Enhanced Oral Bioavailability of Celecoxib Nanocrystalline Solid Dispersion based on Wet Media Milling Technique: Formulation, Optimization and In Vitro/In Vivo Evaluation

Abstract
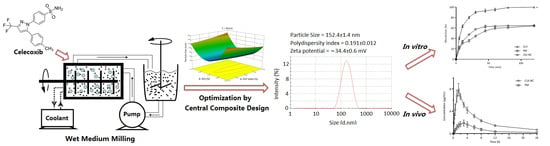
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of Celecoxib Nanocrystalline (CLX-NC) and Physical Mixture
2.3. Characterization of CLX-NC
2.3.1. Particle Size and Zeta Potential
2.3.2. Scanning Electron Microscopy (SEM)
2.3.3. Differential Scanning Calorimetry (DSC)
2.3.4. Powder X-ray Diffraction Analysis (PXRD)
2.4. Experimental Design
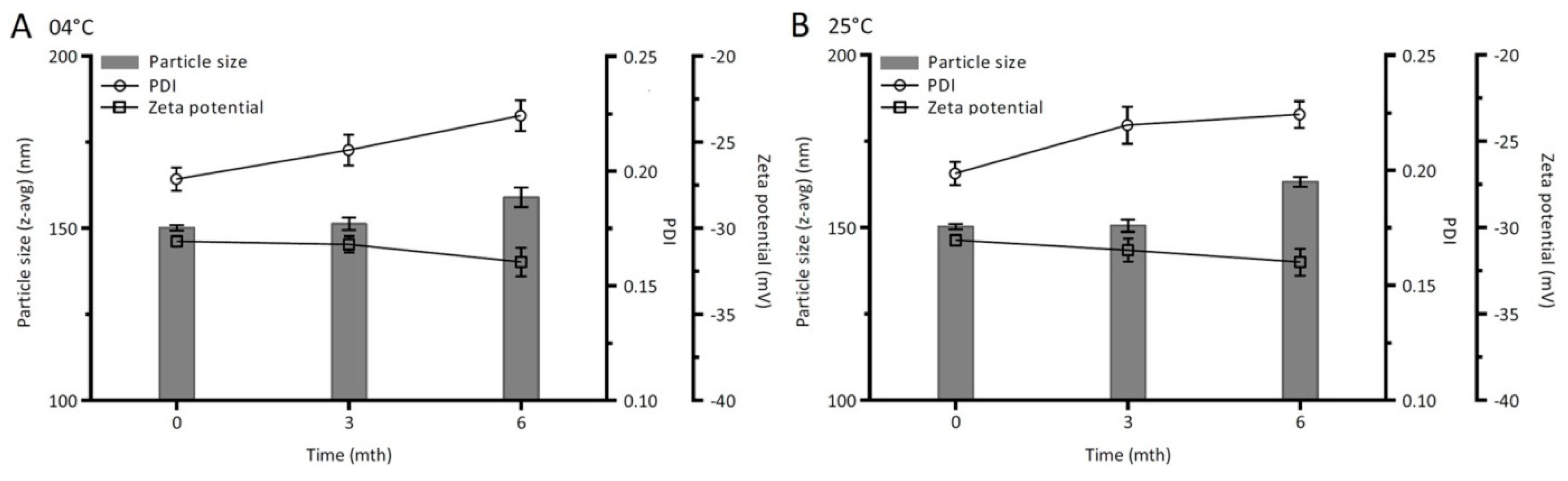
2.5. Storage Stability Study
2.6. High-Performance Liquid Chromatography (HPLC)Analysis of CLX
2.7. Apparent Solubility
2.8. In Vitro Dissolution Study
2.9. In Vivo Oral Bioavailability
2.10. Statistical Analysis
3. Results and Discussions
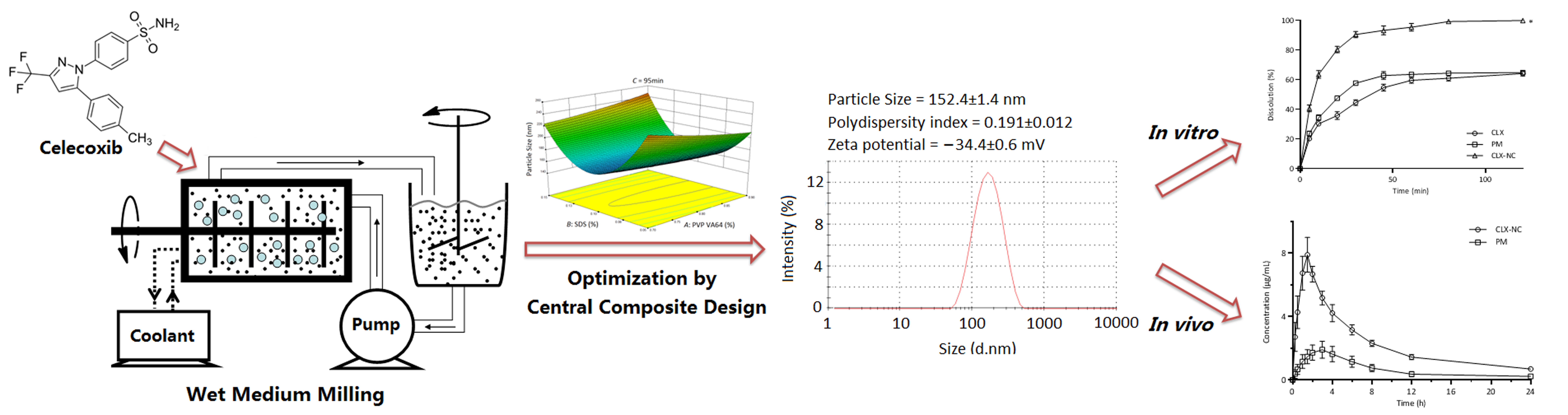
3.1. Screening of Polymer Stabilizers
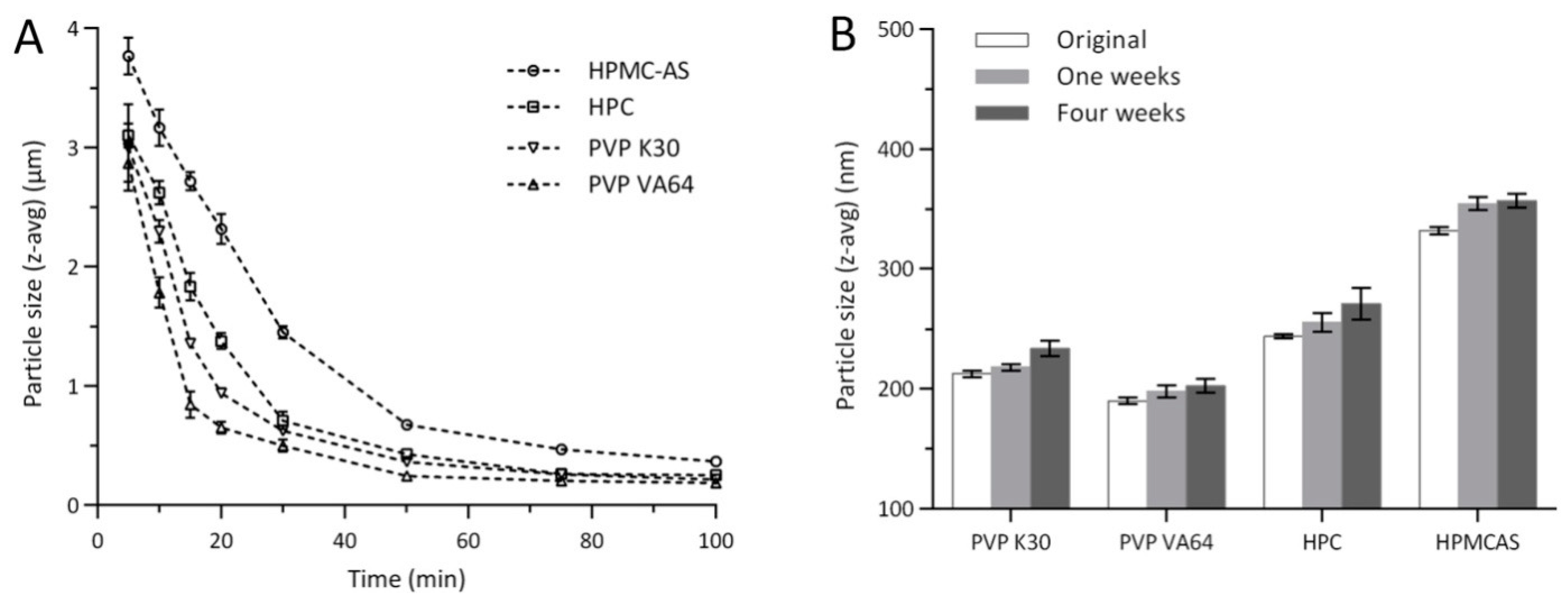
3.2. Optimization of CLX-NC Using Central Composite Design
3.3. Model Validation
3.4. Quality Evaluation of CLX-NC
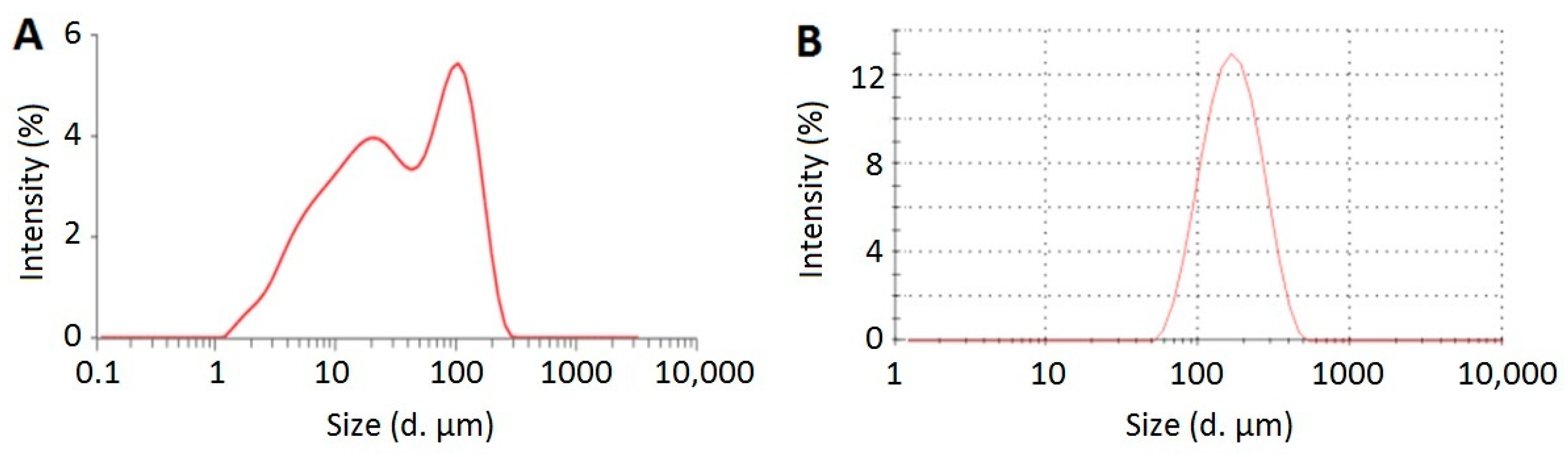
3.4.1. Particle Size and Zeta Potential Analysis
3.4.2. Morphology Evaluation
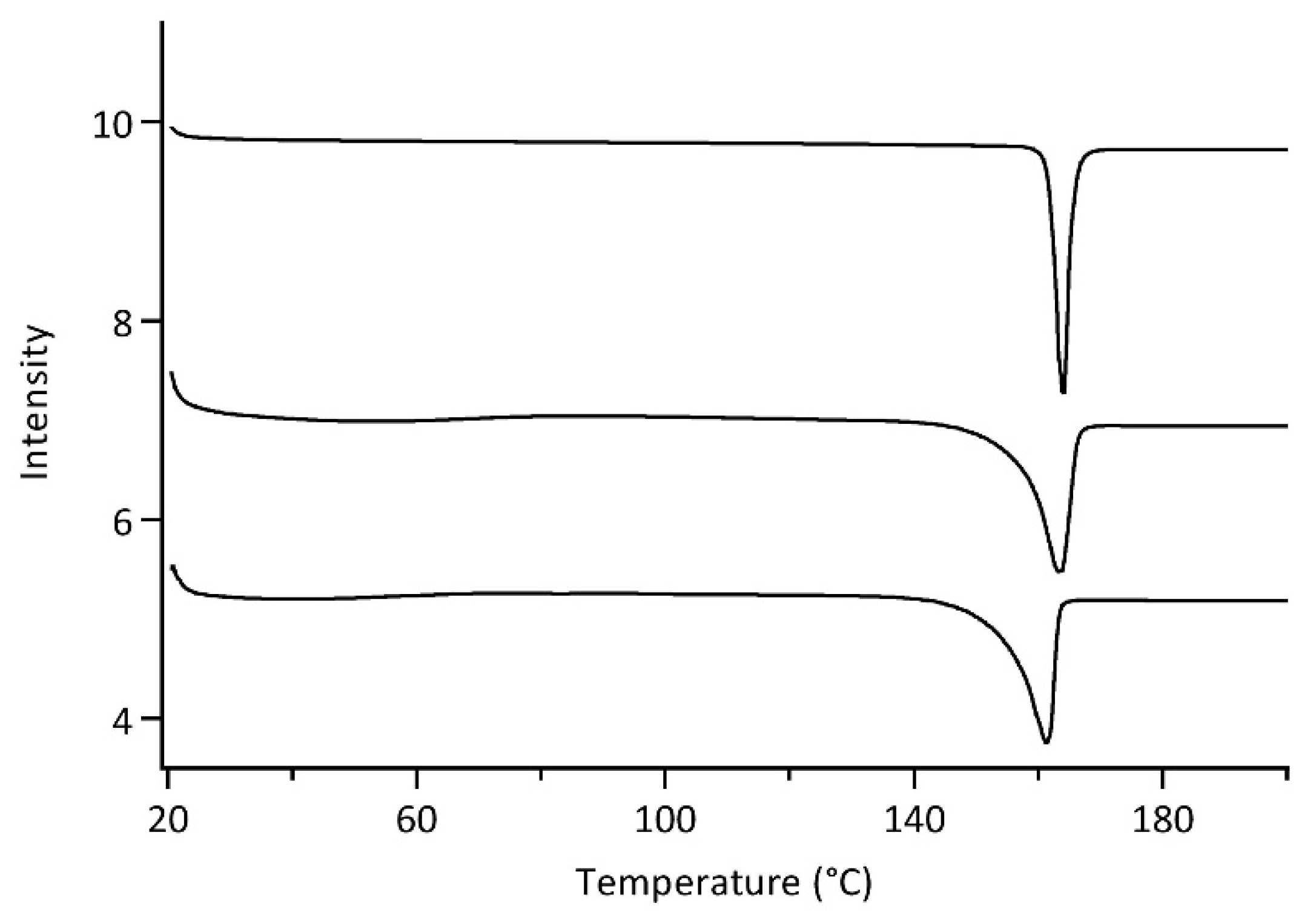
3.4.3. Differential Scanning Calorimetry (DSC)Analysis
3.4.4. Powder X-ray Diffraction (PXRD)Analysis
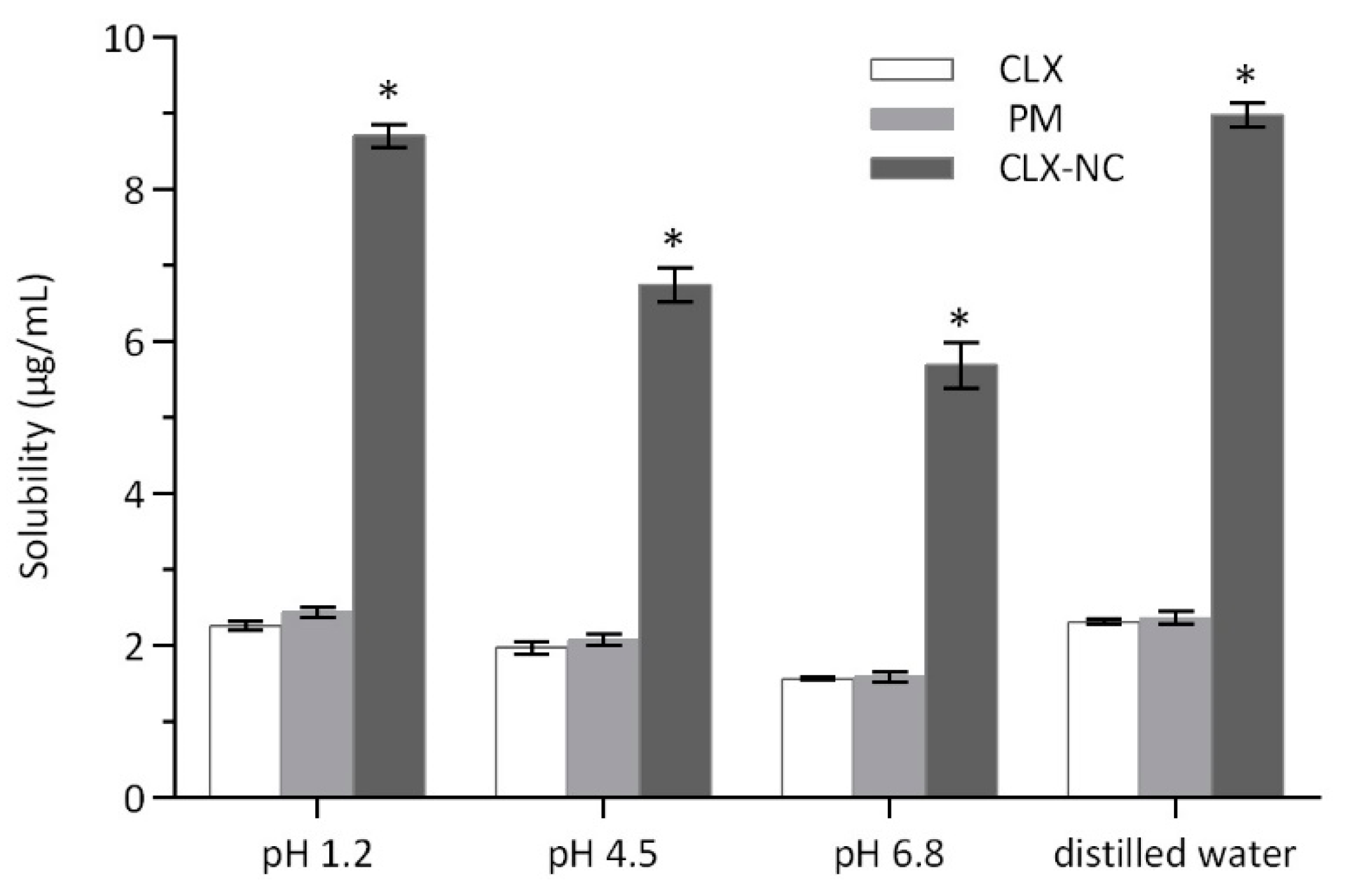
3.4.5. Apparent Solubility Determination
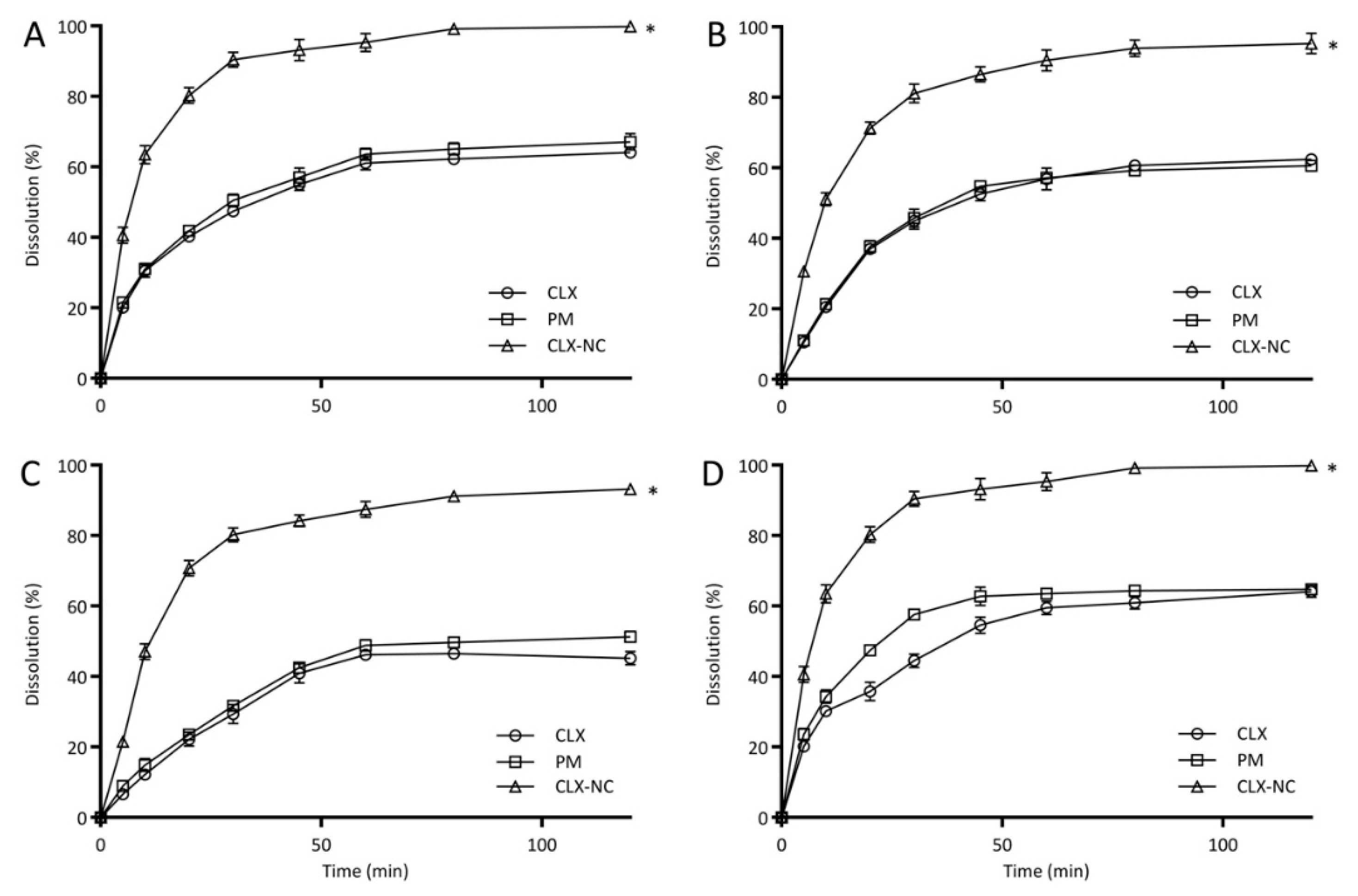
3.4.6. In Vitro Drug Release Study
3.4.7. Storage Stability Study
3.4.8. In Vivo Oral Bioavailability
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Malamatari, M.; Taylor, K.M.G.; Malamataris, S.; Douroumis, D.; Kachrimanis, K. Pharmaceutical nanocrystals: Production by wet milling and applications. Drug Discov. Today 2018, 23, 534–547. [Google Scholar] [CrossRef] [PubMed]
- Merisko-Liversidge, E.M.; Liversidge, G.G. Drug nanoparticles: formulating poorly water-soluble compounds. Toxicol. Pathol. 2008, 36, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Zhao, Y.; Zhao, Y.; Ding, Z.; Fan, Z.; Zhang, H.; Liu, M.; Wang, Z.; Han, J. Effect of HPMCAS on recrystallization inhibition of nimodipine solid dispersions prepared by hot-melt extrusion and dissolution enhancement of nimodipine tablets. Colloids Surf. B Biointerfaces 2018, 172, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Junyaprasert, V.B.; Morakul, B. Nanocrystals for enhancement of oral bioavailability of poorly water-soluble drugs. Asian J. Pharm. Sci. 2015, 10, 13–23. [Google Scholar] [CrossRef] [Green Version]
- Müller, R.H.; Becker, R.; Kruss, B.; Peters, K. Pharmaceutical Nanosuspensions for Medicament Administration as Systems with Increased Saturation Solubility and Rate of Solution. US Patent 5858410, 12 January 1999. [Google Scholar]
- Peltonen, L.; Hirvonen, J. Pharmaceutical nanocrystals by nanomilling: Critical process parameters, particle fracturing and stabilization methods. J. Pharm. Pharmacol. 2010, 62, 1569–1579. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zheng, Y.; Zhang, L.; Wang, Q.; Zhang, D. Stability of nanosuspensions in drug delivery. J. Control Release 2013, 172, 1126–1141. [Google Scholar] [CrossRef]
- Shchekin, A.K.; Rusanov, A.I. Generalization of the Gibbs-Kelvin-Köhler and Ostwald–Freundlich equations for a liquid film on a soluble nanoparticle. J. Chem. Phys. 2008, 129, 154116. [Google Scholar] [CrossRef]
- Mosharraf, M.; Nyström, C. The effect of particle size and shape on the surface specific dissolution rate of microsized practically insoluble drugs. Int. J. Pharm. 1995, 122, 35–47. [Google Scholar] [CrossRef]
- Shegokar, R.; Müller, R.H. Nanocrystals: Industrially feasible multifunctional formulation technology for poorly soluble actives. Int. J. Pharm. 2010, 399, 129–139. [Google Scholar] [CrossRef]
- Möschwitzer, J.P. Drug nanocrystals in the commercial pharmaceutical development process. Int. J. Pharm. 2013, 453, 142–156. [Google Scholar] [CrossRef]
- Li, M.; Azad, M.; Davé, R.; Bilgili, E. Nanomilling of drugs for bioavailability enhancement: A holistic formulation-process perspective. Pharmaceutics 2016, 8, 17. [Google Scholar] [CrossRef] [PubMed]
- Loh, Z.H.; Samanta, A.K.; Heng, P.W.S. Overview of milling techniques for improving the solubility of poorly water-soluble drugs. Asian J. Pharm. Sci. 2015, 10, 255–274. [Google Scholar] [CrossRef] [Green Version]
- Junghanns, J.A.H.; Müller, R.H. Nanocrystal technology: Drug delivery and clinical applications. Int. J. Nanomed. 2008, 3, 295–310. [Google Scholar]
- Iurian, S.; Bogdan, C.; Tomuță, I.; Szabó-Révész, P.; Chvatal, A.; Leucuța, S.E.; Moldovan, M.; Ambrus, R. Development of oral lyophilisates containing meloxicam nanocrystals using QbD approach. Eur. J. Pharm. Sci. 2017, 104, 356–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Qiao, Y.; Wu, Z. Nanosystem trends in drug delivery using quality-by-design concept. J. Control Release 2017, 256, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Peltonen, L.; Hirvonen, J. Drug nanocrystals—Versatile option for formulation of poorly soluble materials. Int. J. Pharm. 2018, 537, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Medarević, D.; Djuriš, J.; Ibrić, S.; Mitrić, M.; Kachrimanis, K. Optimization of formulation and process parameters for the production of carvedilol nanosuspension by wet media milling. Int. J. Pharm. 2018, 540, 150–161. [Google Scholar] [CrossRef] [PubMed]
- Ha, E.S.; Choo, G.H.; Baek, I.W.; Kim, M.S. Formulation, characterization, and in vivo evaluation of celecoxib-PVP solid dispersion nanoparticles using supercritical antisolvent process. Molecules 2014, 19, 20325–20339. [Google Scholar] [CrossRef]
- Rawat, S.; Jain, S.K. Solubility enhancement of celecoxib using beta-cyclodextrin inclusion complexes. Eur. J. Pharm. Biopharm. 2004, 57, 263–267. [Google Scholar] [CrossRef]
- Kwon, H.J.; Heo, E.J.; Kim, Y.H.; Kim, S.; Hwang, Y.H.; Byun, J.M.; Cheon, S.H.; Park, S.Y.; Kim, D.Y.; Cho, K.H.; et al. Development and evaluation of poorly water-soluble celecoxib as solid dispersions containing nonionic surfactants using fluidized-bed granulation. Pharmaceutics 2019, 11, 136. [Google Scholar] [CrossRef]
- Moghimipour, E.; Salimi, A.; Yousefvand, T. Preparation and evaluation of celecoxib nanoemulsion for ocular drug delivery. Asian J. Pharm. 2017, 11, S543–S550. [Google Scholar]
- Mandracchia, D.; Trapani, A.; Perteghella, S.; Sorrenti, M.; Catenacci, L.; Torre, M.L.; Trapani, G.; Tripodo, G. pH-sensitive inulin-based nanomicelles for intestinal site-specific and controlled release of celecoxib. Carbohydr. Polym. 2018, 181, 570–578. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Han, Y.; Xu, G.; Yin, L.; Neubi, M.N.; Zhou, J.; Ding, Y. Preparation and evaluation of celecoxib nanosuspensions for bioavailability enhancement. RSC Adv. 2017, 7, 13053–13064. [Google Scholar] [CrossRef] [Green Version]
- Jeong, S.C.; Kim, D.S.; Jin, S.G.; Youn, Y.S.; Oh, K.T.; Li, D.X.; Yong, C.S.; Oh Kim, J.; Kim, K.S.; Choi, H.G. Development of a novel celecoxib-loaded nanosuspension using a wet media milling process. Pharmazie 2018, 73, 498–502. [Google Scholar] [PubMed]
- Matbou Riahi, M.; Sahebkar, A.; Sadri, K.; Nikoofal-Sahlabadi, S.; Jaafari, M.R. Stable and sustained release liposomal formulations of celecoxib: In vitro and in vivo anti-tumor evaluation. Int. J. Pharm. 2018, 540, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Xie, X.; Zhao, Y.; Gao, Y.; Cai, C.; Zhang, Q.; Ding, Z.; Fan, Z.; Zhang, H.; Liu, M.; et al. Effect of plasticizers on manufacturing ritonavir/copovidone solid dispersions via hot-melt extrusion: Preformulation, physicochemical characterization, and pharmacokinetics in rats. Eur. J. Pharm. Sci. 2019, 127, 60–70. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.D.; Lee, P.I. Probing the mechanisms of drug release from amorphous solid dispersions in medium-soluble and medium-insoluble carriers. J. Control Release 2015, 211, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Ahuja, B.K.; Jena, S.K.; Paidi, S.K.; Bagri, S.; Suresh, S. Formulation, optimization and in vitro–in vivo evaluation of febuxostat nanosuspension. Int. J. Pharm. 2015, 478, 540–552. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Zhang, D.; Chen, M. Drug nanocrystals for the formulation of poorly soluble drugs and its application as a potential drug delivery system. J. Nanoparticle Res. 2008, 10, 845–862. [Google Scholar] [CrossRef]
- Andrews, G.P.; Abu-Diak, O.; Kusmanto, F.; Hornsby, P.; Hui, Z.; Jones, D.S. Physicochemical characterization and drug-release properties of celecoxib hot-melt extruded glass solutions. J. Pharm. Pharmacol. 2010, 62, 1580–1590. [Google Scholar] [CrossRef]
- Jackson, C.L.; McKenna, G.B. The melting behavior of organic materials confined in porous solids. J. Chem. Phys. 1990, 93, 9002–9011. [Google Scholar] [CrossRef]
- Patravale, V.; Date, A.; Kulkarni, R. Nanosuspensions: A promising drug delivery strategy. J. Pharm. Pharmacol. 2004, 56, 827–840. [Google Scholar] [CrossRef] [PubMed]
- Shete, G.; Jain, H.; Punj, D.; Prajapat, H.; Akotiya, P.; Bansal, A.K. Stabilizers used in nano-crystal based drug delivery systems. J. Excip. Food Chem. 2014, 5, 184–209. [Google Scholar]
- Rabinow, B.E. Nanosuspensions in drug delivery. Nat. Rev. Drug Discov. 2004, 3, 785–796. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Wang, Y.; Zhu, L.J.; Liu, Z.Q. Nanocarriers: A general strategy for enhancement of oral bioavailability of poorly absorbed or pre-systemically metabolized drugs. Curr. Drug Metab. 2010, 11, 197–207. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Independent Factors | Design Level | ||
|---|---|---|---|
| Coded | Actual Parameters | Coded Value | Actual Value |
| X1 | Concentration of polymer stabilizer (% w/v) | −1.68 | 0.63 |
| −1 | 0.7 | ||
| 0 | 0.8 | ||
| +1 | 0.9 | ||
| +1.68 | 0.97 | ||
| X2 | Concentration of secondary stabilizer (% w/v) | −1.68 | 0.02 |
| −1 | 0.05 | ||
| 0 | 0.10 | ||
| +1 | 0.15 | ||
| +1.68 | 0.18 | ||
| X3 | Milling times (min) | −1.68 | 32.96 |
| −1 | 50 | ||
| 0 | 75 | ||
| +1 | 100 | ||
| +1.68 | 117.04 | ||
| Run | Independent Factors | Experiment Responses | ||||
|---|---|---|---|---|---|---|
| X1/(% w/v) | X2/(% w/v) | X3/(min) | Particle Size (nm) | PDI | Zeta Potential (mV) | |
| 1 | 0.97 | 0.10 | 75.00 | 152.6 | 0.254 | −20.3 |
| 2 | 0.80 | 0.02 | 75.00 | 342.6 | 0.351 | −17.9 |
| 3 | 0.80 | 0.18 | 75.00 | 371.8 | 0.208 | −33.0 |
| 4 | 0.63 | 0.10 | 75.00 | 171.9 | 0.204 | −30.5 |
| 5 | 0.90 | 0.05 | 100.00 | 217.0 | 0.317 | −19.9 |
| 6 | 0.80 | 0.10 | 117.04 | 160.9 | 0.190 | −33.0 |
| 7 | 0.90 | 0.05 | 50.00 | 217.0 | 0.367 | −22.4 |
| 8 | 0.90 | 0.15 | 50.00 | 252.0 | 0.224 | −30.9 |
| 9 | 0.80 | 0.10 | 75.00 | 162.3 | 0.205 | −34.6 |
| 10 | 0.90 | 0.15 | 100.00 | 248.0 | 0.194 | −23.0 |
| 11 | 0.80 | 0.10 | 75.00 | 153.4 | 0.207 | −34.3 |
| 12 | 0.80 | 0.10 | 75.00 | 156.9 | 0.217 | −34.1 |
| 13 | 0.70 | 0.15 | 50.00 | 243.0 | 0.217 | −35.9 |
| 14 | 0.80 | 0.10 | 75.00 | 158.7 | 0.210 | −35.0 |
| 15 | 0.80 | 0.10 | 75.00 | 156.5 | 0.215 | −34.8 |
| 16 | 0.80 | 0.10 | 32.96 | 176.4 | 0.255 | −34.8 |
| 17 | 0.80 | 0.10 | 75.00 | 157.5 | 0.220 | −34.6 |
| 18 | 0.70 | 0.05 | 100.00 | 248.5 | 0.232 | −26.8 |
| 19 | 0.70 | 0.05 | 50.00 | 263.0 | 0.262 | −20.8 |
| 20 | 0.70 | 0.15 | 100.00 | 224.1 | 0.201 | −36.4 |
| Source | Particle Size(Y1) | PDI (Y2) | Zeta Potential(Y3) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sum of Squares | Degree of Freedom | F-Value | p Value (Prob > F) | Sum of Squares | Degree of Freedom | F-Value | p Value (Prob > F) | Sum of Squares | Degree of Freedom | F-Value | p Value (Prob > F) | |
| Model | 77,170.9 | 9 | 339.2 | <0.0001 | 0.049 | 9 | 89.1 | <0.0001 | 782.3 | 9 | 300.0 | <0.0001 |
| X1 | 434.8 | 1 | 17.2 | 0.0020 | 5.501 × 10−3 | 1 | 90.7 | <0.0001 | 122.2 | 1 | 421.8 | <0.0001 |
| X2 | 366.0 | 1 | 14.4 | 0.0035 | 0.025 | 1 | 409.9 | <0.0001 | 278.7 | 1 | 962.0 | <0.0001 |
| X3 | 294.9 | 1 | 11.6 | 0.0066 | 4.055 × 10−3 | 1 | 66.9 | <0.0001 | 3.5 | 1 | 12.1 | 0.0059 |
| X1 X2 | 1523.5 | 1 | 60.2 | <0.0001 | 4.512 × 10−3 | 1 | 74.4 | <0.0001 | 21.4 | 1 | 74.0 | <0.0001 |
| X1 X3 | 108.0 | 1 | 4.2 | 0.0656 | 1.445 × 10−4 | 1 | 2.3 | 0.1536 | 35.7 | 1 | 123.2 | <0.0001 |
| X2 X3 | 8.8 | 1 | 0.3 | 0.5678 | 1.445 × 10−4 | 1 | 2.3 | 0.1536 | 14.8 | 1 | 51.2 | <0.0001 |
| X12 | 112.2 | 1 | 4.4 | 0.0613 | 7.465 × 10−4 | 1 | 12.3 | 0.0056 | 168.6 | 1 | 582.2 | <0.0001 |
| X22 | 74,120.5 | 1 | 2932.1 | <0.0001 | 9.044 × 10−3 | 1 | 149.2 | <0.0001 | 166.9 | 1 | 576.2 | <0.0001 |
| X32 | 368.0 | 1 | 14.5 | 0.0034 | 3.459 × 10−4 | 1 | 5.7 | 0.0380 | 2.4 | 1 | 8.6 | 0.0149 |
| Residual | 252.7 | 10 | 6.060 × 10−4 | 10 | 2.9 | 10 | ||||||
| Lack of Fit | 210.1 | 5 | 4.9 | 0.0524 | 4.307 × 10−4 | 5 | 2.4 | 0.1732 | 2.3 | 5 | 4.4 | 0.0640 |
| Pure Error | 42.6 | 5 | 1.753 × 10−4 | 5 | 0.5 | 5 | ||||||
| Cor Total | 77,492.4 | 19 | 0.049 | 19 | 785.2 | 19 | ||||||
| Verification Trial | PVP VA64 (%, w/v) | SDS (%, w/v) | Milling Times (min) | Actual by Predicted | Particle Size (nm) | PDI | Zeta Potential (mV) |
|---|---|---|---|---|---|---|---|
| 1 a | 0.75 | 0.11 | 90 | Predicted | 159.3 | 0.194 | −35.8 |
| Actual | 152.4 ± 1.4 | 0.192 ± 0.012 | −34.4 ± 0.6 | ||||
| Error | 4.33 | 1.03 | 3.91 | ||||
| 2 | 0.85 | 0.06 | 55 | Predicted | 196.2 | 0.304 | −27.2 |
| Actual | 191.7 ± 2.9 | 0.291 ± 0.025 | −26.1 ± 1.0 | ||||
| Error | 2.29 | 4.28 | 4.04 | ||||
| 3 | 0.70 | 0.14 | 75 | Predicted | 204.7 | 0.200 | −36.9 |
| Actual | 201.7 ± 3.7 | 0.209 ± 0.014 | −36.8 ± 0.5 | ||||
| Error | 1.47 | 4.50 | 0.27 |
| Parameters | Optimized CLX-NC | Physical Mixture Contain CLX |
|---|---|---|
| Cmax (μg/mL) | 7.88 ± 0.72 | 2.70 ± 0.25 |
| Tmax (h) | 1.50 ± 0.32 | 2.83 ± 0.41 |
| AUC(0–t) (μg·h/mL) | 54.90 ± 0.30 | 16.04 ± 3.36 |
| AUC(0–∞) (μg·h/mL) | 66.75 ± 2.51 | 21.53 ± 3.02 |
| t1/2 (h) | 0.89 ± 0.18 | 0.87 ± 0.09 |
| MRT (h) | 1.37 ± 0.04 | 1.31 ± 0.13 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ding, Z.; Wang, L.; Xing, Y.; Zhao, Y.; Wang, Z.; Han, J. Enhanced Oral Bioavailability of Celecoxib Nanocrystalline Solid Dispersion based on Wet Media Milling Technique: Formulation, Optimization and In Vitro/In Vivo Evaluation. Pharmaceutics 2019, 11, 328. https://doi.org/10.3390/pharmaceutics11070328
Ding Z, Wang L, Xing Y, Zhao Y, Wang Z, Han J. Enhanced Oral Bioavailability of Celecoxib Nanocrystalline Solid Dispersion based on Wet Media Milling Technique: Formulation, Optimization and In Vitro/In Vivo Evaluation. Pharmaceutics. 2019; 11(7):328. https://doi.org/10.3390/pharmaceutics11070328
Chicago/Turabian StyleDing, Zhuang, Lili Wang, Yangyang Xing, Yanna Zhao, Zhengping Wang, and Jun Han. 2019. "Enhanced Oral Bioavailability of Celecoxib Nanocrystalline Solid Dispersion based on Wet Media Milling Technique: Formulation, Optimization and In Vitro/In Vivo Evaluation" Pharmaceutics 11, no. 7: 328. https://doi.org/10.3390/pharmaceutics11070328