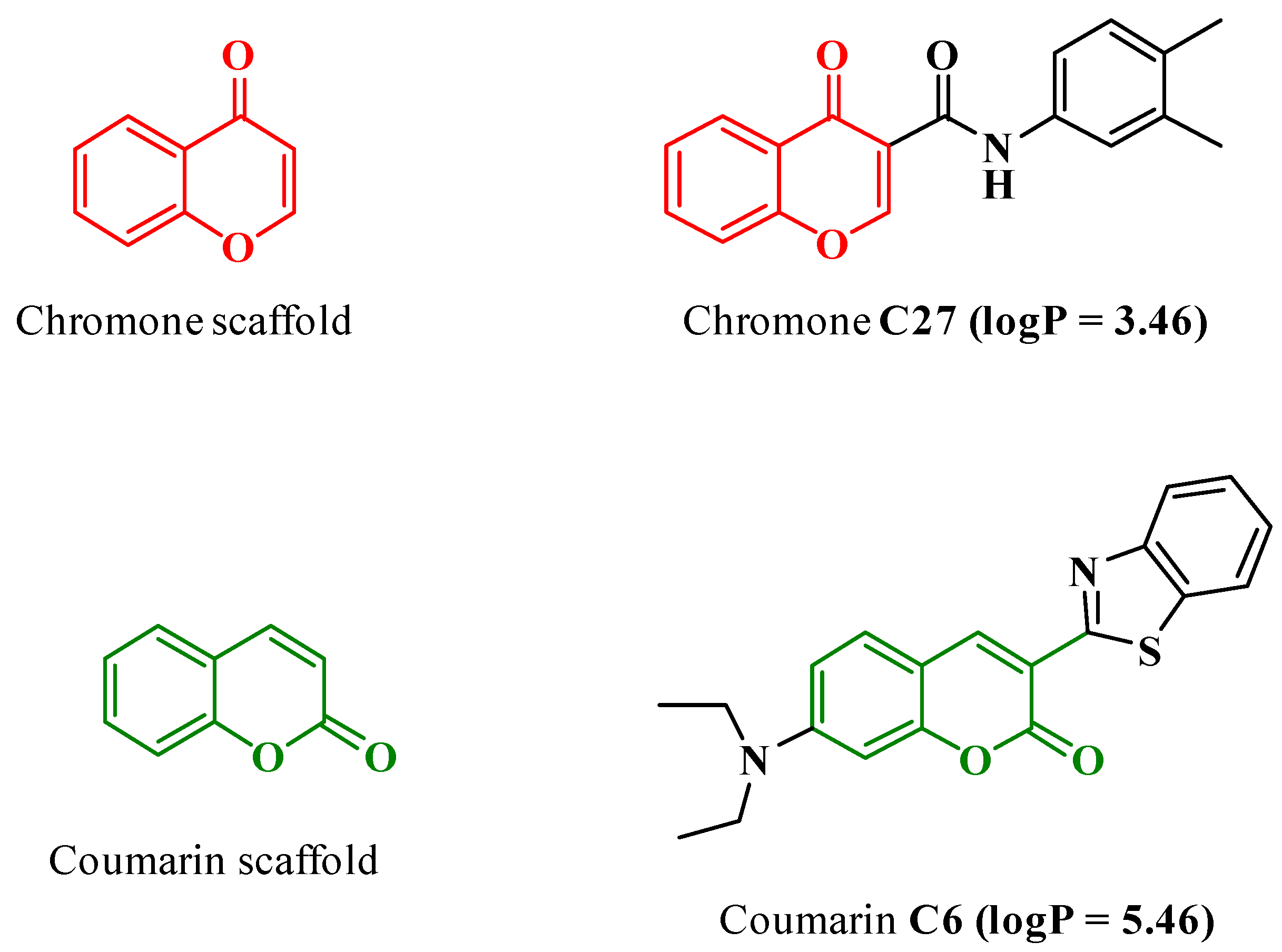
2.2. Synthesis of N-(3′,4′-Dimethylphenyl)-4-oxo-4H-chromene-3-carboxamide (C27)
2.3. Preparation of PEGylated PCL-Based NPs
2.4. Encapsulation and Drug Loading Efficiency
2.5. Spectroscopic Analysis
2.6. Particle Size, Zeta Potential, and Morphology Analysis
2.7. Differential Scanning Calorimetry and Powder X-ray Diffraction
2.8. In Vitro Release Studies
2.9. In Vitro Cellular Studies
2.9.1. Cell Lines and Culture Conditions
2.9.2. Cell Viability Assays
2.9.3. Cellular Uptake and Intracellular Localization Studies
2.9.4. Evaluation of MAO-B Activity by Fluorescence Kynuramine Assay
2.9.5. Protein Quantification
2.9.6. Cellular Permeability Studies
2.9.7. Rhodamine 123 Accumulation Assay
2.10. Statistical Analysis
3. Results
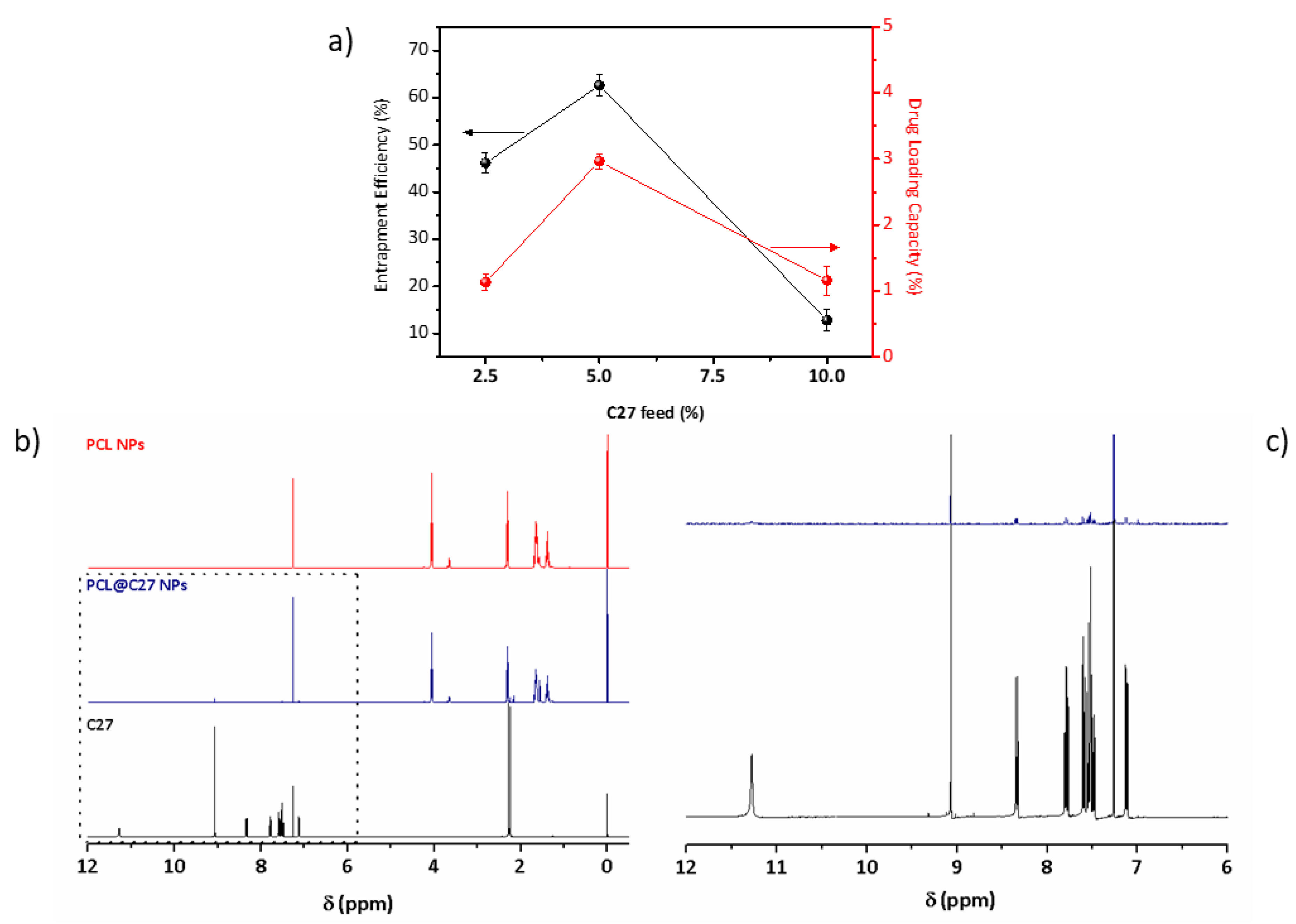
3.1. Preparation and Characterization of PEGylated PCL-Based Nanoformulations
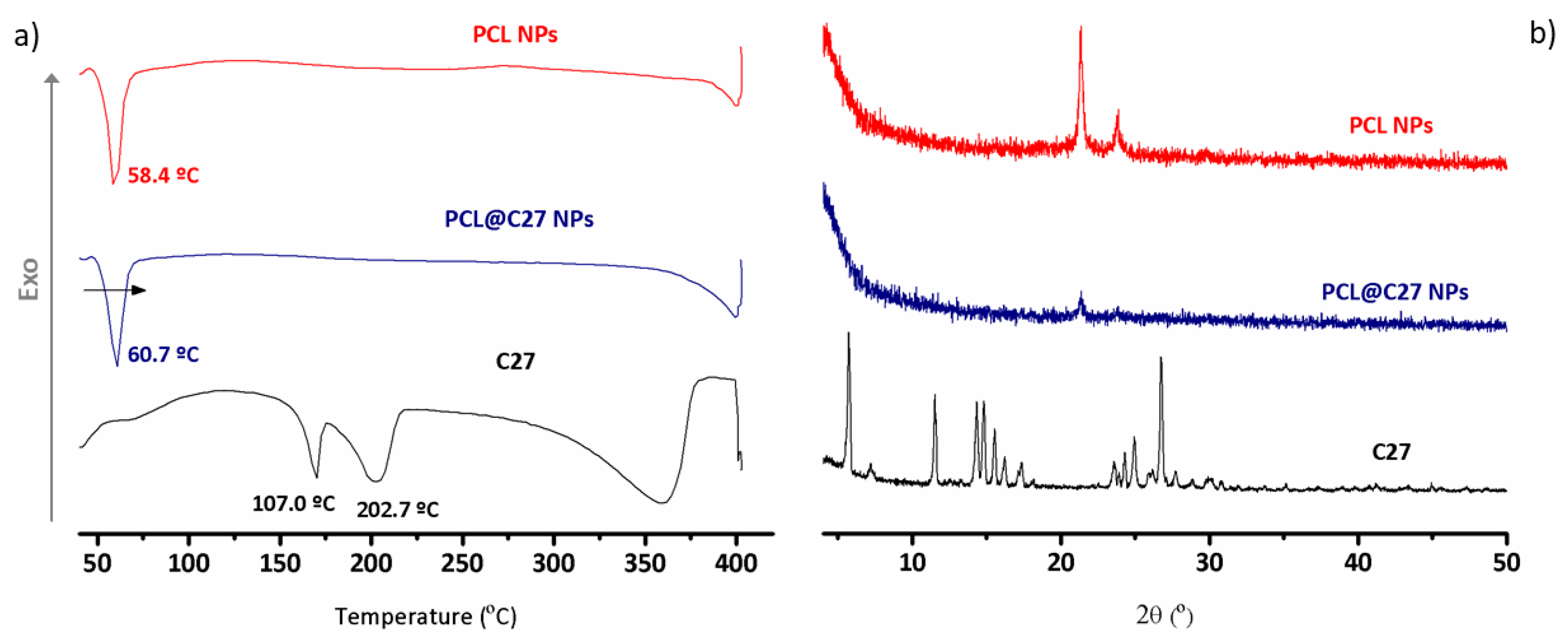
3.2. Differential Scanning Calorimetry and Powder X-ray Diffraction Analysis
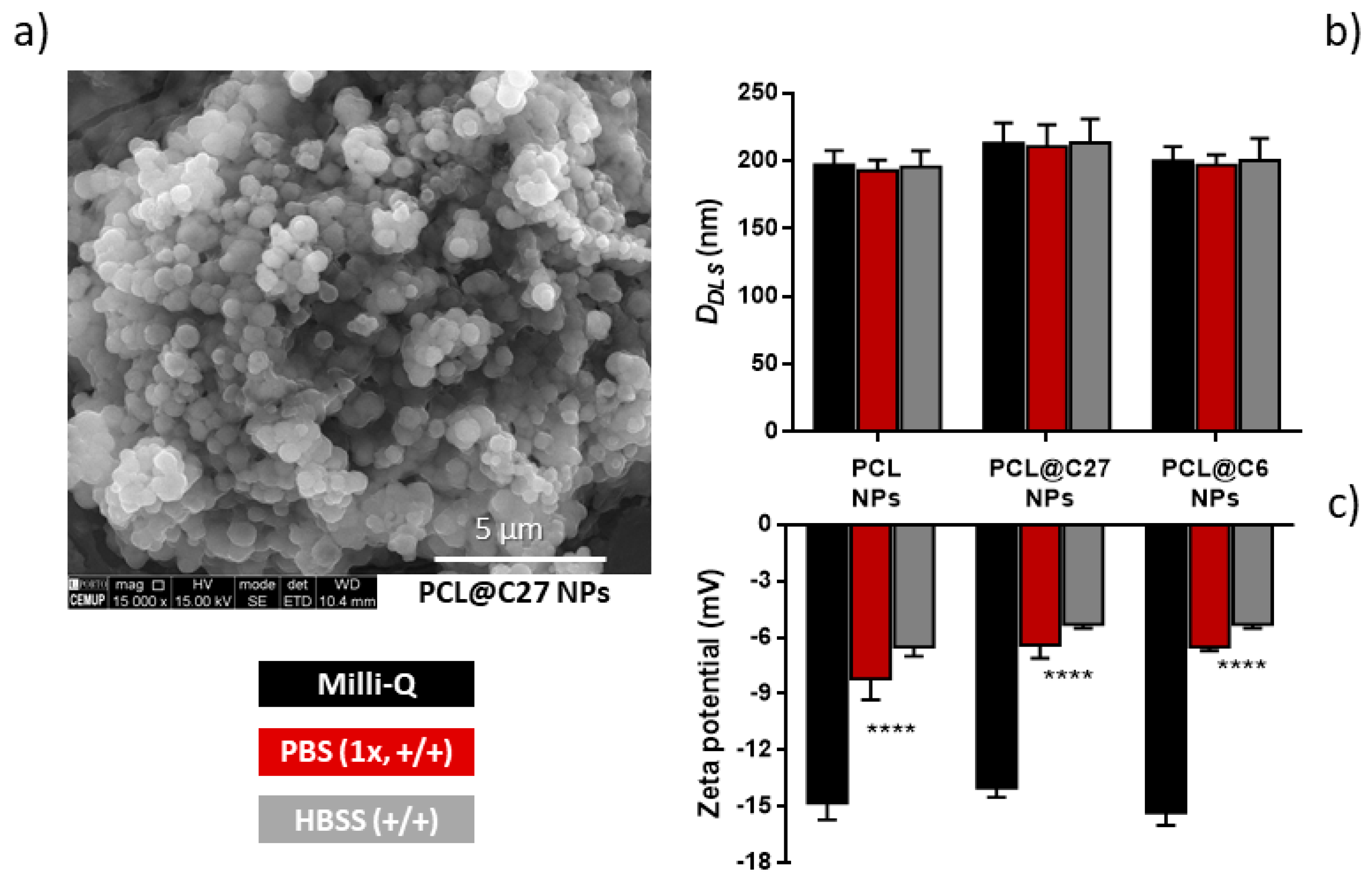
3.3. PEGylated PCL@C27 Particle Size, z-Potential, and Morphology
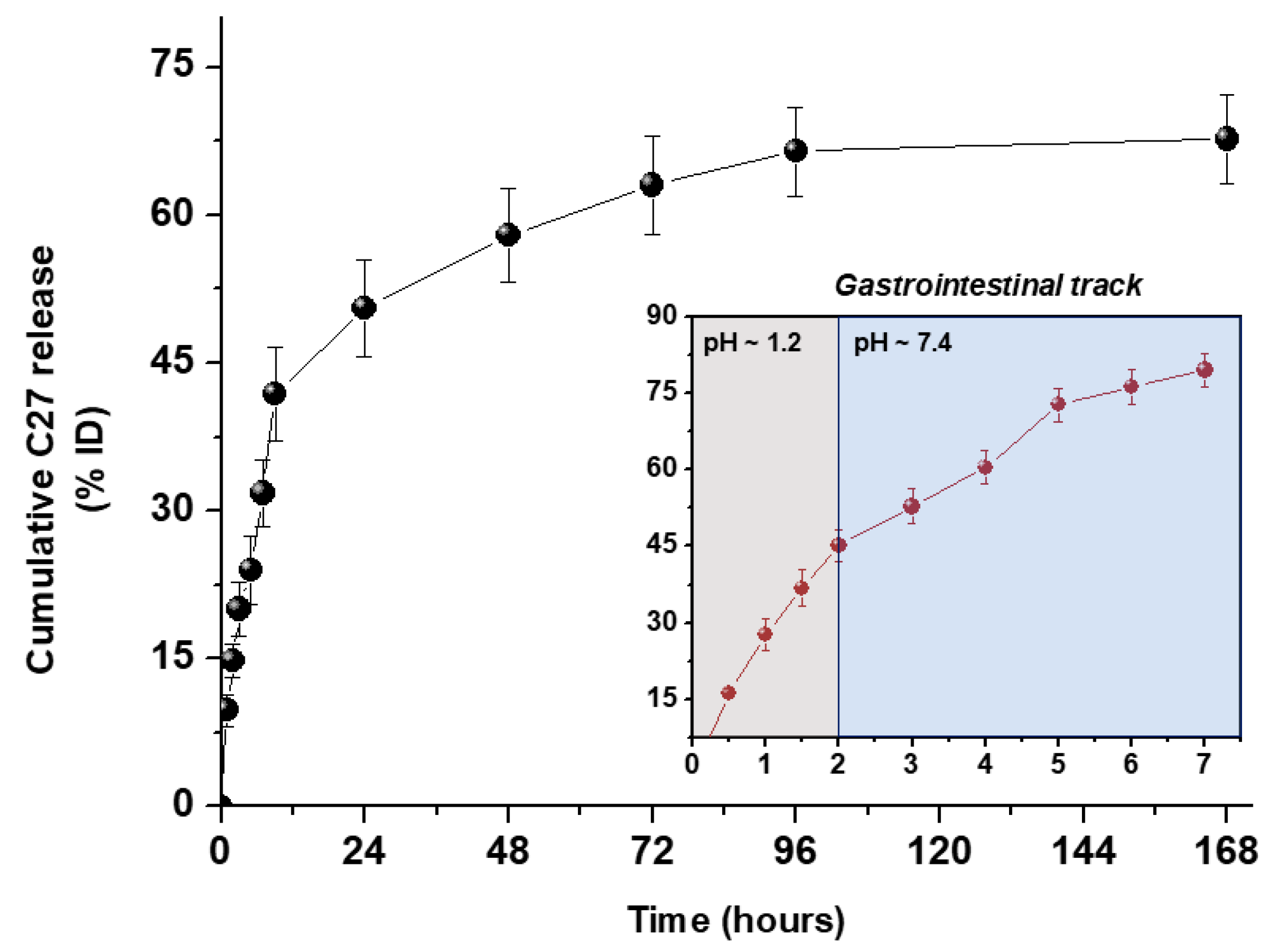
3.4. In Vitro C27 Release Kinetics
3.5. In Vitro Cellular Studies
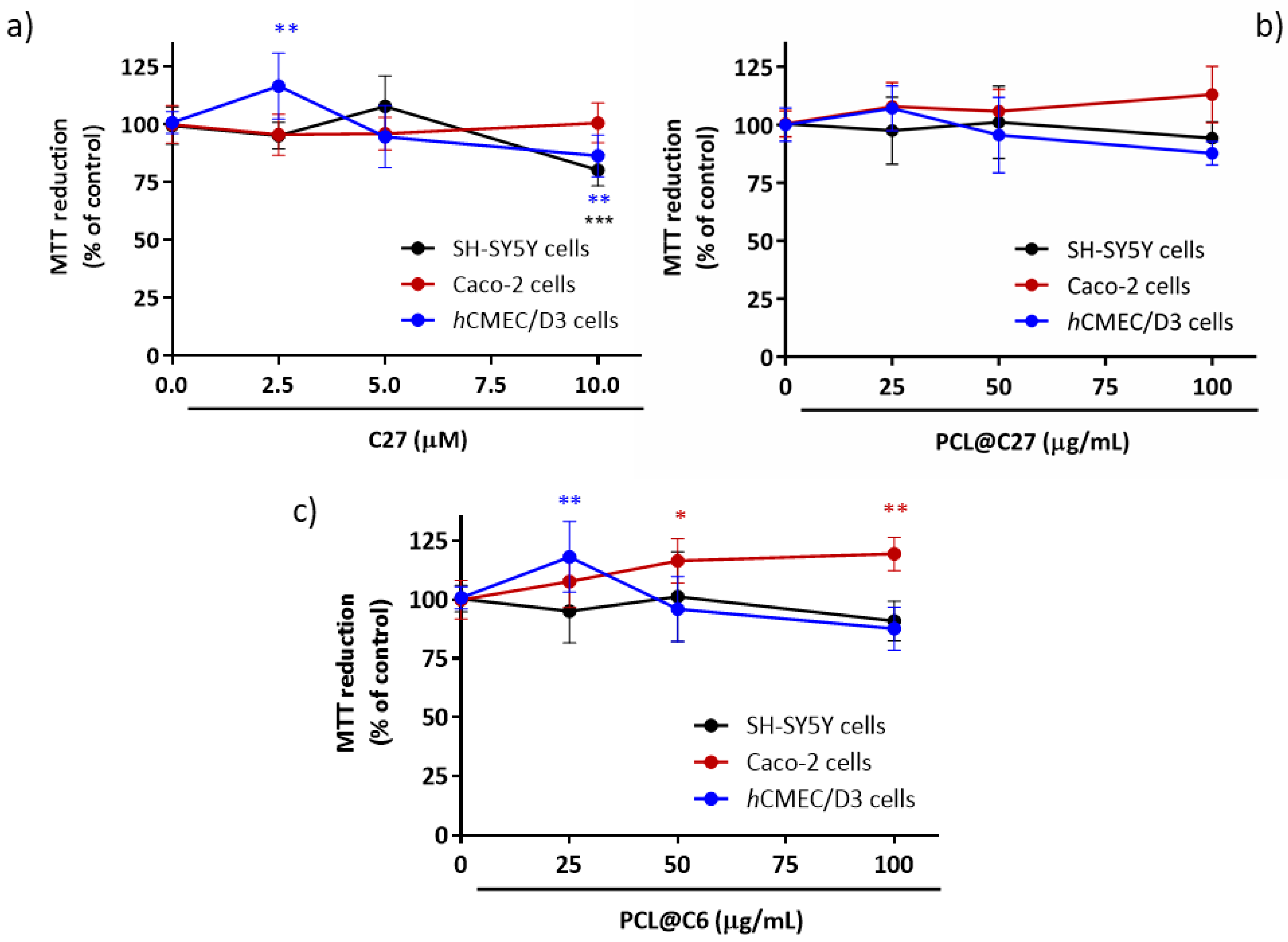
3.5.1. In Vitro Cytotoxicity in Neuronal, Intestinal, and Endothelial Cells
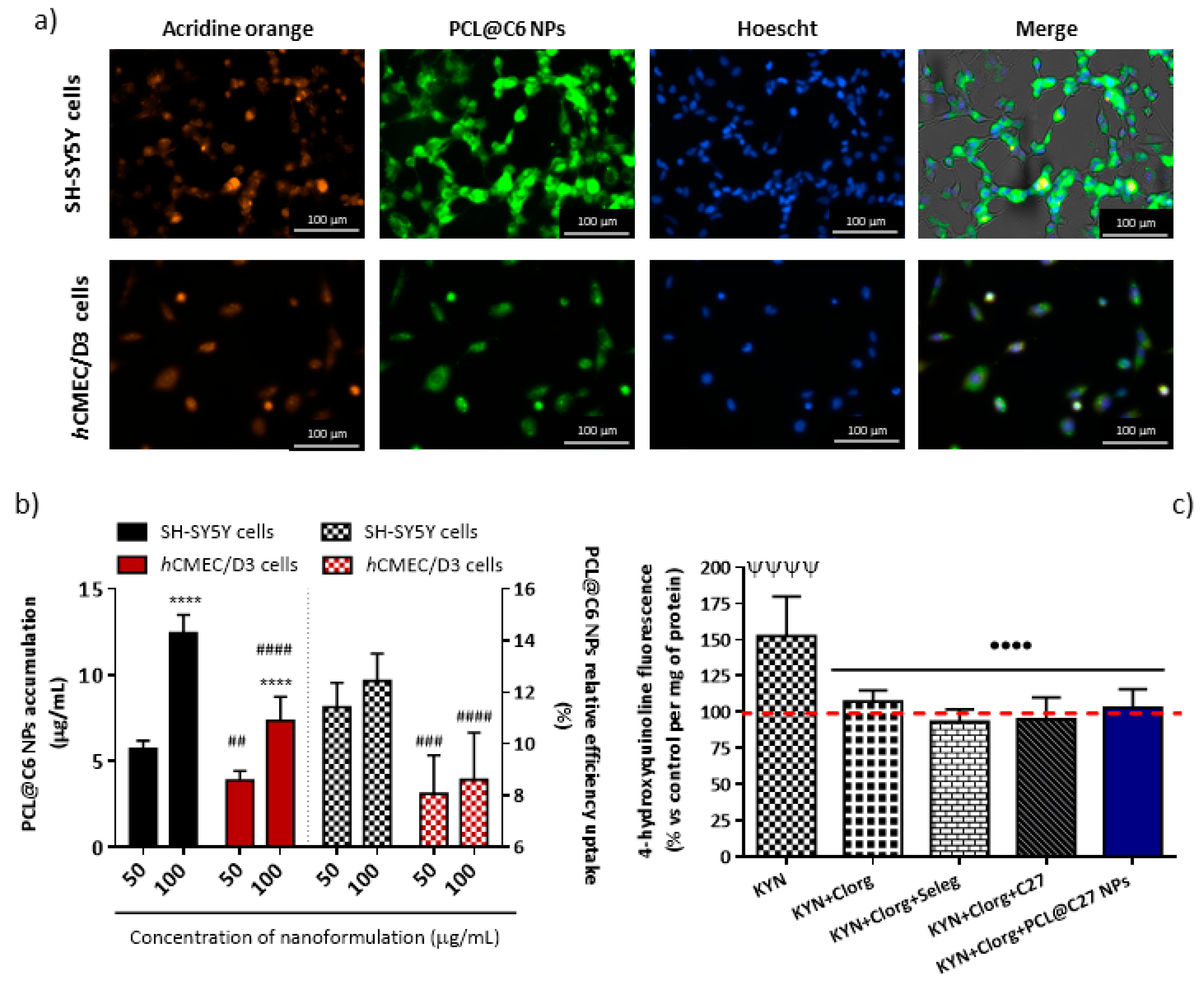
3.5.2. In Vitro Cellular Uptake and Intracellular Localization in Neuronal and Endothelial Cells
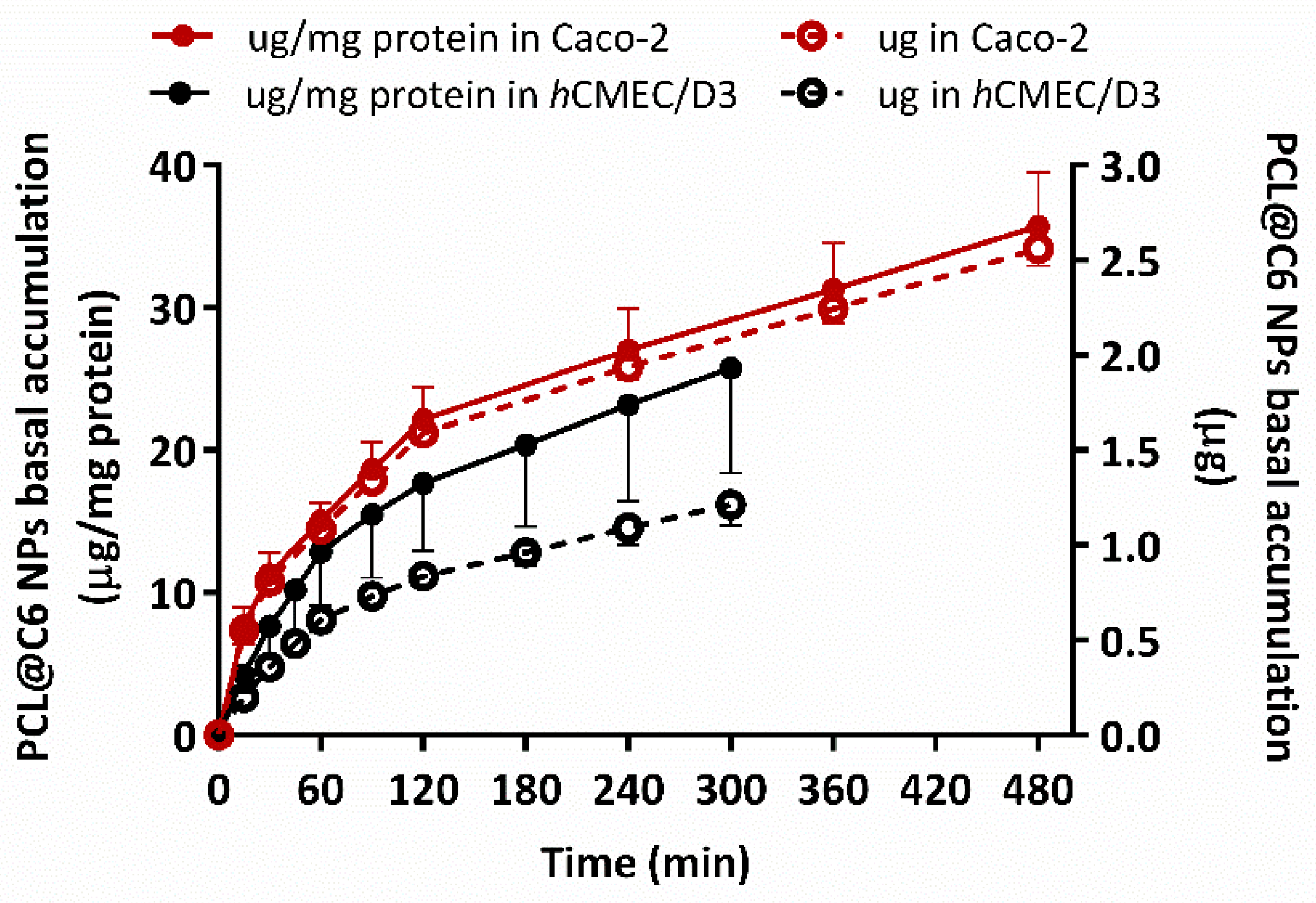
3.5.3. In Vitro Permeability Studies in Epithelial and Endothelial Cells
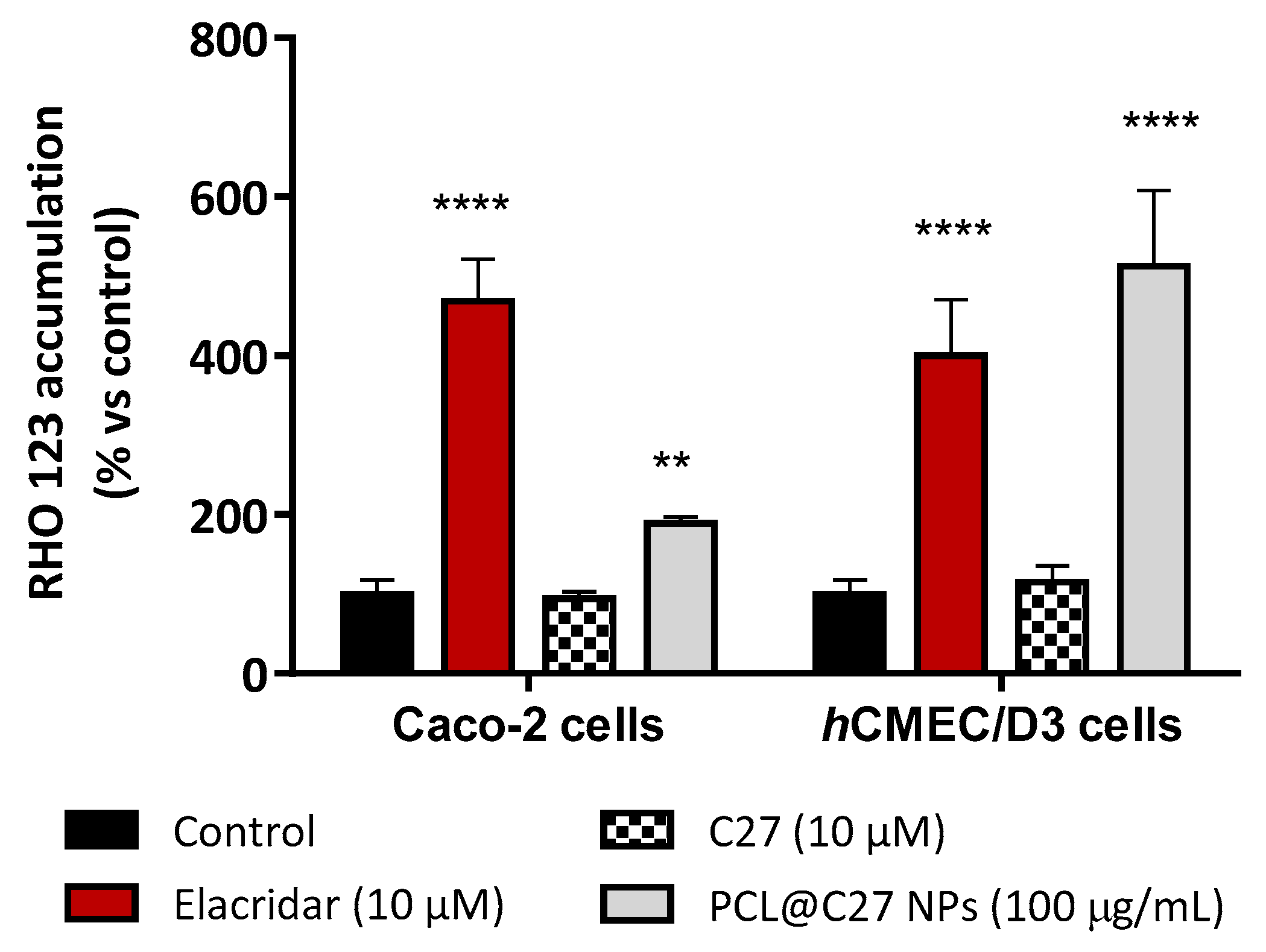
3.5.4. Rhodamine 123 Accumulation in Epithelial and Endothelial Cells
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Taylor, J.P.; Hardy, J.; Fischbeck, K.H. Toxic Proteins in Neurodegenerative Disease. Science 2002, 296, 1991–1995. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P. Apoptosis in neurodegenerative disorders. Nat. Rev. Mol. Cell Biol. 2000, 1, 120. [Google Scholar] [CrossRef] [PubMed]
- Fernández, M.; Barcia, E.; Fernández-Carballido, A.; Garcia, L.; Slowing, K.; Negro, S. Controlled release of rasagiline mesylate promotes neuroprotection in a rotenone-induced advanced model of Parkinson’s disease. Int. J. Pharm. 2012, 438, 266–278. [Google Scholar] [CrossRef] [PubMed]
- Massano, J.; Bhatia, K.P. Clinical Approach to Parkinson’s disease: Features, Diagnosis, and Principles of Management. Cold Spring Harb. Perspect. Med. 2012, 2, a008870. [Google Scholar] [CrossRef] [PubMed]
- Reis, J.; Cagide, F.; Chavarria, D.; Silva, T.; Fernandes, C.; Gaspar, A.; Uriarte, E.; Remião, F.; Alcaro, S.; Ortuso, F.; et al. Discovery of New Chemical Entities for Old Targets: Insights on the Lead Optimization of Chromone-Based Monoamine Oxidase B (MAO-B) Inhibitors. J. Med. Chem. 2016, 59, 5879–5893. [Google Scholar] [CrossRef] [PubMed]
- Gaspar, A.; Matos, M.J.; Garrido, J.; Uriarte, E.; Borges, F. Chromone: A Valid Scaffold in Medicinal Chemistry. Chem. Rev. 2014, 114, 4960–4992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reis, J.; Gaspar, A.; Milhazes, N.; Borges, F. Chromone as a Privileged Scaffold in Drug Discovery: Recent Advances. J. Med. Chem. 2017, 60, 7941–7957. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, C.; Martins, C.; Fonseca, A.; Nunes, R.; Matos, M.J.; Silva, R.; Garrido, J.; Sarmento, B.; Remião, F.; Otero-Espinar, F.J.; et al. PEGylated PLGA Nanoparticles As a Smart Carrier to Increase the Cellular Uptake of a Coumarin-Based Monoamine Oxidase B Inhibitor. ACS Appl. Mater. Interfaces 2018, 10, 39557–39569. [Google Scholar] [CrossRef] [PubMed]
- Molinspiration. Molinspiration Cheminformatics. Available online: http://www.molinspiration.com (accessed on 2 March 2017).
- Singh, R.; Lillard, J.W. Nanoparticle-based targeted drug delivery. Exp. Mol. Pathol. 2009, 86, 215–223. [Google Scholar] [CrossRef] [Green Version]
- Ekladious, I.; Colson, Y.L.; Grinstaff, M.W. Polymer–drug conjugate therapeutics: Advances, insights and prospects. Nat. Rev. Drug Discov. 2018. [Google Scholar] [CrossRef]
- Seju, U.; Kumar, A.; Sawant, K.K. Development and evaluation of olanzapine-loaded PLGA nanoparticles for nose-to-brain delivery: In vitro and in vivo studies. Acta Biomater. 2011, 7, 4169–4176. [Google Scholar] [CrossRef] [PubMed]
- Mei, L.; Zhang, Y.; Zheng, Y.; Tian, G.; Song, C.; Yang, D.; Chen, H.; Sun, H.; Tian, Y.; Liu, K.; et al. A Novel Docetaxel-Loaded Poly (ε-Caprolactone)/Pluronic F68 Nanoparticle Overcoming Multidrug Resistance for Breast Cancer Treatment. Nanoscale Res. Lett. 2009, 4, 1530–1539. [Google Scholar] [CrossRef] [PubMed]
- Nance, E.A.; Woodworth, G.F.; Sailor, K.A.; Shih, T.-Y.; Xu, Q.; Swaminathan, G.; Xiang, D.; Eberhart, C.; Hanes, J. A Dense Poly (Ethylene Glycol) Coating Improves Penetration of Large Polymeric Nanoparticles Within Brain Tissue. Sci. Transl. Med. 2012, 4, 119–149. [Google Scholar] [CrossRef] [PubMed]
- Calvo, P.; Gouritin, B.; Chacun, H.; Desmaële, D.; D’Angelo, J.; Noel, J.-P.; Georgin, D.; Fattal, E.; Andreux, J.P.; Couvreur, P. Long-Circulating PEGylated Polycyanoacrylate Nanoparticles as New Drug Carrier for Brain Delivery. Pharm. Res. 2001, 18, 1157–1166. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, S.A.A.; Saleh, A.M. Applications of nanoparticle systems in drug delivery technology. Saudi Pharm. J. 2018, 26, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Suk, J.S.; Xu, Q.; Kim, N.; Hanes, J.; Ensign, L.M. PEGylation as a strategy for improving nanoparticle-based drug and gene delivery. Adv. Drug Deliv. Rev. 2016, 99, 28–51. [Google Scholar] [CrossRef]
- Wilson, B.; Samanta, M.K.; Santhi, K.; Kumar, K.P.S.; Paramakrishnan, N.; Suresh, B. Poly(n-butylcyanoacrylate) nanoparticles coated with polysorbate 80 for the targeted delivery of rivastigmine into the brain to treat Alzheimer’s disease. Brain Res. 2008, 1200, 159–168. [Google Scholar] [CrossRef]
- Wilson, B.; Samanta, M.K.; Santhi, K.; Kumar, K.P.S.; Paramakrishnan, N.; Suresh, B. Targeted delivery of tacrine into the brain with polysorbate 80-coated poly(n-butylcyanoacrylate) nanoparticles. Eur. J. Pharm. Biopharm. 2008, 70, 75–84. [Google Scholar] [CrossRef]
- Ma, Y.; Zheng, Y.; Zeng, X.; Jiang, L.; Chen, H.; Liu, R.; Huang, L.; Mei, L. Novel docetaxel-loaded nanoparticles based on PCL-Tween 80 copolymer for cancer treatment. Int. J. Nanomed. 2011, 6, 2679–2688. [Google Scholar] [CrossRef] [Green Version]
- Kurakhmaeva, K.B.; Djindjikhashvili, I.A.; Petrov, V.E.; Balabanyan, V.U.; Voronina, T.A.; Trofimov, S.S.; Kreuter, J.; Gelperina, S.; Begley, D.; Alyautdin, R.N. Brain targeting of nerve growth factor using poly(butyl cyanoacrylate) nanoparticles. J. Drug Target. 2009, 17, 564–574. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, C.; Gong, C.; Wang, Y.; Guo, G.; Luo, F.; Qian, Z. Polysorbate 80 coated poly (ɛ-caprolactone)–poly (ethylene glycol)–poly (ɛ-caprolactone) micelles for paclitaxel delivery. Int. J. Pharm. 2012, 434, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Saraiva, C.; Praça, C.; Ferreira, R.; Santos, T.; Ferreira, L.; Bernardino, L. Nanoparticle-mediated brain drug delivery: Overcoming blood–brain barrier to treat neurodegenerative diseases. J. Control. Release 2016, 235, 34–47. [Google Scholar] [CrossRef] [PubMed]
- Le Droumaguet, B.; Souguir, H.; Brambilla, D.; Verpillot, R.; Nicolas, J.; Taverna, M.; Couvreur, P.; Andrieux, K. Selegiline-functionalized, PEGylated poly(alkyl cyanoacrylate) nanoparticles: Investigation of interaction with amyloid-β peptide and surface reorganization. Int. J. Pharm. 2011, 416, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Gomes, M.J.; Fernandes, C.; Martins, S.; Borges, F.; Sarmento, B. Tailoring Lipid and Polymeric Nanoparticles as siRNA Carriers towards the Blood-Brain Barrier—From Targeting to Safe Administration. J. Neuroimmune Pharmacol. 2017, 12, 107–119. [Google Scholar] [CrossRef] [PubMed]
- Gou, P.-F.; Zhu, W.-P.; Shen, Z.-Q. Synthesis, Self-Assembly, and Drug-Loading Capacity of Well-Defined Cyclodextrin-Centered Drug-Conjugated Amphiphilic A14B7 Miktoarm Star Copolymers Based on Poly(ε-caprolactone) and Poly(ethylene glycol). Biomacromolecules 2010, 11, 934–943. [Google Scholar] [CrossRef] [PubMed]
- Gu, C.; Huang, N.; Xu, F.; Gao, J.; Jiang, D. Cascade exciton-pumping engines with manipulated speed and efficiency in light-harvesting porous π-network films. Sci. Rep. 2015, 5, 8867. Available online: https://www.nature.com/articles/srep08867#supplementary-information (accessed on 11 July 2019). [CrossRef] [PubMed]
- Teixeira, J.; Oliveira, C.; Amorim, R.; Cagide, F.; Garrido, J.; Ribeiro, J.A.; Pereira, C.M.; Silva, A.F.; Andrade, P.B.; Oliveira, P.J.; et al. Development of hydroxybenzoic-based platforms as a solution to deliver dietary antioxidants to mitochondria. Sci. Rep. 2017, 7, 6842. [Google Scholar] [CrossRef]
- Santillo, M.F.; Liu, Y.; Ferguson, M.; Vohra, S.N.; Wiesenfeld, P.L. Inhibition of monoamine oxidase (MAO) by β-carbolines and their interactions in live neuronal (PC12) and liver (HuH-7 and MH1C1) cells. Toxicol. In Vitro 2014, 28, 403–410. [Google Scholar] [CrossRef]
- Van Breemen, R.B.; Li, Y. Caco-2 cell permeability assays to measure drug absorption. Expert Opin. Drug Metab. Toxicol. 2005, 1, 175–185. [Google Scholar] [CrossRef]
- Jouan, E.; Le Vée, M.; Mayati, A.; Denizot, C.; Parmentier, Y.; Fardel, O. Evaluation of P-Glycoprotein Inhibitory Potential Using a Rhodamine 123 Accumulation Assay. Pharmaceutics 2016, 8, 12. [Google Scholar] [CrossRef]
- Gao, X.; Wang, B.; Wei, X.; Rao, W.; Ai, F.; Zhao, F.; Men, K.; Yang, B.; Liu, X.; Huang, M.; et al. Preparation, characterization and application of star-shaped PCL/PEG micelles for the delivery of doxorubicin in the treatment of colon cancer. Int. J. Nanomed. 2013, 8, 971–982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bensaid, F.; du Boullay, O.T.; Amgoune, A.; Pradel, C.; Reddy, L.H.; Didier, E.; Sable, S.; Louit, G.; Bazile, D.; Bourissou, D. Y-Shaped mPEG-PLA Cabazitaxel Conjugates: Well-Controlled Synthesis by Organocatalytic Approach and Self-Assembly into Interface Drug-Loaded Core-Corona Nanoparticles. Biomacromolecules 2013, 14, 1189–1198. [Google Scholar] [CrossRef] [PubMed]
- Gamisans, F.; Lacoulonche, F.; Chauvet, A.; Espina, M.; Garcı́a, M.L.; Egea, M.A. Flurbiprofen-loaded nanospheres: Analysis of the matrix structure by thermal methods. Int. J. Pharm. 1999, 179, 37–48. [Google Scholar] [CrossRef]
- Calvo, P.; Vila-Jato, J.L.; Alonso, M.J. Comparative in vitro evaluation of several colloidal systems, nanoparticles, nanocapsules, and nanoemulsions, as ocular drug carriers. J. Pharm. Sci. 1996, 85, 530–536. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.; Wu, F.; Mu, Y.; Hu, Y.; Zhao, X.; Meng, W.; Giesy, J.P.; Lin, Y. Characterization of organic matter of plants from lakes by thermal analysis in a N2 atmosphere. Sci. Rep. 2016, 6, 22877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Tang, L.; Sun, L.; Bao, J.; Song, C.; Huang, L.; Liu, K.; Tian, Y.; Tian, G.; Li, Z.; et al. A novel paclitaxel-loaded poly (ε-caprolactone)/Poloxamer 188 blend nanoparticle overcoming multidrug resistance for cancer treatment. Acta Biomater. 2010, 6, 2045–2052. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Lopez, E.; Egea, M.A.; Cano, A.; Espina, M.; Calpena, A.C.; Ettcheto, M.; Camins, A.; Souto, E.B.; Silva, A.M.; Garcia, M.L. PEGylated PLGA nanospheres optimized by design of experiments for ocular administration of dexibuprofen-in vitro, ex vivo and in vivo characterization. Colloids Surf. B Biointerfaces 2016, 145, 241–250. [Google Scholar] [CrossRef]
- Panyam, J.; Williams, D.; Dash, A.; Leslie-Pelecky, D.; Labhasetwar, V. Solid-state solubility influences encapsulation and release of hydrophobic drugs from PLGA/PLA nanoparticles. J. Pharm. Sci. 2004, 93, 1804–1814. [Google Scholar] [CrossRef]
- Hombreiro Pérez, M.; Zinutti, C.; Lamprecht, A.; Ubrich, N.; Astier, A.; Hoffman, M.; Bodmeier, R.; Maincent, P. The preparation and evaluation of poly (ϵ-caprolactone) microparticles containing both a lipophilic and a hydrophilic drug. J. Control. Release 2000, 65, 429–438. [Google Scholar] [CrossRef]
- Silveira, N.; Longuinho, M.M.; Leitão, S.G.; Silva, R.S.; Lourenço, M.C.; Silva, P.E.; Maria do Carmo, F.R.; Abraçado, L.G.; Finotelli, P.V. Synthesis and characterization of the antitubercular phenazine lapazine and development of PLGA and PCL nanoparticles for its entrapment. Mater. Sci. Eng. C 2016, 58, 458–466. [Google Scholar] [CrossRef]
- Deshmukh, R.K.; Naik, J.B. Aceclofenac microspheres: Quality by design approach. Mater. Sci. Eng. C 2014, 36, 320–328. [Google Scholar] [CrossRef] [PubMed]
- Pinkerton, N.M.; Grandeury, A.; Fisch, A.; Brozio, J.; Riebesehl, B.U.; Prud’homme, R.K. Formation of Stable Nanocarriers by in Situ Ion Pairing during Block-Copolymer-Directed Rapid Precipitation. Mol. Pharm. 2013, 10, 319–328. [Google Scholar] [CrossRef] [PubMed]
- Li, J.W.; Zhang, C.; Li, J.; Fan, L.; Jiang, X.G.; Chen, J.; Pang, Z.Q.; Zhang, Q.Z. Brain Delivery of NAP with PEG-PLGA Nanoparticles Modified with Phage Display Peptides. Pharm. Res. 2013, 30, 1813–1823. [Google Scholar] [CrossRef] [PubMed]
- Shilo, M.; Motiei, M.; Hana, P.; Popovtzer, R. Transport of nanoparticles through the blood-brain barrier for imaging and therapeutic applications. Nanoscale 2014, 6, 2146–2152. [Google Scholar] [CrossRef] [PubMed]
- Massella, D.; Celasco, E.; Salaün, F.; Ferri, A.; Barresi, A. Overcoming the Limits of Flash Nanoprecipitation: Effective Loading of Hydrophilic Drug into Polymeric Nanoparticles with Controlled Structure. Polymers 2018, 10, 1092. [Google Scholar] [CrossRef] [PubMed]
- Fornaguera, C.; Dols-Perez, A.; Calderó, G.; García-Celma, M.J.; Camarasa, J.; Solans, C. PLGA nanoparticles prepared by nano-emulsion templating using low-energy methods as efficient nanocarriers for drug delivery across the blood–brain barrier. J. Control. Release 2015, 211, 134–143. [Google Scholar] [CrossRef] [PubMed]
- Pfeiffer, C.; Rehbock, C.; Hühn, D.; Carrillo-Carrion, C.; de Aberasturi, D.J.; Merk, V.; Barcikowski, S.; Parak, W.J. Interaction of colloidal nanoparticles with their local environment: The (ionic) nanoenvironment around nanoparticles is different from bulk and determines the physico-chemical properties of the nanoparticles. J. R. Soc. Interface 2014, 11. [Google Scholar] [CrossRef] [PubMed]
- Sanna, V.; Siddiqui, I.A.; Sechi, M.; Mukhtar, H. Resveratrol-Loaded Nanoparticles Based on Poly(epsilon-caprolactone) and Poly(d,l-lactic-co-glycolic acid)–Poly(ethylene glycol) Blend for Prostate Cancer Treatment. Mol. Pharm. 2013, 10, 3871–3881. [Google Scholar] [CrossRef] [PubMed]
- Kamaly, N.; Yameen, B.; Wu, J.; Farokhzad, O.C. Degradable Controlled-Release Polymers and Polymeric Nanoparticles: Mechanisms of Controlling Drug Release. Chem. Rev. 2016, 116, 2602–2663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sriamornsak, P.; Thirawong, N.; Weerapol, Y.; Nunthanid, J.; Sungthongjeen, S. Swelling and erosion of pectin matrix tablets and their impact on drug release behavior. Eur. J. Pharm. Biopharm. 2007, 67, 211–219. [Google Scholar] [CrossRef]
- Riederer, P.; Laux, G. MAO-inhibitors in Parkinson’s disease. Exp. Neurobiol. 2011, 20, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Schneider, L.; Giordano, S.; Zelickson, B.R.; Johnson, M.; Benavides, G.; Ouyang, X.; Fineberg, N.; Darley-Usmar, V.M.; Zhang, J. Differentiation of SH-SY5Y cells to a neuronal phenotype changes cellular bioenergetics and the response to oxidative stress. Free Radic. Biol. Med. 2011, 51, 2007–2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopes, F.M.; Schröder, R.; da Frota Júnior, M.L.C.; Zanotto-Filho, A.; Müller, C.B.; Pires, A.S.; Meurer, R.T.; Colpo, G.D.; Gelain, D.P.; Kapczinski, F.; et al. Comparison between proliferative and neuron-like SH-SY5Y cells as an in vitro model for Parkinson disease studies. Brain Res. 2010, 1337, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Je, H.J.; Kim, E.S.; Lee, J.-S.; Lee, H.G. Release Properties and Cellular Uptake in Caco-2 Cells of Size-Controlled Chitosan Nanoparticles. J. Agric. Food Chem. 2017, 65, 10899–10906. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Chen, J.; Huang, H.; Zhou, M.; Zhu, Q.; Yao, S.Q.; Chai, Z.; Hu, Y. Iron modulates the activity of monoamine oxidase B in SH-SY5Y cells. BioMetals 2017, 30, 599–607. [Google Scholar] [CrossRef] [PubMed]
- Shawahna, R.; Uchida, Y.; Declèves, X.; Ohtsuki, S.; Yousif, S.; Dauchy, S.; Jacob, A.; Chassoux, F.; Daumas-Duport, C.; Couraud, P.-O.; et al. Transcriptomic and Quantitative Proteomic Analysis of Transporters and Drug Metabolizing Enzymes in Freshly Isolated Human Brain Microvessels. Mol. Pharm. 2011, 8, 1332–1341. [Google Scholar] [CrossRef] [PubMed]
- Geldenhuys, W.; Mbimba, T.; Bui, T.; Harrison, K.; Sutariya, V. Brain-targeted delivery of paclitaxel using glutathione-coated nanoparticles for brain cancers. J. Drug Target. 2011, 19, 837–845. [Google Scholar] [CrossRef]
- Stanimirovic, D.B.; Bani-Yaghoub, M.; Perkins, M.; Haqqani, A.S. Blood–brain barrier models: In vitro to in vivo translation in preclinical development of CNS-targeting biotherapeutics. Expert Opin. Drug Discov. 2015, 10, 141–155. [Google Scholar] [CrossRef]
- Veszelka, S.; Tóth, A.; Walter, F.R.; Tóth, A.E.; Gróf, I.; Mészáros, M.; Bocsik, A.; Hellinger, É.; Vastag, M.; Rákhely, G.; et al. Comparison of a Rat Primary Cell-Based Blood-Brain Barrier Model with Epithelial and Brain Endothelial Cell Lines: Gene Expression and Drug Transport. Front. Mol. Neurosci. 2018, 11, 166. [Google Scholar] [CrossRef]
- Artursson, P.; Palm, K.; Luthman, K. Caco-2 monolayers in experimental and theoretical predictions of drug transport1PII of original article: S0169-409X(96)00415-2. The article was originally published in Advanced Drug Delivery Reviews 22 (1996) 67–84.1. Adv. Drug Deliv. Rev. 2001, 46, 27–43. [Google Scholar] [CrossRef]
- Weksler, B.; Romero, I.A.; Couraud, P.-O. The hCMEC/D3 cell line as a model of the human blood brain barrier. Fluids Barriers CNS 2013, 10, 16. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, C.; Pinto, M.; Martins, C.; Gomes, M.J.; Sarmento, B.; Oliveira, P.J.; Remião, F.; Borges, F. Development of a PEGylated-Based Platform for Efficient Delivery of Dietary Antioxidants Across the Blood–Brain Barrier. Bioconj. Chem. 2018, 29, 1677–1689. [Google Scholar] [CrossRef] [PubMed]
- Sugano, K.; Kansy, M.; Artursson, P.; Avdeef, A.; Bendels, S.; Di, L.; Ecker, G.F.; Faller, B.; Fischer, H.; Gerebtzoff, G.; et al. Coexistence of passive and carrier-mediated processes in drug transport. Nat. Rev. Drug Discov. 2010, 9, 597–614. [Google Scholar] [CrossRef] [PubMed]
- Win, K.Y.; Feng, S.S. Effects of particle size and surface coating on cellular uptake of polymeric nanoparticles for oral delivery of anticancer drugs. Biomaterials 2005, 26, 2713–2722. [Google Scholar] [CrossRef] [PubMed]
- Murugan, K.; Choonara, Y.E.; Kumar, P.; Bijukumar, D.; du Toit, L.C.; Pillay, V. Parameters and characteristics governing cellular internalization and trans-barrier trafficking of nanostructures. Int. J. Nanomed. 2015, 10, 2191–2206. [Google Scholar] [CrossRef]
- Deli, M.A. Potential use of tight junction modulators to reversibly open membranous barriers and improve drug delivery. Biochim. Biophys. Acta Biomembr. 2009, 1788, 892–910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhavna Md, S.; Ali, M.; Baboota, S.; Sahni, J.K.; Bhatnagar, A.; Ali, J. Preparation, characterization, in vivo biodistribution and pharmacokinetic studies of donepezil-loaded PLGA nanoparticles for brain targeting. Drug Dev. Ind. Pharm. 2014, 40, 278–287. [Google Scholar] [CrossRef]
- Zhu, Q.; Song, W.; Xia, D.; Fan, W.; Yu, M.; Guo, S.; Zhu, C.; Gan, Y. A poly-l-glutamic acid functionalized nanocomplex for improved oral drug absorption. J. Mater. Chem. B 2015, 3, 8508–8517. [Google Scholar] [CrossRef]
- Poller, B.; Gutmann, H.; Krähenbühl, S.; Weksler, B.; Romero, I.; Couraud, P.-O.; Tuffin, G.; Drewe, J.; Huwyler, J. The human brain endothelial cell line hCMEC/D3 as a human blood-brain barrier model for drug transport studies. J. Neurochem. 2008, 107, 1358–1368. [Google Scholar] [CrossRef]
- Amin, M.L. P-glycoprotein Inhibition for Optimal Drug Delivery. Drug Target Insights 2013, 7, 27–34. [Google Scholar] [CrossRef]
- Werle, M. Natural and Synthetic Polymers as Inhibitors of Drug Efflux Pumps. Pharm. Res. 2008, 25, 500–511. [Google Scholar] [CrossRef] [PubMed]
- Shen, Q.; Lin, Y.; Handa, T.; Doi, M.; Sugie, M.; Wakayama, K.; Okada, N.; Fujita, T.; Yamamoto, A. Modulation of intestinal P-glycoprotein function by polyethylene glycols and their derivatives by in vitro transport and in situ absorption studies. Int. J. Pharm. 2006, 313, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Friche, E.; Jensen, P.; Sehested, M.; Demant, E.J.; Nissen, N.N. The solvents Cremophor EL and Tween 80 modulate daunorubicin resistance in the multidrug resistant Ehrlich ascites tumor. Cancer Commun. 1990, 2, 297–303. [Google Scholar] [PubMed]
- Hugger, E.D.; Audus, K.L.; Borchardt, R.T. Effects of Poly(ethylene glycol) on Efflux Transporter Activity in Caco-2 Cell Monolayers. J. Pharm. Sci. 2002, 91, 1980–1990. [Google Scholar] [CrossRef] [PubMed]
- Netsomboon, K.; Laffleur, F.; Suchaoin, W.; Bernkop-Schnürch, A. Novel in vitro transport method for screening the reversibility of P-glycoprotein inhibitors. Eur. J. Pharm. Biopharm. 2016, 100, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.-H.; Lin, X.-N.; Wei, F.; Feng, W.; Huang, Z.-C.; Wang, P.; Ren, L.; Diao, Y. Enhanced brain targeting of temozolomide in polysorbate-80 coated polybutylcyanoacrylate nanoparticles. Int. J. Nanomed. 2011, 6, 445–452. [Google Scholar] [CrossRef] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Medium pH | Korsmeyer–Peppas | ||
|---|---|---|---|
| R2 | n | K (h−1) | |
| 7.4 | 0.972 | 0.464 | 11.2 |
| 1.2–7.4 | 0.985 | 0.594 | 26.9 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pinto, M.; Fernandes, C.; Martins, E.; Silva, R.; Benfeito, S.; Cagide, F.; Mendes, R.F.; Almeida Paz, F.A.; Garrido, J.; Remião, F.; et al. Boosting Drug Discovery for Parkinson’s: Enhancement of the Delivery of a Monoamine Oxidase-B Inhibitor by Brain-Targeted PEGylated Polycaprolactone-Based Nanoparticles. Pharmaceutics 2019, 11, 331. https://doi.org/10.3390/pharmaceutics11070331
Pinto M, Fernandes C, Martins E, Silva R, Benfeito S, Cagide F, Mendes RF, Almeida Paz FA, Garrido J, Remião F, et al. Boosting Drug Discovery for Parkinson’s: Enhancement of the Delivery of a Monoamine Oxidase-B Inhibitor by Brain-Targeted PEGylated Polycaprolactone-Based Nanoparticles. Pharmaceutics. 2019; 11(7):331. https://doi.org/10.3390/pharmaceutics11070331
Chicago/Turabian StylePinto, Miguel, Carlos Fernandes, Eva Martins, Renata Silva, Sofia Benfeito, Fernando Cagide, Ricardo F. Mendes, Filipe A. Almeida Paz, Jorge Garrido, Fernando Remião, and et al. 2019. "Boosting Drug Discovery for Parkinson’s: Enhancement of the Delivery of a Monoamine Oxidase-B Inhibitor by Brain-Targeted PEGylated Polycaprolactone-Based Nanoparticles" Pharmaceutics 11, no. 7: 331. https://doi.org/10.3390/pharmaceutics11070331