Polymeric Mesoporous Silica Nanoparticles for Enhanced Delivery of 5-Fluorouracil In Vitro



Abstract
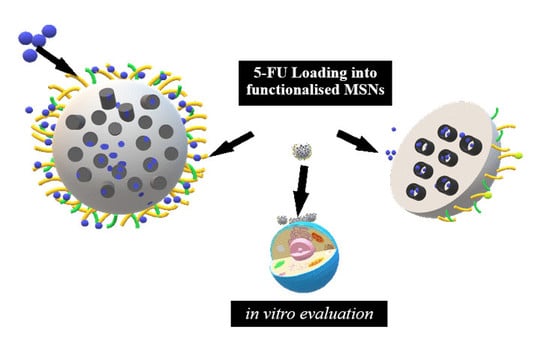
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis of Mesoporous Silica Nanoparticles (MSNs)
2.3. Chitosan Functionalisation
2.4. Functionalisation with 2% and 5% PEG
2.5. Formation of 5-Fluorouracil:MSN Nanoconjugates
2.6. 5-FU Release
2.7. Electron Microscopy
2.8. Nitrogen Adsorption and Desorptio
2.9. Nanoparticle Tracking Analysis (NTA)
2.10. Cytotoxicity
2.11. Apoptosis
2.12. Cell Cycle Analysis
2.13. Statistical Analyses
3. Results
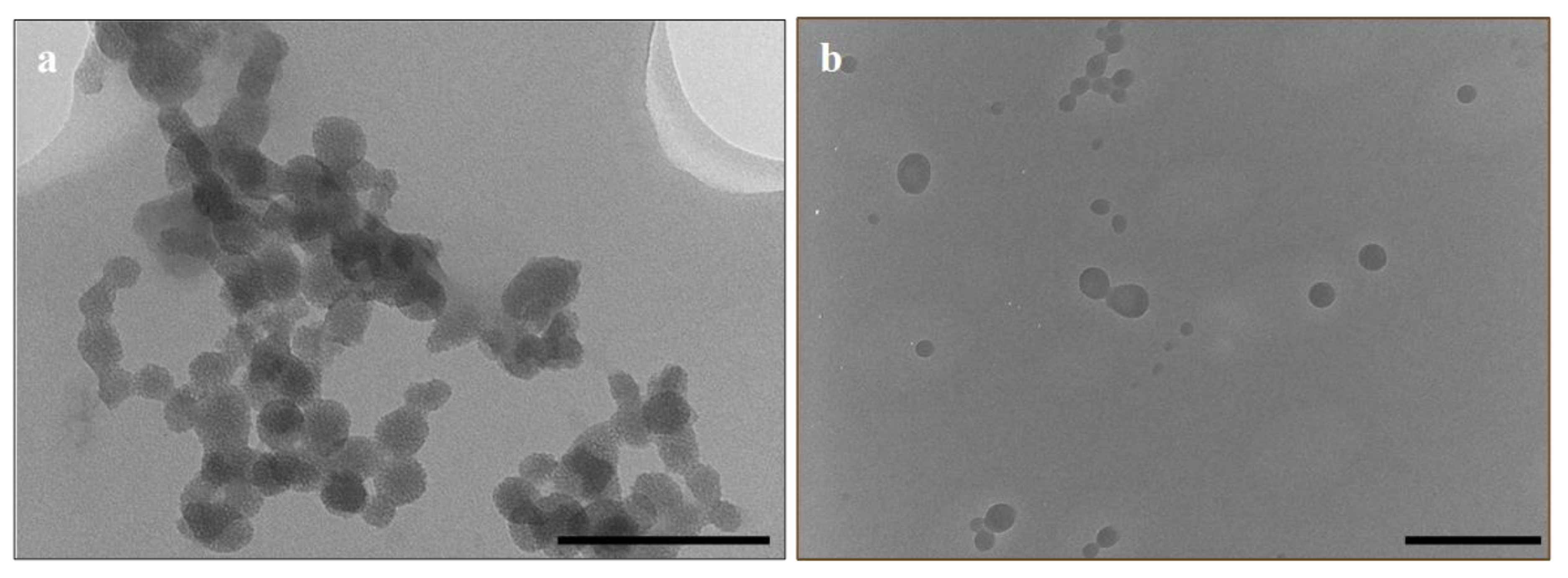
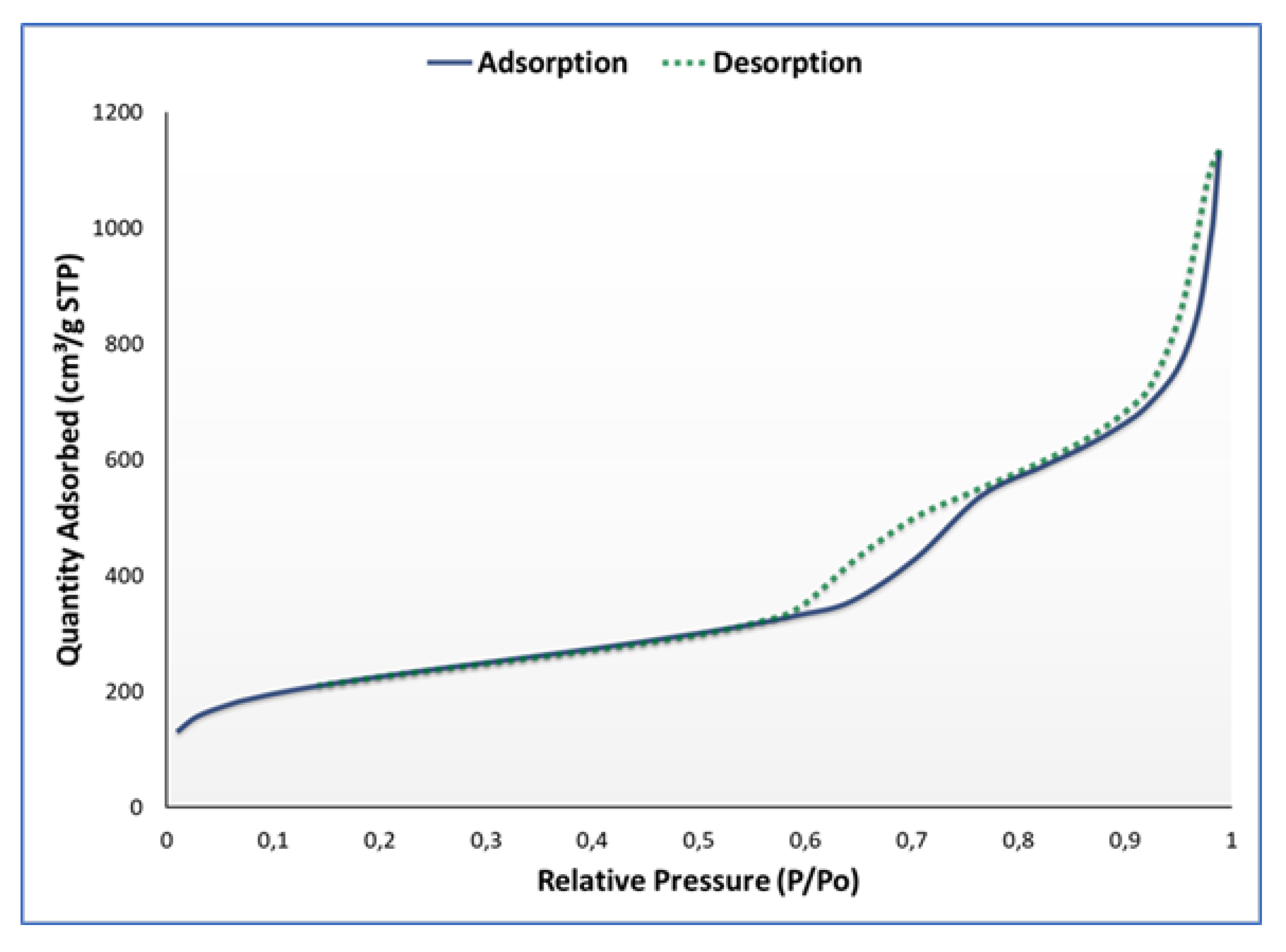
3.1. Synthesis and Characterisation
3.2. Drug Loading Efficiency
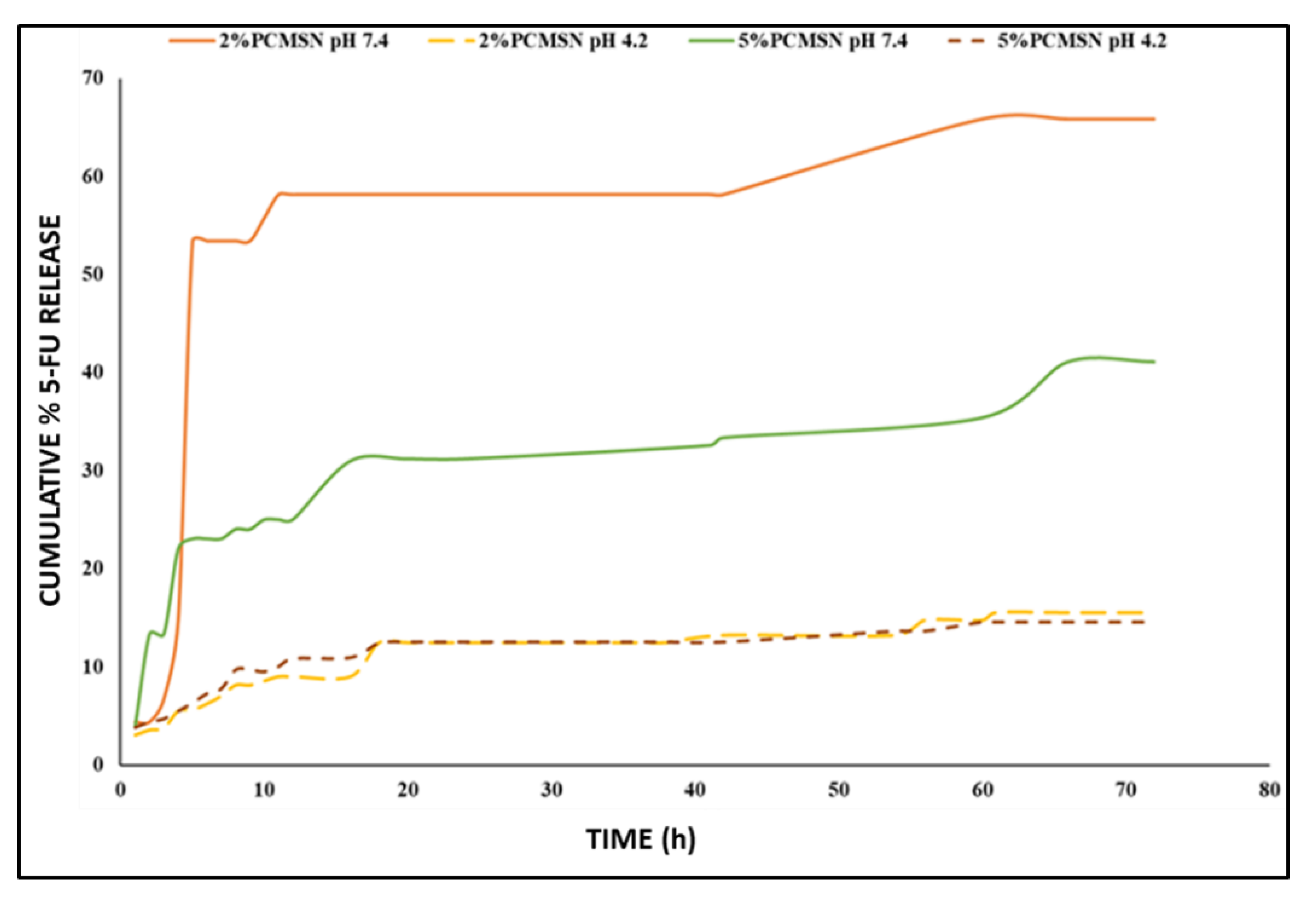
3.3. 5-FU Release and Kinetics
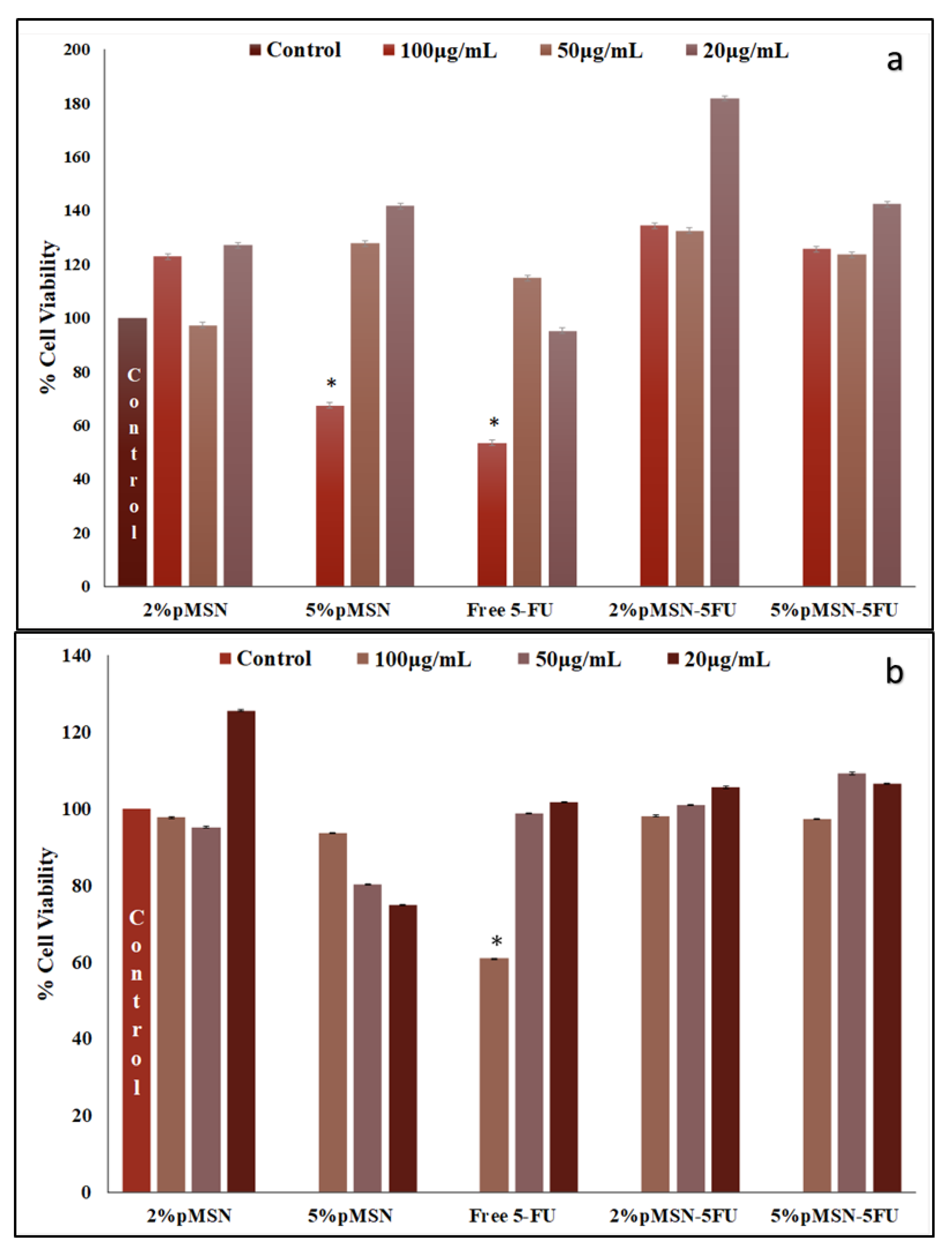
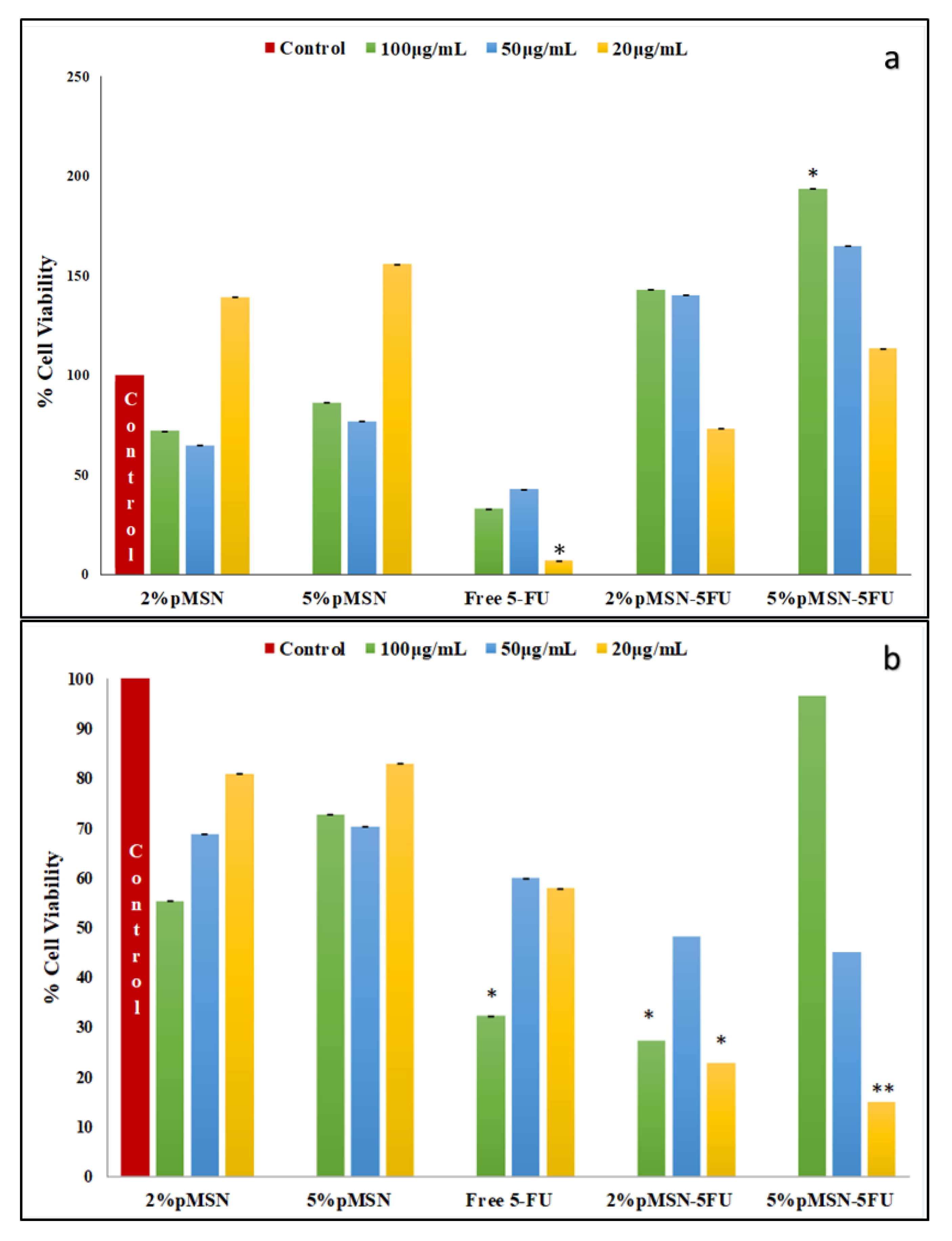
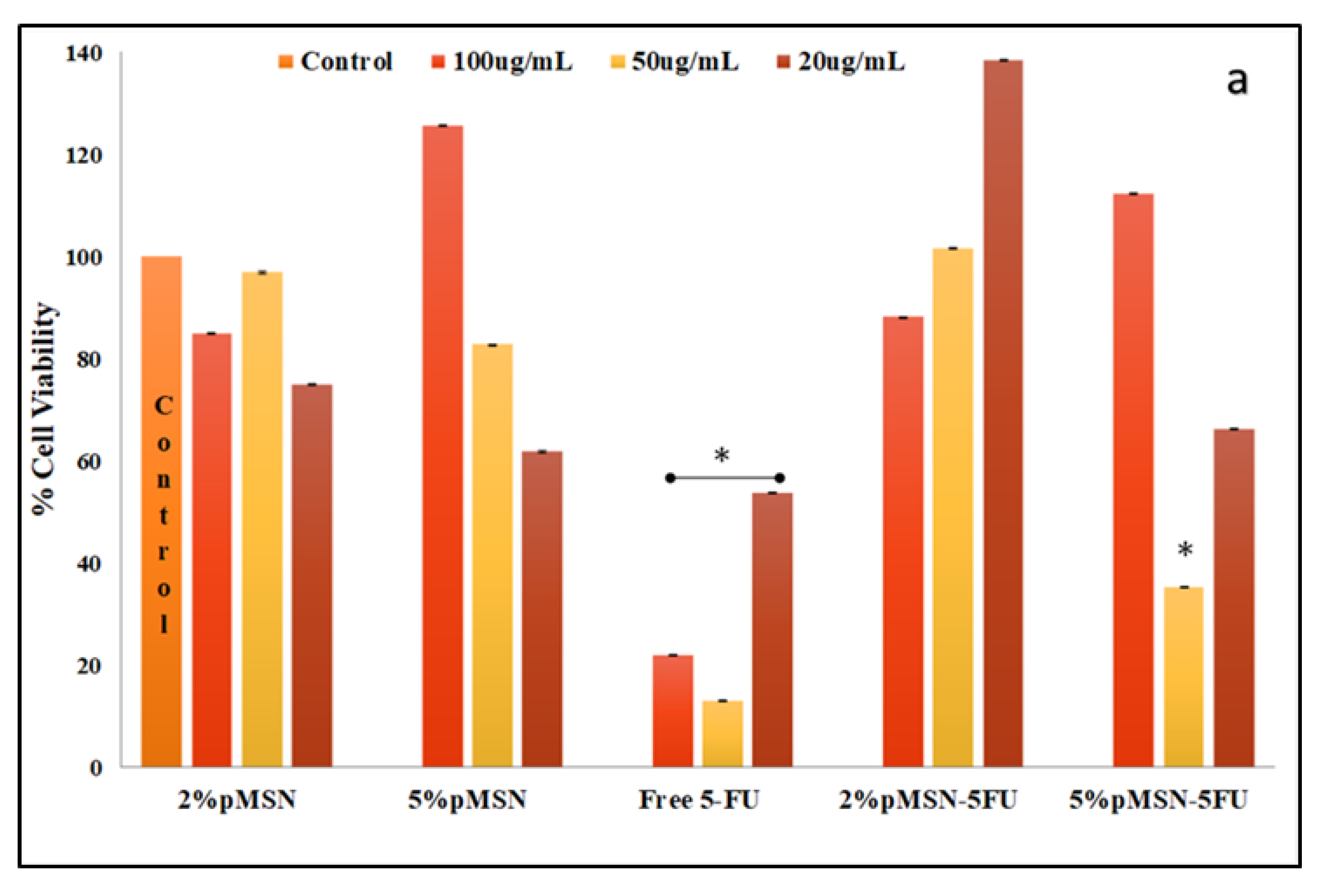
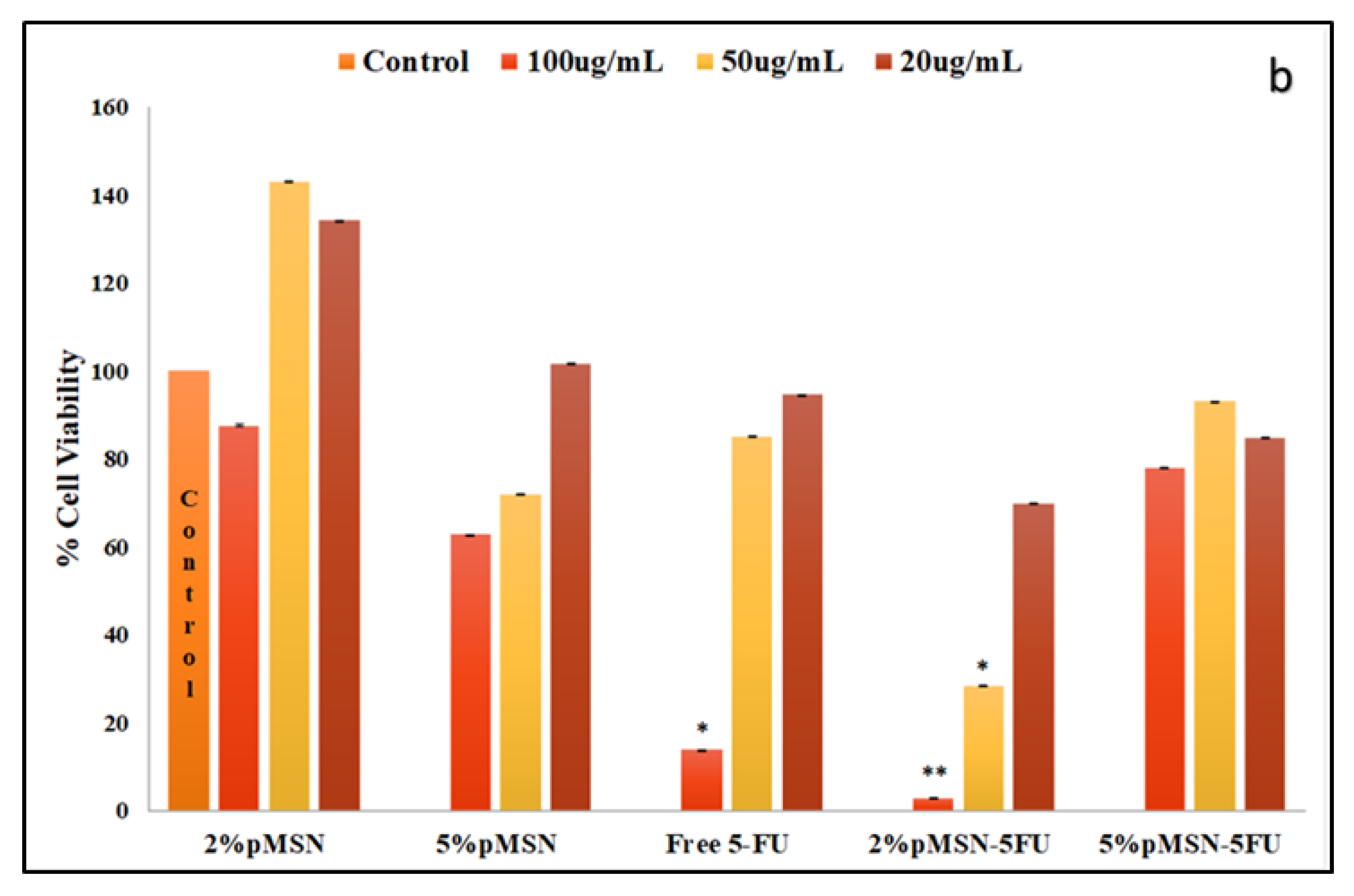
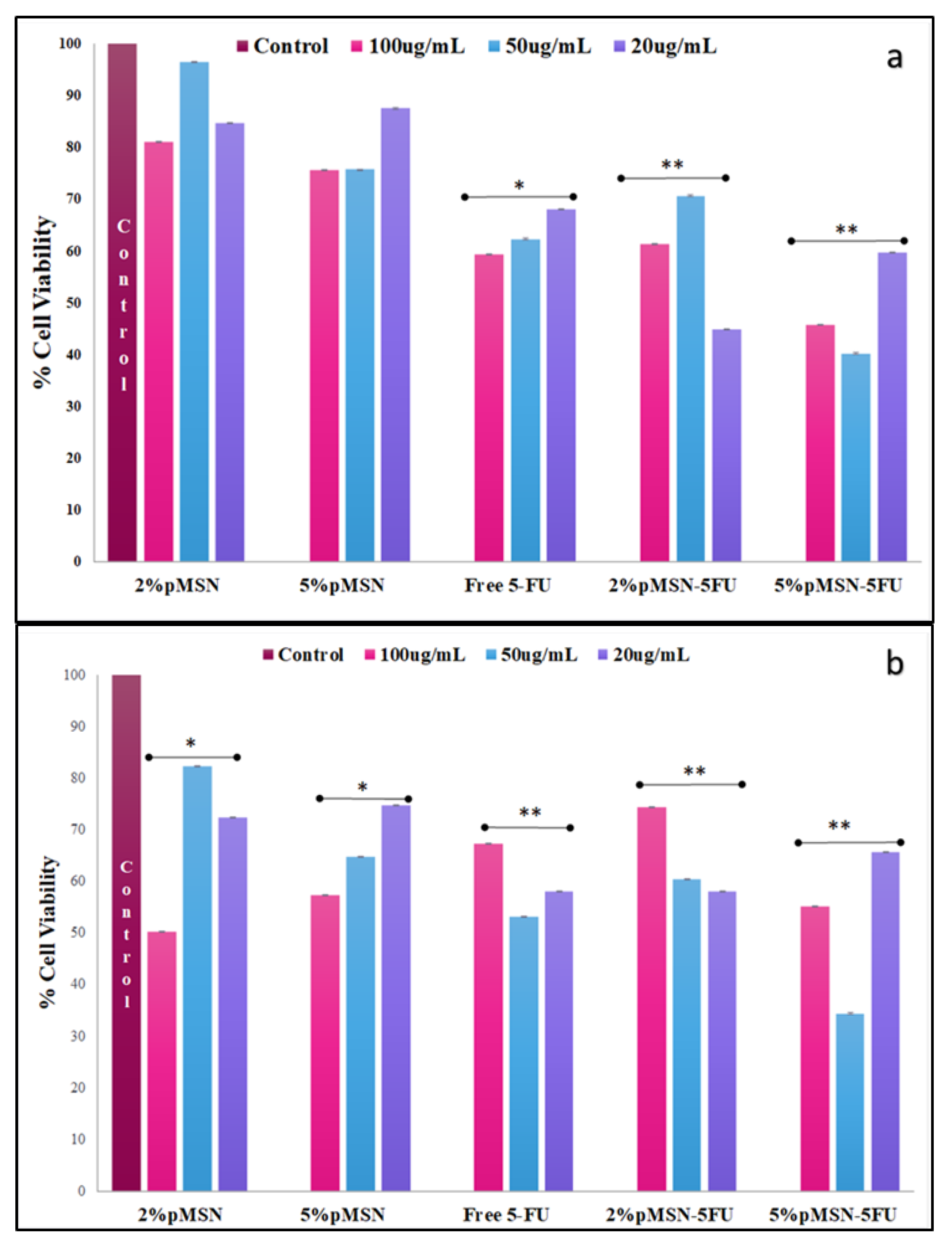
3.4. Cytotoxicity in Vitro
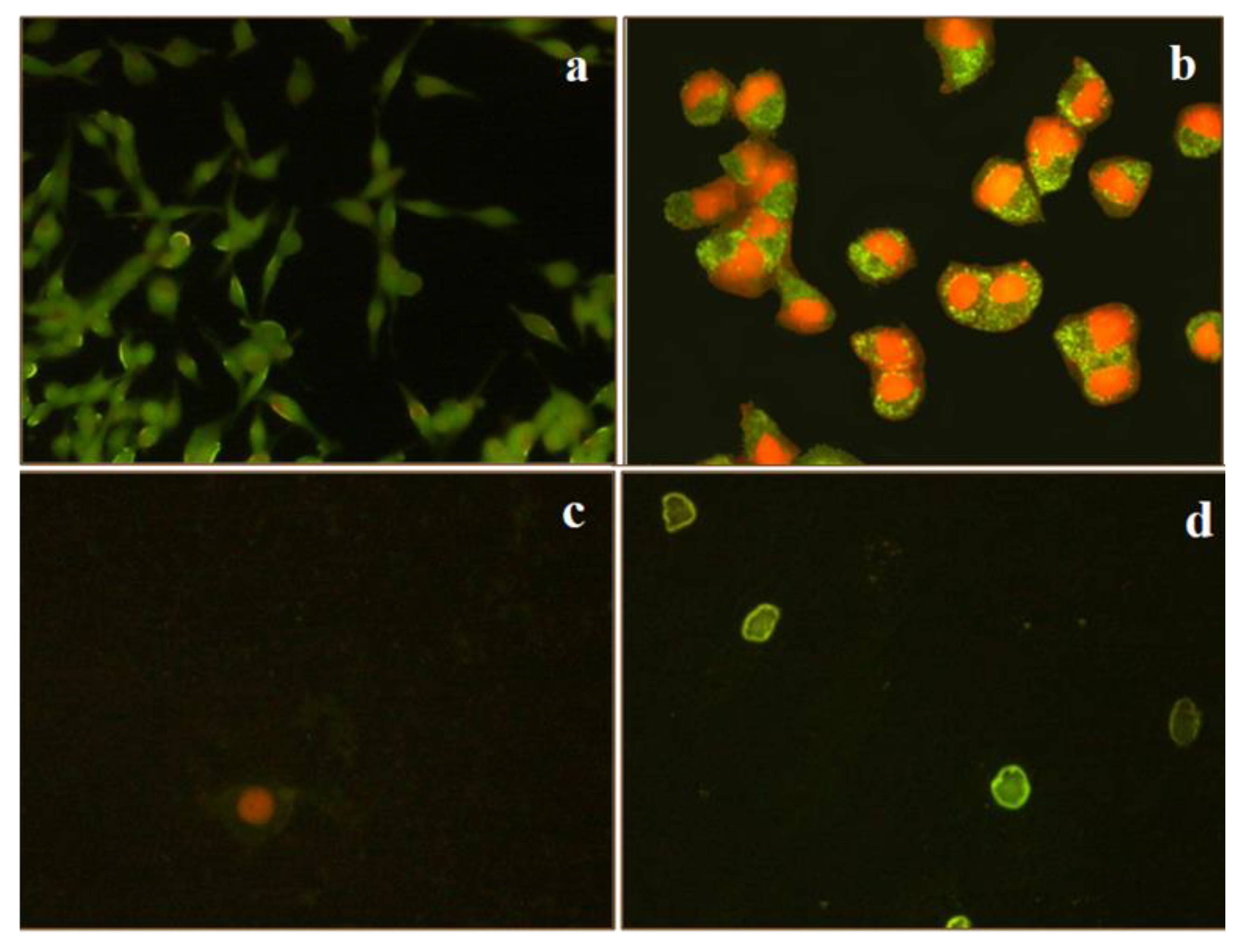
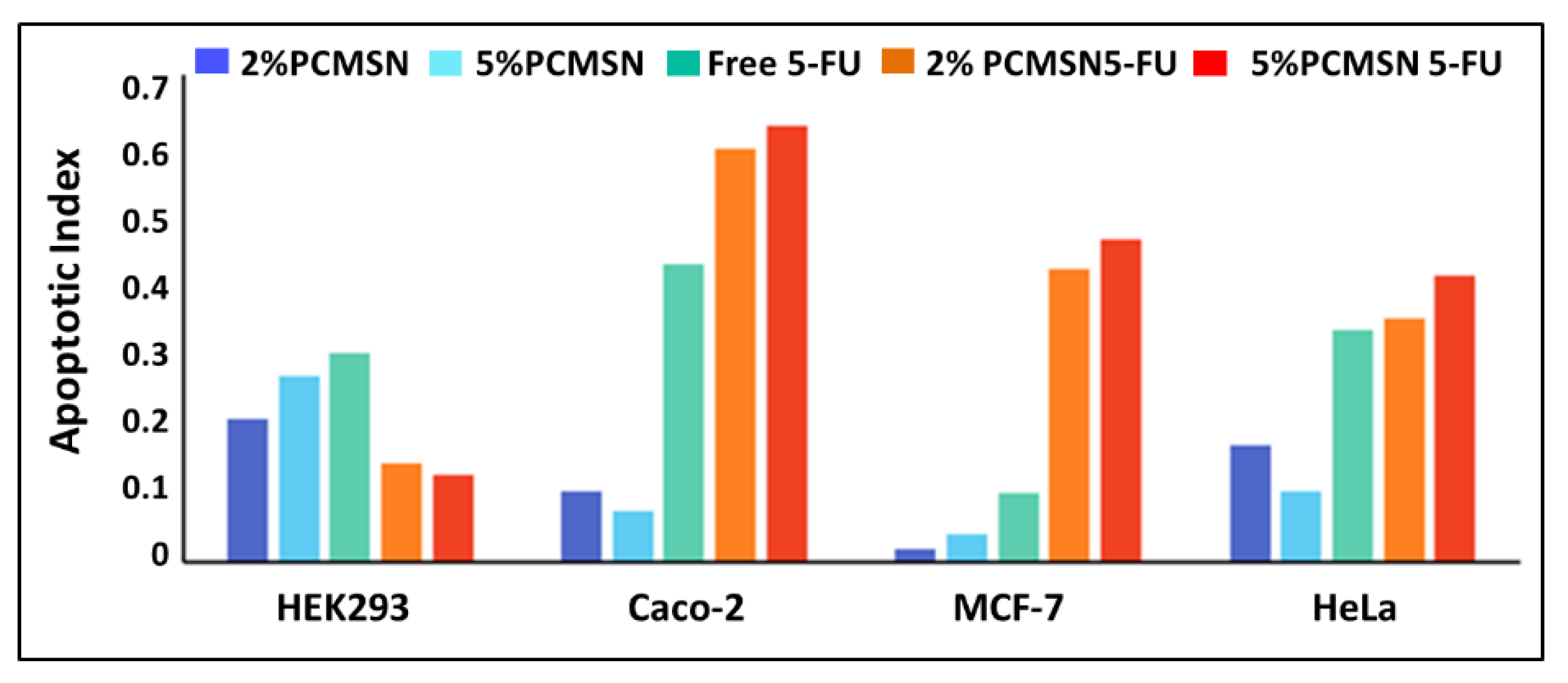
3.5. Apoptosis
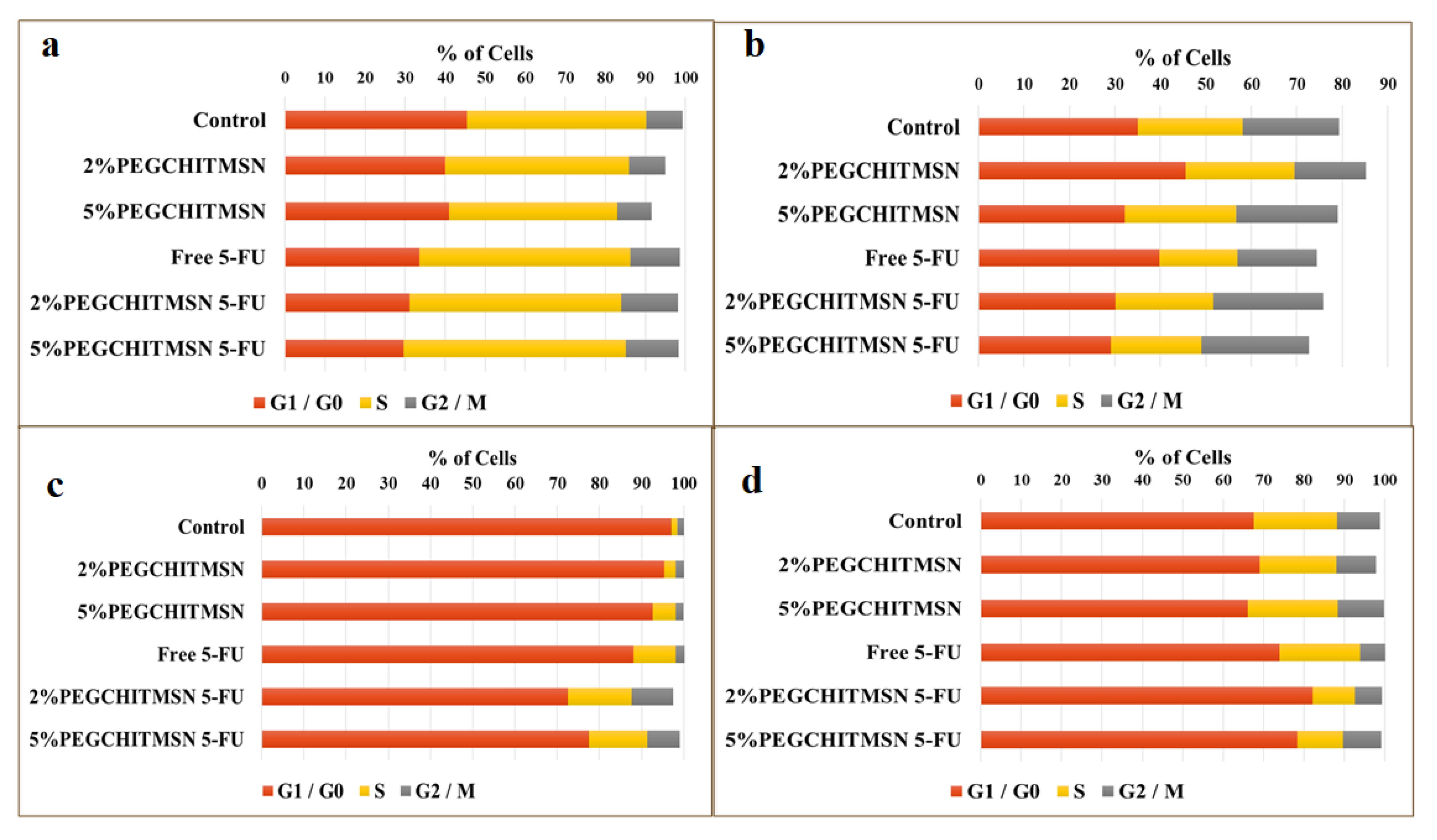
3.6. Cell Cycle Analysis
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Diasio, R.B.; Harris, B.E. Clinical Pharmacology of 5-Fluorouracil. Clin. Pharmacokinet. 1989, 16, 215–237. [Google Scholar] [CrossRef] [PubMed]
- Zoli, W.; Ulivi, P.; Tesei, A.; Fabbri, F.; Rosetti, M.; Maltoni, R.; Giunchi, D.C.; Ricotti, L.; Brigliadori, G.; Vannini, I.; et al. Addition of 5-fluorouracil to doxorubicin-paclitaxel sequence increases caspase-dependent apoptosis in breast cancer cell lines. Breast Cancer Res. 2005, 7, R681–R689. [Google Scholar] [CrossRef] [PubMed]
- Groves, T.R.; Farris, R.; Anderson, J.E.; Alexander, T.C.; Kiffer, F.; Carter, G.; Wang, J.; Boerma, M.; Allen, A.R. 5-Fluorouracil chemotherapy upregulates cytokines and alters hippocampal dendritic complexity in aged mice. Behav. Brain Res. 2017, 316, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Akinyelu, J.; Singh, M. Folate-tagged chitosan functionalized gold nanoparticles for enhanced delivery of 5-fluorouracil to cancer cells. Appl. Nanosci. 2019, 9, 7–17. [Google Scholar]
- Yoshikawa, R.; Kusunoki, M.; Yanagi, H.; Noda, M.; Furuyama, J.I.; Yamamura, T.; Hashimoto-Tamaoki, T. Dual antitumor effects of 5-fluorouracil on the cell cycle in colorectal carcinoma cells: A novel target mechanism concept for pharmacokinetic modulating chemotherapy. Cancer Res. 2001, 61, 1029–1037. [Google Scholar] [PubMed]
- Martin, M.; Villar, A.; Sole-Calvo, A.; Gonzalez, R.; Massuti, B.; Lizon, J.; Camps, C.; Carrato, A.; Casado, A.; Candel, M.T.; et al. Doxorubicin in combination with fluorouracil and cyclophosphamide (i.v. FAC regimen, day 1, 21) versus methotrexate in combination with fluorouracil and cyclophosphamide (i.v. CMF regimen, day 1, 21) as adjuvant chemotherapy for operable breast cancer: A study by the GEICAM group. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2003, 14, 833–842. [Google Scholar]
- Lopez, M.; Papaldo, P.; Di Lauro, L.; Vici, P.; Carpano, S.; Conti, E.M.S. 5-Fluorouracil, Adriamycin, Cyclophosphamide (FAC) vs. 5-Fluorouracil, Epirubicin, Cyclophosphamide (FEC) in Metastatic Breast Cancer. Oncology 1989, 46, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Van Kuilenburg, A.B.P. Dihydropyrimidine dehydrogenase and the efficacy and toxicity of 5-fluorouracil. Eur. J. Cancer 2004, 40, 939–950. [Google Scholar] [CrossRef]
- Wang, A.Z.; Langer, R.; Farokhzad, O.C. Nanoparticle Delivery of Cancer Drugs. Annu. Rev. Med. 2012, 63, 185–198. [Google Scholar] [CrossRef]
- Anselmo, A.C.; Mitragotri, S. A Review of Clinical Translation of Inorganic Nanoparticles. AAPS J. 2015, 17, 1041–1054. [Google Scholar] [CrossRef] [Green Version]
- Jahangirian, H.; Lemraski, E.G.; Webster, T.J.; Rafiee-Moghaddam, R.; Abdollahi, Y. A review of drug delivery systems based on nanotechnology and green chemistry: Green nanomedicine. Int. J. Nanomed. 2017, 12, 2957–2978. [Google Scholar] [CrossRef]
- Tang, F.; Li, L.; Chen, D. Mesoporous silica nanoparticles: Synthesis, biocompatibility and drug delivery. Adv. Mater. 2012, 24, 1504–1534. [Google Scholar] [CrossRef]
- Kwon, S.; Singh, R.K.; Perez, R.A.; Abou Neel, E.A.; Kim, H.-W.; Chrzanowski, W. Silica-based mesoporous nanoparticles for controlled drug delivery. J. Tissue Eng. 2013, 4, 2041731413503357. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhao, Q.; Han, N.; Bai, L.; Li, J.; Liu, J.; Che, E.; Hu, L.; Zhang, Q.; Jiang, T.; et al. Mesoporous silica nanoparticles in drug delivery and biomedical applications. Nanomed. Nanotechnol. Biol. Med. 2015, 11, 313–327. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Ye, M.; Park, K. Biodegradable polymers for microencapsulation of drugs. Molecules 2005, 10, 146–161. [Google Scholar] [CrossRef] [PubMed]
- Barrett, W.E.; Bianchine, J.R. The bioavailability of ultramicrosize griseofulvin (Gris-PEG) tablets in man. Curr. Ther. Res. Clin. Exp. 1975, 18, 501–509. [Google Scholar] [PubMed]
- Kumari, A.; Yadav, S.K.; Yadav, S.C. Biodegradable polymeric nanoparticles based drug delivery systems. Coll. Surf. B Biointerf. 2010, 75, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Näkki, S.; Rytkönen, J.; Nissinen, T.; Florea, C.; Riikonen, J.; Ek, P.; Zhang, H.; Santos, H.A.; Närvänen, A.; Xu, W.; et al. Improved stability and biocompatibility of nanostructured silicon drug carrier for intravenous administration. Acta Biomater. 2015, 13, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Verhoef, J.J.F.; Anchordoquy, T.J. Questioning the Use of PEGylation for Drug Delivery. Drug. Deliv. Transl. Res. 2013, 3, 499–503. [Google Scholar] [CrossRef]
- Feng, W.; Zhou, X.; He, C.; Qiu, K.; Nie, W.; Chen, L.; Wang, H.; Mo, X.; Zhang, Y. Polyelectrolyte multilayer functionalized mesoporous silica nanoparticles for pH-responsive drug delivery: Layer thickness-dependent release profiles and biocompatibility. J. Mater. Chem. B 2013, 1, 5886. [Google Scholar] [CrossRef]
- Zhao, Y.; Sun, X.; Zhang, G.; Trewyn, B.G.; Slowing, I.I.; Lin, V.S.-Y. Interaction of Mesoporous Silica Nanoparticles with Human Red Blood Cell Membranes: Size and Surface Effects. ACS Nano 2011, 5, 1366–1375. [Google Scholar] [CrossRef] [Green Version]
- Gref, R.; Lück, M.; Quellec, P.; Marchand, M.; Dellacherie, E.; Harnisch, S.; Blunk, T.; Müller, R.H. “Stealth” corona-core nanoparticles surface modified by polyethylene glycol (PEG): Influences of the corona (PEG chain length and surface density) and of the core composition on phagocytic uptake and plasma protein adsorption. Coll. Surf. B. Biointerf. 2000, 18, 301–313. [Google Scholar] [CrossRef]
- Yin Win, K.; Feng, S.-S. Effects of particle size and surface coating the cellular uptake of polymeric nanoparticles. Biomaterials 2005, 26, 2713–2722. [Google Scholar] [CrossRef] [PubMed]
- Lu, F.; Wu, S.-H.; Hung, Y.; Mou, C.-Y. Size Effect on Cell Uptake in Well-Suspended, Uniform Mesoporous Silica Nanoparticles. Small 2009, 5, 1408–1413. [Google Scholar] [CrossRef] [PubMed]
- Tourne-Peteilh, C.; Bégu, S.; Lerner, D.; Galarneau, A.; Lafont, U.; Devoisselle, J.-M. Sol–gel one-pot synthesis in soft conditions of mesoporous silica materials ready for drug delivery system. J. Sol-Gel Sci. Technol. 2012, 61, 455–462. [Google Scholar] [CrossRef]
- Vazquez, N.I.; Gonzalez, Z.; Ferrari, B.; Castro, Y. Synthesis of mesoporous silica nanoparticles by sol–gel as nanocontainer for future drug delivery applications. Boletín la Soc. Española Cerámica y Vidr. 2017, 56, 139–145. [Google Scholar] [CrossRef]
- Hu, Y.; Ke, L.; Chen, H.; Zhuo, M.; Yang, X.; Zhao, D.; Zeng, S.; Xiao, X. Natural material-decorated mesoporous silica nanoparticle container for multifunctional membrane-controlled targeted drug delivery. Int. J. Nanomed. 2017, 12, 8411–8426. [Google Scholar] [CrossRef]
- Wang, J.; Liu, H.; Leng, F.; Zheng, L.; Yang, J.; Wang, W.; Huang, C.Z. Autofluorescent and pH-responsive mesoporous silica for cancer-targeted and controlled drug release. Microporous Mesoporous Mater. 2014, 186, 187–193. [Google Scholar] [CrossRef]
- She, X.; Chen, L.; Velleman, L.; Li, C.; Zhu, H.; He, C.; Wang, T.; Shigdar, S.; Duan, W.; Kong, L. Fabrication of high specificity hollow mesoporous silica nanoparticles assisted by Eudragit for targeted drug delivery. J. Coll. Interf. Sci. 2015, 445, 151–160. [Google Scholar] [CrossRef]
- Barrett, E.P.; Joyner, L.G.; Halenda, P. The determination of pore volume and area distribution in porous substances. Vol. Area Distrib. Porous Subst. 1951, 73, 373–380. [Google Scholar]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Dash, S.; Murthy, P.; Nath, L.; Chowdhury, P. Kinetic modeling on drug release from controlled drug delivery systems. Acta Pol. Pharm. 2010, 67, 217–223. [Google Scholar] [PubMed]
- Higuchi, T. Mechanism of sustained-action medication. Theoretical analysis of rate of release of solid drugs dispersed in solid matrices. J. Pharm. Sci. 1963, 52, 1145–1149. [Google Scholar] [CrossRef] [PubMed]
- Hixson, A.W.; Crowell, J.H. Dependence of Reaction Velocity upon surface and Agitation. Ind. Eng. Chem. 1931, 23, 923–931. [Google Scholar] [CrossRef]
- Korsmeyer, R.W.; Lustig, S.R.; Peppas, N.A. Solute and penetrant diffusion in swellable polymers. I. Mathematical modeling. J. Polym. Sci. Part B Polym. Phys. 1986, 24, 395–408. [Google Scholar] [CrossRef]
- Kopcha, M.; Tojo, K.; Lordi, N.G. Evaluation of methodology for assessing release characteristics of thermosoftening vehicles. J. Pharm. Pharmacol. 1990, 42, 745–751. [Google Scholar] [CrossRef] [PubMed]
- Sankalia, J.M.; Sankalia, M.G.; Mashru, R.C. Drug release and swelling kinetics of directly compressed glipizide sustained-release matrices: Establishment of level A IVIVC. J. Control. Rel. 2008, 129, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Mulye, N.V.; Turco, S.J. A Simple Model Based on First Order Kinetics to Explain Release of Highly Water Soluble Drugs from Porous Dicalcium Phosphate Dihydrate Matrices. Drug Dev. Ind. Pharm. 1995, 21, 943–953. [Google Scholar] [CrossRef]
- Korsmeyer, R.W.; Gurny, R.; Doelker, E.; Buri, P.; Peppas, N.A. Mechanisms of solute release from porous hydrophilic polymers. Int. J. Pharm. 1983, 15, 25–35. [Google Scholar] [CrossRef]
- Kopcha, M.; Lordi, N.G.; Tojo, K.J. Evaluation of Release from Selected Thermosoftening Vehicles. J. Pharm. Pharmacol. 1991, 43, 382–387. [Google Scholar] [CrossRef]
- Anirudhan, T.S.; Vasantha, C.S.; Sasidharan, A.V. Layer-by-layer assembly of hyaluronic acid/carboxymethylchitosan polyelectrolytes on the surface of aminated mesoporous silica for the oral delivery of 5-fluorouracil. Eur. Polym. J. 2017, 93, 572–589. [Google Scholar] [CrossRef]
- Werschler, W.P. Considerations for use of Fluorouracil cream 0.5% for the treatment of actinic keratosis in elderly patients. J. Clin. Aesthet. Dermatol. 2008, 1, 22–27. [Google Scholar] [PubMed]
- Grem, J.L. 5-Fluorouracil: Forty-Plus and Still Ticking. A Review of its Preclinical and Clinical Development. Invest. New Drugs 2000, 18, 299–313. [Google Scholar] [CrossRef] [PubMed]
- Montagnoli, A.; Valsasina, B.; Croci, V.; Menichincheri, M.; Rainoldi, S.; Marchesi, V.; Tibolla, M.; Tenca, P.; Brotherton, D.; Albanese, C.; et al. A Cdc7 kinase inhibitor restricts initiation of DNA replication and has antitumor activity. Nat. Chem. Biol. 2008, 4, 357–365. [Google Scholar] [CrossRef] [PubMed]
- Scartozzi, M.; Maccaroni, E.; Giampieri, R.; Pistelli, M.; Bittoni, A.; Del Prete, M.; Berardi, R.; Cascinu, S. 5-fluorouracil pharmacogenomics: Still rocking after all these years? Pharmacogenomics 2011, 12, 251–265. [Google Scholar] [CrossRef] [PubMed]
- Jarzembska, K.N.; Kubsik, M.; Kamiński, R.; Woźniak, K.; Dominiak, P.M. From a Single Molecule to Molecular Crystal Architectures: Structural and Energetic Studies of Selected Uracil Derivatives. Cryst. Growth Des. 2012, 12, 2508–2524. [Google Scholar] [CrossRef]
- Barnett, S.A.; Hulme, A.T.; Tocher, D.A. 5-Fluorouracil and thymine form a crystalline solid solution. Acta Crystallogr. Sect. C Cryst. Struct. Commun. 2006, 62, o412–o415. [Google Scholar] [CrossRef] [Green Version]
- Hulme, A.T.; Price, S.L.; Tocher, D.A. A New Polymorph of 5-Fluorouracil Found Following Computational Crystal Structure Predictions. J. Am. Chem. Soc. 2005, 127, 1116–1117. [Google Scholar] [CrossRef]
- Fischer, J.; Ganellin, C.R. Analogue-Based Drug Discovery; Wiley-VCH: Weinheim, Germany, 2006. [Google Scholar]
- Pullarkat, S.T.; Stoehlmacher, J.; Ghaderi, V.; Xiong, Y-P.; Ingles, S.A.; Sherrod, A.; Warren, R.; Tsao-Wei, D.; Groshen, S.; Lenz, H-J. Thymidylate synthase gene polymorphism determines response and toxicity of 5-FU chemotherapy. Pharmacogenomics J. 2001, 1, 65–70. [Google Scholar] [CrossRef] [Green Version]
- Sarker, D. Engineering of Nanoemulsions for Drug Delivery. Curr. Drug Deliv. 2005, 2, 297–310. [Google Scholar] [CrossRef]
- Elsabahy, M.; Wooley, K.L. Design of polymeric nanoparticles for biomedical delivery applications. Chem. Soc. Rev. 2012, 41, 2545–2561. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Thanou, M. Targeting nanoparticles to cancer. Pharmacol. Res. 2010, 62, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Shi, J.; Shen, W.; Dong, X.; Feng, J.; Ruan, M.; Li, Y. Stimuli-Responsive Controlled Drug Release from a Hollow Mesoporous Silica Sphere/Polyelectrolyte Multilayer Core–Shell Structure. Angewandte Chemie. 2005, 44, 5083–5087. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Yu, H.; Xiao, C. pH-Sensitive Cationic Guar Gum/Poly (Acrylic Acid) Polyelectrolyte Hydrogels: Swelling and In Vitro Drug Release. Carbohydrate Polymers. 2007, 69, 774–783. [Google Scholar] [CrossRef]
- Yang, Y.; Tao, X.; Hou, Q.; Ma, Y.; Chen, X.-L.; Chen, J.-F. Mesoporous Silica Nanotubes Coated with Multilayered Polyelectrolytes for pH-Controlled Drug Release. Acta Biomaterialia 2010, 6, 3092–3100. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, A.R.; Soppimath, K.S.; Aminabhavi, T.M.; Rudzinski, W.E. In-vitro release kinetics of cefadroxil-loaded sodium alginate interpenetrating network beads. Eur. J. Pharm. Biopharm. 2001, 51, 127–133. [Google Scholar] [CrossRef]
- Barzegar-Jalali, M.; Adibkia, K.; Valizadeh, H.; Reza, M.; Shadbad, S.; Nokhodchi, A.; Omidi, Y.; Mohammadi, G.; Nezhadi, S.H.; Hasan, M. Kinetic Analysis of Drug Release from Nanoparticles. J. Pharm. Pharm. Sci. 2008, 11, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Kao, W.J. Drug release kinetics and transport mechanisms of non-degradable and degradable polymeric delivery systems. Expert Opin. Drug Deliv. 2010, 7, 429–444. [Google Scholar] [CrossRef]
- Costa, P.; Sousa Lobo, J.M. Evaluation of mathematical models describing drug release from estradiol transdermal systems. Drug Dev. Ind. Pharm. 2003, 29, 89–97. [Google Scholar] [CrossRef]
- Ritger, P.L.; Peppas, N.A. A simple equation for description of solute release II. Fickian and anomalous release from swellable devices. J. Control. Rel. 1987, 5, 37–42. [Google Scholar] [CrossRef]
- Chen, T.; Wu, W.; Xiao, H.; Chen, Y.; Chen, M.; Li, J. Intelligent Drug Delivery System Based on Mesoporous Silica Nanoparticles Coated with an Ultra-pH-Sensitive Gatekeeper and Poly(ethylene glycol). ACS Macro Lett. 2016, 5, 55–58. [Google Scholar] [CrossRef]
- Gbureck, U.; Vorndran, E.; Müller, F.A.; Barralet, J.E. Low temperature direct 3D printed bioceramics and biocomposites as drug release matrices. J. Control. Rel. 2007, 122, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Watermann, A.; Brieger, J. Mesoporous Silica Nanoparticles as Drug Delivery Vehicles in Cancer. Nanomaterials 2017, 7, 189. [Google Scholar] [CrossRef] [PubMed]
- Kusaczuk, M.; Krętowski, R.; Naumowicz, M.; Stypułkowska, A.; Cechowska-Pasko, M. Silica nanoparticle-induced oxidative stress and mitochondrial damage is followed by activation of intrinsic apoptosis pathway in glioblastoma cells. Int. J. Nanomed. 2018, 13, 2279–2294. [Google Scholar] [CrossRef] [PubMed]
- Babaei, M.; Abnous, K.; Taghdisi, S.M.; Amel Farzad, S.; Peivandi, M.T.; Ramezani, M.; Alibolandi, M. Synthesis of theranostic epithelial cell adhesion molecule targeted mesoporous silica nanoparticle with gold gatekeeper for hepatocellular carcinoma. Nanomedicine 2017, 12, 1261–1279. [Google Scholar] [CrossRef] [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nanoparticle | Mean Diameter (TEM) (nm ± Standard Deviation) | PDI (SD/mean)2 | Hydrodynamic Size (Nanoparticle Tracking Analysis) (nm ± SD) | Zeta Potential (mV) |
|---|---|---|---|---|
| MSN | 36.09 ± 7.08 | 0.0385 | 188 ± 51.6 | −9.8 ± 1 |
| CHITOSAN (CHIT)-MSN | 39.43 ± 7.22 | 0.0335 | 62.2 ± 16 | 32.4 ± 0.4 |
| 2% Polyethylene glycol (PEG)-CHIT-MSN | 40.75 ± 7.11 | 0.0422 | 12 ± 3.3 | 17.0 ± 16.5 |
| 5% PEG-CHIT-MSN | 40.37 ± 7.70 | 0.0364 | 54.8 ± 2.1 | 7.4 ± 0.7 |
| 2% PEG-CHIT-MSN-5FU | 48.32 ± 8.20 | 0.0287 | 54.8 ± 40.7 | 11.2 ± 7.0 |
| 5% PEG-CHIT-MSN-5FU | 64.54 ± 25.11 | 0.1514 | 47.4 ± 5.5 | 3.4 ± 0.0 |
| Surface Area | |
| Brunauer-Emmet-Teller (BET) surface area | 809.4447 m2/g |
| t-Plot micropore area | 162.2294 m2/g |
| t-Plot external surface area | 647.2153 m2/g |
| Barrett-Joyner-Halenda (BJH) adsorption cumulative surface area of pores between 17.000 Å and 3000.000 Å diameter | 680.013 m2/g |
| BJH desorption cumulative surface area of pores between 17.000 Å and 3000.000 Å diameter | 710.3616 m2/g |
| Pore Volume | |
| Single point adsorption total pore volume of pores less than 1047.206 Å diameter at P/Po = 0.981156675 | 1.529764 cm3/g |
| t-Plot micropore volume | 0.066025 cm3/g |
| BJH adsorption cumulative volume of pores between 17.000 Å and 3000.000 Å diameter | 1.726356 cm3/g |
| BJH desorption cumulative volume of pores between 17.000 Å and 3000.000 Å diameter | 1.743321 cm3/g |
| Pore Size | |
| Adsorption average pore width (4V/A by BET) | 75.5957 Å |
| BJH adsorption average pore diameter (4V/A) | 101.548 Å |
| BJH desorption average pore diameter (4V/A) | 98.165 Å |
| Average particle size | 74.125 Å |
| 5-FU Loaded MSNs | ||
|---|---|---|
| 5% PEG-CHIT-MSN | 2% PEG-CHIT-MSN | |
| Loading capacity (%) | 18.02 | 15.02 |
| Loading capacity (mg 5-FU/mg f-MSN) | 0.1802 | 0.1502 |
| pH | Zero-Order | First-Order | Higuchi’s | Hixson-Crowell’s | Korsmeyer-Peppas’ | Kopcha’s |
|---|---|---|---|---|---|---|
| Correlation value (R2) | ||||||
| 4.2 | 0.81 | 0.55 | 0.91 | 0.27 | 0.95 | 0.98 |
| 7.4 | 0.35 | 0.26 | 0.48 | 0.64 | 0.65 | 0.16 |
| pH | Zero-Order | First-Order | Higuchi’s | Hixson-Crowell’s | Korsmeyer-Peppas’ | Kopcha’s |
|---|---|---|---|---|---|---|
| Correlation value (R2) | ||||||
| 4.2 | 0.72 | 0.44 | 0.85 | 0.72 | 0.91 | 0.97 |
| 7.4 | 0.69 | 0.29 | 0.81 | 0.51 | 0.75 | 0.70 |
| pH 4.2 | ||||
| Multidrug Formulation | Korsmeyer-Peppas Model | Kopcha Model | ||
| n-value | A | B | A/B | |
| 2% PEG-CHIT-MSN | 0.39 | 2.97 | 0.13 | 22.85 |
| 5% PEG-CHIT-MSN | 0.32 | 3.76 | 0.27 | 13.93 |
| pH 7.4 | ||||
| Multidrug Formulation | Korsmeyer-Peppas Model | Kopcha Model | ||
| n-value | A | B | A/B | |
| 2% PEG-CHIT-MSN | 0.74 | 5.16 | 2.02 | 2.55 |
| 5% PEG-CHIT-MSN | 0.35 | 6.87 | 0.16 | 42.94 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moodley, T.; Singh, M. Polymeric Mesoporous Silica Nanoparticles for Enhanced Delivery of 5-Fluorouracil In Vitro. Pharmaceutics 2019, 11, 288. https://doi.org/10.3390/pharmaceutics11060288
Moodley T, Singh M. Polymeric Mesoporous Silica Nanoparticles for Enhanced Delivery of 5-Fluorouracil In Vitro. Pharmaceutics. 2019; 11(6):288. https://doi.org/10.3390/pharmaceutics11060288
Chicago/Turabian StyleMoodley, Thashini, and Moganavelli Singh. 2019. "Polymeric Mesoporous Silica Nanoparticles for Enhanced Delivery of 5-Fluorouracil In Vitro" Pharmaceutics 11, no. 6: 288. https://doi.org/10.3390/pharmaceutics11060288