Recent Progress in the Development of Poly(lactic-co-glycolic acid)-Based Nanostructures for Cancer Imaging and Therapy



Abstract
:1. Introduction
2. Preparation and Modification of PLGA-Based NPs
2.1. Preparation of PLGA NPs
2.1.1. Emulsification-Evaporation Method
2.1.2. Nanoprecipitation Method
2.1.3. Spray-Drying Method
2.1.4. Microfluidics Method
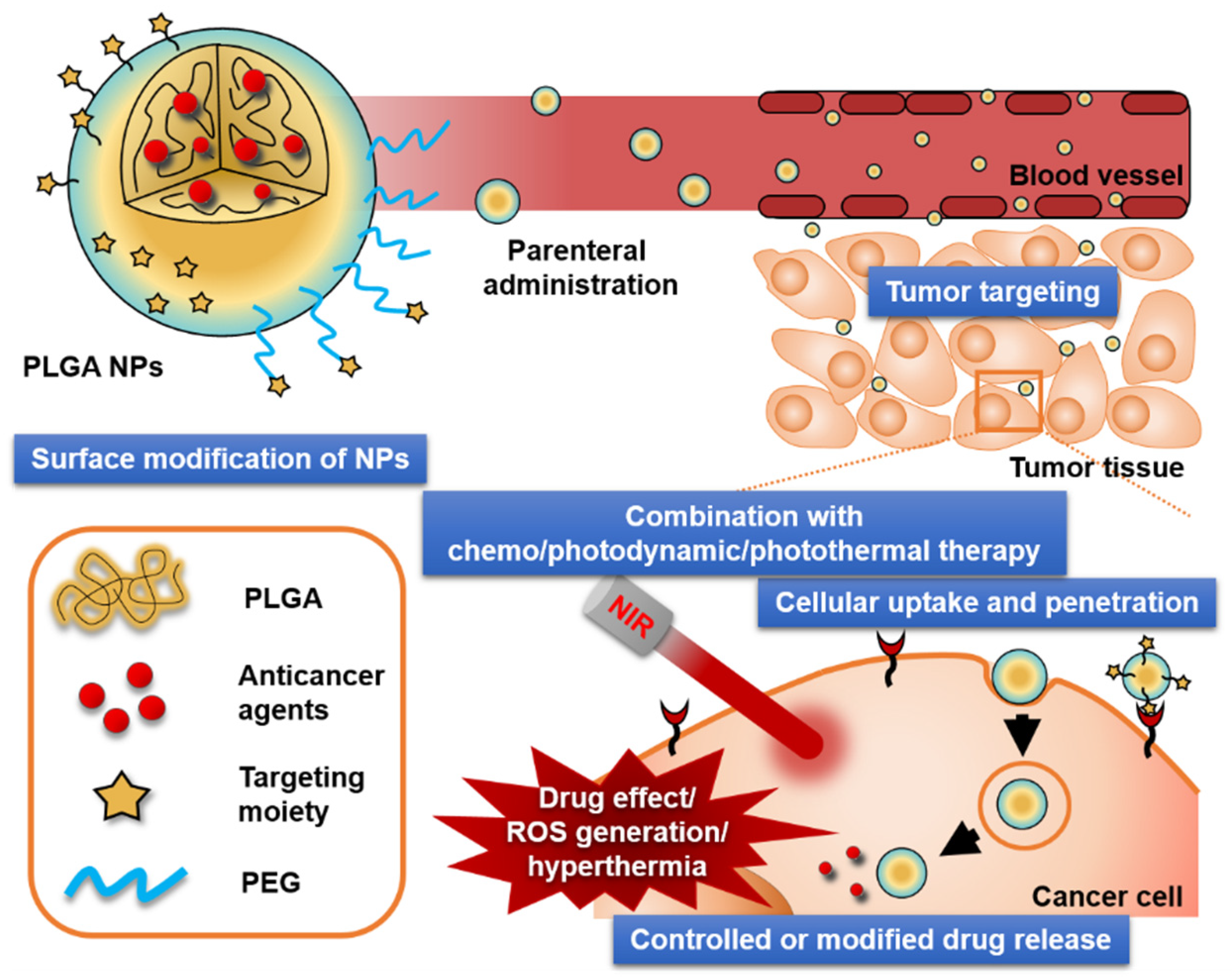
2.2. Surface Modification of PLGA NPs for Tumor Targeting
2.2.1. Physicochemical and Biological Properties of PLGA
2.2.2. Surface Engineering of PLGA-Based NPs
3. Uptake of PLGA NPs into Cancer Cells
3.1. Receptor-Mediated Endocytosis
3.2. Carrier-Mediated Endocytosis
3.3. Adsorption-Mediated Endocytosis
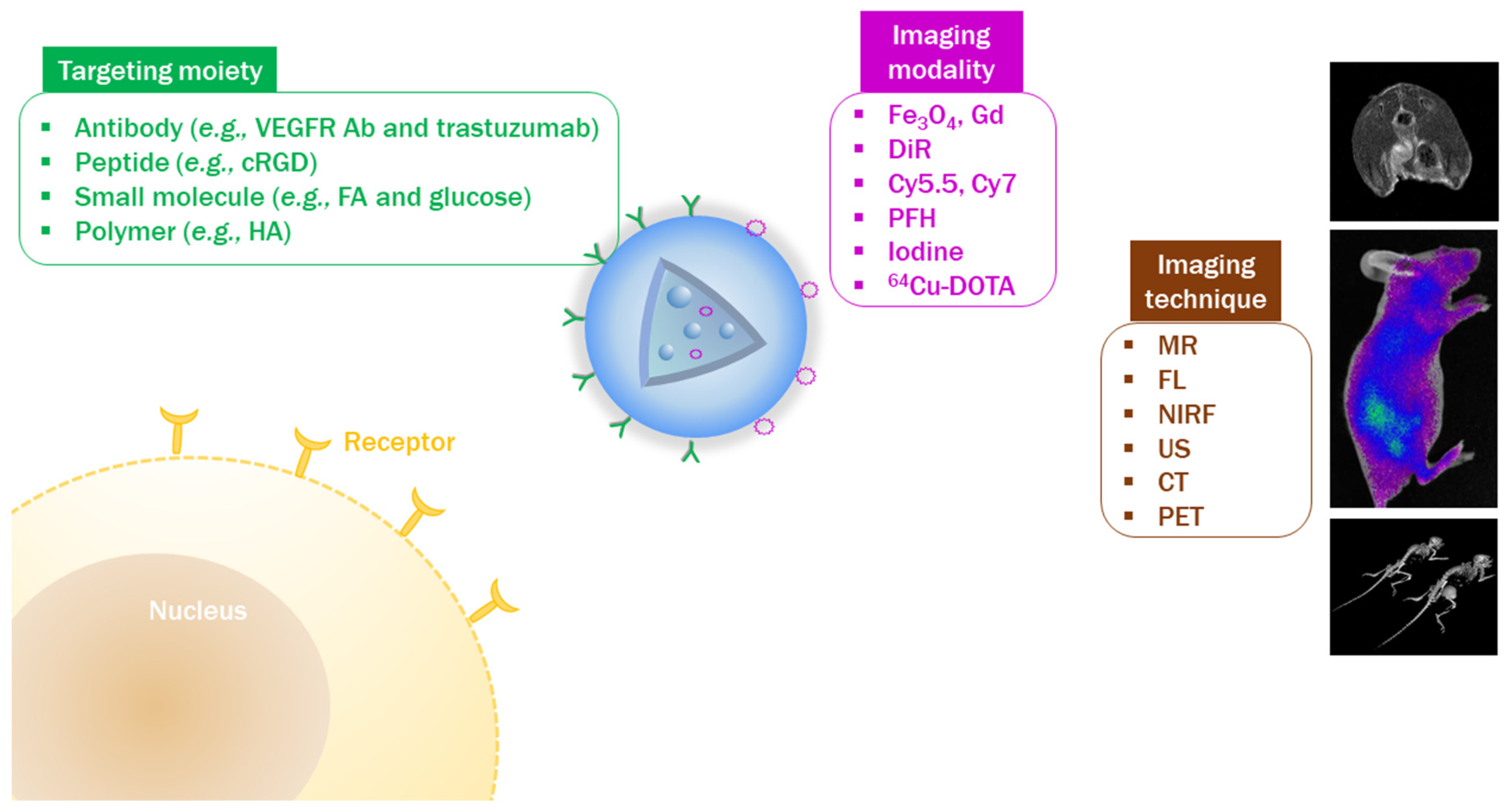
4. Visualization of Tumor Targeting by In Vivo Imaging
4.1. Design of NPs with Imaging Modalities
4.2. Introduction of Tumor-Targeting Ligands to PLGA NPs
4.3. Verification of Tumor Targetability by In Vivo Imaging
5. In Vivo Anticancer Activities of PLGA-based NPs
5.1. Parameters for Assessing In Vivo Antitumor Activity
5.2. Key Factors for Improved Antitumor Efficacy
5.2.1. Controlled Release and EPR Effect
5.2.2. Targeted Delivery
5.2.3. Cellular Uptake and Penetration
5.2.4. Combination with Photodynamic Therapy (PDT) and/or PTT
6. In Vivo Pharmacokinetics of PLGA-Based NPs
7. Current Status and Challenges of PLGA-Based Formulations for Clinical Applications
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Choi, Y.H.; Han, H.K. Nanomedicines: Current status and future perspectives in aspect of drug delivery and pharmacokinetics. J. Pharm. Investig. 2018, 48, 43–60. [Google Scholar] [CrossRef] [PubMed]
- Elzoghby, A.O.; Hemasa, A.L.; Freag, M.S. Hybrid protein-inorganic nanoparticles: From tumor-targeted drug delivery to cancer imaging. J. Control. Release 2016, 243, 303–322. [Google Scholar] [CrossRef] [PubMed]
- Fan, W.; Yung, B.C.; Chen, X. Stimuli-responsive NO release for on-demand gas-sensitized synergistic cancer therapy. Angew. Chem. Int. Ed. 2018, 57, 8383–8394. [Google Scholar] [CrossRef] [PubMed]
- Fang, R.H.; Jiang, Y.; Fang, J.C.; Zhang, L. Cell membrane-derived nanomaterials for biomedical applications. Biomaterials 2017, 128, 69–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, M.; Yu, F.; Lv, C.; Choo, J.; Chen, L. Fluorescent chemical probes for accurate tumor diagnosis and targeting therapy. Chem. Soc. Rev. 2017, 46, 2237–2271. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.; Zhu, S.; Yan, L.; Zhao, F.; Zhao, Y. Graphene-based smart platforms for combined cancer therapy. Adv. Mater. 2019, 31, 1800662. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; You, J. Near infrared light-controlled therapeutic molecules release of nanocarriers in cancer therapy. J. Pharm. Investig. 2017, 47, 297–316. [Google Scholar] [CrossRef]
- Hu, Y.; Mignani, S.; Majoral, J.P.; Shen, M.; Shi, X. Construction of iron oxide nanoparticle-based hybrid platforms for tumor imaging and therapy. Chem. Soc. Rev. 2018, 47, 1874–1900. [Google Scholar] [CrossRef]
- Guan, Q.; Li, Y.A.; Li, W.Y.; Dong, Y.B. Photodynamic therapy based on nanoscale metal-organic frameworks: From material design to cancer nanotherapeutics. Chem. Asian J. 2018, 13, 3122–3149. [Google Scholar] [CrossRef]
- Jeon, G.; Ko, Y.T. Enhanced photodyamic therapy via photosensitizer-loaded nanoparticles for cancer treatment. J. Pharm. Investig. 2019, 49, 1–8. [Google Scholar] [CrossRef]
- Le, Q.V.; Choi, J.; Oh, Y.K. Nano delivery systems and cancer immunotherapy. J. Pharm. Investig. 2018, 48, 527–539. [Google Scholar] [CrossRef]
- Lee, M.K. Clinical usefulness of liposomal formulations in cancer therapy: Lessons from the experiences of doxorubicin. J. Pharm. Investig. 2019, 49, 203–214. [Google Scholar] [CrossRef]
- Liu, Y.; Bhattarai, P.; Dai, Z.; Chen, X. Photothermal therapy and photoacoustic imaging via nanotheranostics in fighting cancer. Chem. Soc. Rev. 2019, 48, 2053–2108. [Google Scholar] [CrossRef] [PubMed]
- Raichur, V.; Devi, V.K. Formulation and evaluation of osteotropic drug delivery system of methotrexate with a potential for passive bone targeting. J. Pharm. Investig. 2017, 47, 335–347. [Google Scholar] [CrossRef]
- Sobh, R.A.; Nasr, H.E.; Moustafa, A.B.; Mohamed, W.S. Tailoring of anticancer drugs loaded in MWCNT/Poly(MMA-co-HEMA) nanosphere composite by using in situ microemulsion polymerization. J. Pharm. Investig. 2019, 49, 45–55. [Google Scholar] [CrossRef]
- Sun, Q.; Barz, M.; De Geest, B.G.; Diken, M.; Hennink, W.E.; Kiessling, F.; Lammers, T.; Shi, Y. Nanomedicine and macroscale materials in immuno-oncology. Chem. Soc. Rev. 2019, 48, 351–381. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Zheng, Y.; Chen, Y. Materials chemistry of nanoultrasonic biomedicine. Adv. Mater. 2017, 29, 1604105. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.L.; Tang, W.H.; Li, S.D. Cancer theranostic applications of lipid-based nanoparticles. Drug Discov. Today 2018, 23, 1159–1166. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Li, X.; Tse, B.W.; Yang, H.; Thorling, C.A.; Liu, Y.; Touraud, M.; Chouane, J.B.; Liu, X.; Roberts, M.S.; et al. Indocyanine green-incorporating nanoparticles for cancer theranostics. Theranostics 2018, 8, 1227–1242. [Google Scholar] [CrossRef]
- Youn, Y.S.; Kwag, D.S.; Lee, E.S. Multifunctional nano-sized fullerenes for advanced tumor therapy. J. Pharm. Investig. 2017, 47, 1–10. [Google Scholar] [CrossRef]
- Golombek, S.K.; May, J.N.; Theek, B.; Appold, L.; Drude, N.; Kiessling, F.; Lammers, T. Tumor targeting via EPR: Strategies to enhance patient responses. Adv. Drug Deliv. Rev. 2018, 130, 17–38. [Google Scholar] [CrossRef] [PubMed]
- Maeda, H. Macromolecular therapeutics in cancer treatment: The EPR effect and beyond. J. Control. Release 2012, 164, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Maeda, H.; Wu, J.; Sawa, T.; Matsumura, Y.; Hori, K. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: A review. J. Control. Release 2000, 65, 271–284. [Google Scholar] [CrossRef]
- Nichols, J.W.; Bae, Y.H. EPR: Evidence and fallacy. J. Control. Release 2014, 190, 451–464. [Google Scholar] [CrossRef] [PubMed]
- Maeda, H.; Nakamura, H.; Fang, J. The EPR effect for macromolecular drug delivery to solid tumors: Improvement of tumor uptake, lowering of systemic toxicity, and distinct tumor imaging in vivo. Adv. Drug Deliv. Rev. 2013, 65, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Nakamura, H.; Maeda, H. The EPR effect: Unique features of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect. Adv. Drug Deliv. Rev. 2011, 63, 136–151. [Google Scholar] [CrossRef] [PubMed]
- Danhier, F.; Feron, O.; Préat, V. To exploit the tumor microenvironment: Passive and active tumor targeting of nanocarriers for anti-cancer drug delivery. J. Control. Release 2010, 148, 135–146. [Google Scholar] [CrossRef]
- Jeong, J.Y.; Hong, E.H.; Lee, S.Y.; Lee, J.Y.; Song, J.H.; Ko, S.H.; Shim, J.S.; Choe, S.; Kim, D.D.; Ko, H.J.; et al. Boronic acid-tethered amphiphilic hyaluronic acid derivative-based nanoassemblies for tumor targeting and penetration. Acta Biomater. 2017, 53, 414–426. [Google Scholar] [CrossRef]
- Lee, J.Y.; Chung, S.J.; Cho, H.J.; Kim, D.D. Phenylboronic acid-decorated chondroitin sulfate A-based theranostic nanoparticles for enhanced tumor targeting and penetration. Adv. Funct. Mater. 2015, 25, 3705–3717. [Google Scholar] [CrossRef]
- Lee, S.Y.; Cho, H.J. Amine-functionalized poly(lactic-co-glycolic acid) nanoparticles for improved cellular uptake and tumor penetration. Colloids Surf. B Biointerfaces 2016, 148, 85–94. [Google Scholar] [CrossRef]
- Lee, S.Y.; Cho, H.J. Mitochondria targeting and destabilizing hyaluronic acid derivative-based nanoparticles for the delivery of lapatinib to triple-negative breast cancer. Biomacromolecules 2019, 20, 835–845. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y.; Ko, S.H.; Shim, J.S.; Kim, D.D.; Cho, H.J. Tumor targeting and lipid rafts disrupting hyaluronic acid-cyclodextrin-based nanoassembled structure for cancer therapy. ACS Appl. Mater. Interfaces 2018, 10, 36628–36640. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y.; Koo, J.S.; Yang, M.; Cho, H.J. Application of temporary agglomeration of chitosan-coated nanoparticles for the treatment of lung metastasis of melanoma. J. Colloid Interface Sci. 2019, 544, 266–275. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y.; Park, J.H.; Ko, S.H.; Shim, J.S.; Kim, D.D.; Cho, H.J. Mussel-inspired hyaluronic acid derivative nanostructures for improved tumor targeting and penetration. ACS Appl. Mater. Interfaces 2017, 9, 22308–22320. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Zhang, Y.; Chen, G.; Hu, F.; Zhao, K.; Wang, Q. Engineered multifunctional nanomedicine for simultaneous stereotactic chemotherapy and inhibited osteolysis in an orthotopic model of bone metastasis. Adv. Mater. 2017, 29, 1605754. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Ojha, T.; Kiessling, F.; Lammers, T.; Shi, Y. Enhancing tumor penetration of nanomedicines. Biomacromolecules 2017, 18, 1449–1459. [Google Scholar] [CrossRef] [PubMed]
- Tian, B.; Li, J.; Pang, R.; Dai, S.; Li, T.; Weng, Y.; Jin, Y.; Hua, Y. Gold nanoparticles biosynthesized and functionalized using a hydroxylated tetraterpenoid trigger gene expression changes and apoptosis in cancer cells. ACS Appl. Mater. Interfaces 2018, 10, 37353–37363. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Li, L.; Kopeček, J. Biorecognition: A key to drug-free macromolecular therapeutics. Biomaterials 2019, 190–191, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Bhushan, B.; Khanadeev, V.; Khlebtsov, B.; Khlebtsov, N.; Gopinath, P. Impact of albumin based approaches in nanomedicine: Imaging, targeting and drug delivery. Adv. Colloid Interface Sci. 2017, 246, 13–39. [Google Scholar] [CrossRef]
- De Matteis, V.; Cascione, M.; Toma, C.C.; Leporatti, S. Silver nanoparticles: Synthetic routes, in vitro toxicity and theranostic applications for cancer disease. Nanomaterials 2018, 8, 319. [Google Scholar] [CrossRef]
- Dykman, L.A.; Khlebtsov, N.G. Multifunctional gold-based nanocomposites for theranostics. Biomaterials 2016, 108, 13–34. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.H.; Choi, Y. Mesoporous silica-based nanoplatforms for the delivery of photodynamic therapy agents. J. Pharm. Investig. 2018, 48, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.S.; Lee, D.Y. Photothermal therapy with gold nanoparticles as an anticancer medication. J. Pharm. Investig. 2017, 47, 19–26. [Google Scholar] [CrossRef]
- Kim, S.; Lee, S.Y.; Cho, H.J. Berberine and zinc oxide-based nanoparticles for the chemo-photothermal therapy of lung adenocarcinoma. Biochem. Biophys. Res. Commun. 2018, 501, 765–770. [Google Scholar] [CrossRef] [PubMed]
- Koo, J.S.; Lee, S.Y.; Azad, M.O.K.; Kim, M.; Hwang, S.J.; Nam, S.; Kim, S.; Chae, B.J.; Kang, W.S.; Cho, H.J. Development of iron(II) sulfate nanoparticles produced by hot-melt extrusion and their therapeutic potentials for colon cancer. Int. J. Pharm. 2019, 558, 388–395. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.J.; Lee, S.Y.; Park, J.H.; Kim, D.D.; Cho, H.J. Cholesterol-modified poly(lactide-co-glycolide) nanoparticles for tumor-targeted drug delivery. Int. J. Pharm. 2016, 509, 483–491. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Youn, Y.H.; Kwon, I.K.; Ko, N.R. Recent advances in quantum dots for biomedical applications. J. Pharm. Investig. 2018, 48, 209–214. [Google Scholar] [CrossRef]
- Lee, J.Y.; Chung, S.J.; Cho, H.J.; Kim, D.D. Iodinated hyaluronic acid oligomer-based nanoassemblies for tumor-targeted drug delivery and cancer imaging. Biomaterials 2016, 85, 218–231. [Google Scholar] [CrossRef]
- Lee, J.Y.; Park, J.H.; Lee, J.J.; Lee, S.Y.; Chung, S.J.; Cho, H.J.; Kim, D.D. Polyethylene glycol-conjugated chondroitin sulfate A derivative nanoparticles for tumor-targeted delivery of anticancer drugs. Carbohydr. Polym. 2016, 151, 68–77. [Google Scholar] [CrossRef]
- Lee, J.Y.; Termsarasab, U.; Lee, M.Y.; Kim, D.H.; Lee, S.Y.; Kim, J.S.; Cho, H.J.; Kim, D.D. Chemosensitizing indomethacin-conjugated chitosan oligosaccharide nanoparticles for tumor-targeted drug delivery. Acta Biomater. 2017, 57, 262–273. [Google Scholar] [CrossRef]
- Lee, J.Y.; Termsarasab, U.; Park, J.H.; Lee, S.Y.; Ko, S.H.; Shim, J.S.; Chung, S.J.; Cho, H.J.; Kim, D.D. Dual CD44 and folate receptor-targeted nanoparticles for cancer diagnosis and anticancer drug delivery. J. Control. Release 2016, 236, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y.; Cho, H.J. An α-tocopheryl succinate enzyme-based nanoassembly for cancer imaging and therapy. Drug Deliv. 2018, 25, 738–749. [Google Scholar] [CrossRef] [PubMed]
- Oberli, M.A.; Reichmuth, A.M.; Dorkin, J.R.; Mitchell, M.J.; Fenton, O.S.; Jaklenec, A.; Anderson, D.G.; Langer, R.; Blankschtein, D. Lipid nanoparticle assisted mRNA delivery for potent cancer immunotherapy. Nano Lett. 2017, 17, 1326–1335. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Cho, H.J.; Yoon, H.Y.; Yoon, I.S.; Ko, S.H.; Shim, J.S.; Cho, J.H.; Park, J.H.; Kim, K.; Kwon, I.C.; et al. Hyaluronic acid derivative-coated nanohybrid liposomes for cancer imaging and drug delivery. J. Control. Release 2014, 174, 98–108. [Google Scholar] [CrossRef] [PubMed]
- Sarisozen, C.; Pan, J.; Dutta, I.; Torchilin, V.P. Polymers in the co-delivery of siRNA and anticancer drugs to treat multidrug-resistant tumors. J. Pharm. Investig. 2017, 47, 37–49. [Google Scholar] [CrossRef]
- Sim, T.; Lim, C.; Hoang, N.H.; Oh, K.T. Recent advance of pH-sensitive nanocarriers targeting solid tumors. J. Pharm. Investig. 2017, 47, 383–394. [Google Scholar] [CrossRef]
- Tasciotti, E. Smart cancer therapy with DNA orgami. Nat. Biotechnol. 2018, 36, 234–235. [Google Scholar] [CrossRef]
- Zhu, B.; Wang, L.; Li, J.; Fan, C. Precisely tailored DNA nanostructures and their theranostic applications. Chem. Rec. 2017, 17, 1213–1230. [Google Scholar] [CrossRef]
- Martins, C.; Sousa, F.; Araújo, F.; Sarmento, B. Functionalizing PLGA and PLGA derivatives for drug delivery and tissue regeneration applications. Adv. Healthc. Mater. 2018, 7, 1701035. [Google Scholar] [CrossRef]
- Swider, E.; Koshkina, O.; Tel, J.; Cruz, L.J.; de Vries, I.J.M.; Srinivas, M. Customizing poly(lactic-co-glycolic acid) particles for biomedical applications. Acta Biomater. 2018, 73, 38–51. [Google Scholar] [CrossRef]
- Danhier, F.; Ansorena, E.; Silva, J.M.; Coco, R.; Le Breton, A.; Préat, V. PLGA-based nanoparticles: An overview of biomedical applications. J. Control. Release 2012, 161, 505–522. [Google Scholar] [CrossRef] [PubMed]
- Mir, M.; Ahmed, N.; Rehman, A.U. Recent applications of PLGA based nanostructures in drug delivery. Colloids Surf. B Biointerfaces 2017, 159, 217–231. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Li, F.; Yao, Y.; Wang, Z.; Zhang, Z.; Tan, N. Redox dual-responsive and O2-evolving theranostic nanosystem for highly selective chemotherapy against hypoxic tumors. Theranostics 2019, 9, 90–103. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Liang, C.; Song, X.; Chen, Q.; Jin, Q.; Wang, C.; Liu, Z. Erythrocyte-membrane-enveloped perfluorocarbon as nanoscale artificial red blood cells to relieve tumor hypoxia and enhance cancer radiotherapy. Adv. Mater. 2017, 29, 1701429. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Lee, J.Y.; Termsarasab, U.; Yoon, I.S.; Ko, S.H.; Shim, J.S.; Cho, H.J.; Kim, D.D. Development of poly(lactic-co-glycolic) acid nanoparticles-embedded hyaluronic acid–ceramide-based nanostructure for tumor-targeted drug delivery. Int. J. Pharm. 2014, 473, 426–433. [Google Scholar] [CrossRef] [PubMed]
- Saneja, A.; Arora, D.; Kumar, R.; Dubey, R.D.; Panda, A.K.; Gupta, P.N. CD44 targeted PLGA nanomedicines for cancer chemotherapy. Eur. J. Pharm. Sci. 2018, 121, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Acharya, S.; Sahoo, S.K. PLGA nanoparticles containing various anticancer agents and tumour delivery by EPR effect. Adv. Drug Deliv. Rev. 2011, 63, 170–183. [Google Scholar] [CrossRef]
- Ding, D.; Zhu, Q. Recent advances of PLGA micro/nanoparticles for the delivery of biomacromolecular therapeutics. Mater. Sci. Eng. C Mater. Biol. Appl. 2018, 92, 1041–1060. [Google Scholar] [CrossRef]
- Lee, S.Y.; Cho, H.J. Dopamine-conjugated poly(lactic-co-glycolic acid) nanoparticles for protein delivery to macrophages. J. Colloid Interface Sci. 2017, 490, 391–400. [Google Scholar] [CrossRef]
- Park, J.H.; Cho, H.J.; Kim, D.D. Poly((d,l)lactic-glycolic)acid–star glucose nanoparticles for glucose transporter and hypoglycemia-mediated tumor targeting. Int. J. Nanomed. 2017, 12, 7453–7467. [Google Scholar] [CrossRef]
- Allahyari, M.; Mohit, E. Peptide/protein vaccine delivery system based on PLGA particles. Hum. Vaccin. Immunother. 2016, 12, 806–828. [Google Scholar] [CrossRef] [PubMed]
- Rezvantalab, S.; Drude, N.I.; Moraveji, M.K.; Guvener, N.; Koons, E.K.; Shi, Y.; Lammers, T.; Kiessling, F. PLGA-based nanoparticles in cancer treatment. Front. Pharmacol. 2018, 9, 1260. [Google Scholar] [CrossRef] [PubMed]
- Dinarvand, R.; Sepehri, N.; Manoochehri, S.; Rouhani, H.; Atyabi, F. Polylactide-co-glycolide nanoparticles for controlled delivery of anticancer agents. Int. J. Nanomed. 2011, 6, 877–895. [Google Scholar] [CrossRef] [PubMed]
- Booysen, L.L.; Kalombo, L.; Brooks, E.; Hansen, R.; Gilliland, J.; Gruppo, V.; Lungenhofer, P.; Semete-Makokotlela, B.; Swai, H.S.; Kotze, A.F.; et al. In vivo/in vitro pharmacokinetic and pharmacodynamic study of spray-dried poly-(dl-lactic-co-glycolic) acid nanoparticles encapsulating rifampicin and isoniazid. Int. J. Pharm. 2013, 444, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, D.N.; Bhatia, A.; Kaur, R.; Sharma, R.; Kaur, G.; Dhawan, S. PLGA: A unique polymer for drug delivery. Ther. Deliv. 2015, 6, 41–58. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Kim, J.S.; Cho, H.J.; Kim, D.D. Poly(styrene)-b-poly(dl-lactide) copolymer-based nanoparticles for anticancer drug delivery. Int. J. Nanomed. 2014, 9, 2803–2813. [Google Scholar]
- Rafiei, P.; Haddadi, A. Docetaxel-loaded PLGA and PLGA-PEG nanoparticles for intravenous application: Pharmacokinetics and biodistribution profile. Int. J. Nanomed. 2017, 12, 935–947. [Google Scholar] [CrossRef] [PubMed]
- Sanna, V.; Roggio, A.M.; Posadino, A.M.; Cossu, A.; Marceddu, S.; Mariani, A.; Alzari, V.; Uzzau, S.; Pintus, G.; Sechi, M. Novel docetaxel-loaded nanoparticles based on poly(lactide-co-caprolactone) and poly(lactide-co-glycolide-co-caprolactone) for prostate cancer treatment: Formulation, characterization, and cytotoxicity studies. Nanoscale Res. Lett. 2011, 6, 260. [Google Scholar] [CrossRef]
- Ashjari, M.; Khoee, S.; Mahdavian, A.R.; Rahmatolahzadeh, R. Self-assembled nanomicelles using PLGA-PEG amphiphilic block copolymer for insulin delivery: A physicochemical investigation and determination of CMC values. J. Mater. Sci. Mater. Med. 2012, 23, 943–953. [Google Scholar] [CrossRef]
- Chen, X.; Chen, J.; Li, B.; Yang, X.; Zeng, R.; Liu, Y.; Li, T.; Ho, R.J.Y.; Shao, J. PLGA-PEG-PLGA triblock copolymeric micelles as oral drug delivery system: In vitro drug release and in vivo pharmacokinetics assessment. J. Colloid Interface Sci. 2017, 490, 542–552. [Google Scholar] [CrossRef]
- Cai, Q.; Wang, L.; Deng, G.; Liu, J.; Chen, Q.; Chen, Z. Systemic delivery to central nervous system by engineered PLGA nanoparticles. Am. J. Transl. Res. 2016, 8, 749–764. [Google Scholar] [PubMed]
- Chung, Y.I.; Kim, J.C.; Kim, Y.H.; Tae, G.; Lee, S.Y.; Kim, K.; Kwon, I.C. The effect of surface functionalization of PLGA nanoparticles by heparin- or chitosan-conjugated Pluronic on tumor targeting. J. Control. Release 2010, 143, 374–382. [Google Scholar] [CrossRef] [PubMed]
- Hwang, H.S.; Shin, H.; Na, K. Combination of photodynamic therapy (PDT) and anti-tumor immunity in cancer therapy. J. Pharm. Investig. 2018, 48, 143–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kydd, J.; Jadia, R.; Velpurisiva, P.; Gad, A.; Paliwal, S.; Rai, P. Targeting strategies for the combination treatment of cancer using drug delivery systems. Pharmaceutics 2017, 9, 46. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Ding, D.; Huo, D.; Pu, K.Y.; Thao, N.N.P.; Hu, Y.; Li, Z.; Liu, B. Conjugated polymer based nanoparticles as dual-modal probes for targeted in vivo fluorescence and magnetic resonance imaging. Adv. Funct. Mater. 2012, 22, 3107–3115. [Google Scholar] [CrossRef]
- Chen, J.; Wu, Q.; Luo, L.; Wang, Y.; Zhong, Y.; Dai, H.B.; Sun, D.; Luo, M.L.; Wu, W.; Wang, G.X. Dual tumor-targeted poly(lactic-co-glycolic acid)-polyethylene glycol-folic acid nanoparticles: A novel biodegradable nanocarrier for secure and efficient antitumor drug delivery. Int. J. Nanomed. 2017, 12, 5745–5760. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.J.; Li, L.; Liu, X.J.; Li, Y.; Zhao, C.Y.; Wang, R.Q.; Zhen, Y.S. Mithramycin-loaded mPEG-PLGA nanoparticles exert potent antitumor efficacy against pancreatic carcinoma. Int. J. Nanomed. 2017, 12, 5255–5269. [Google Scholar] [CrossRef] [PubMed]
- Lu, B.; Lv, X.; Le, Y. Chitosan-modified PLGA nanoparticles for control-released drug delivery. Polymers 2019, 11, 304. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.; Chaudhari, K.; Dantuluri, P.; Murthy, R.S.; Das, S. Paclitaxel-loaded PLGA nanoparticles surface modified with transferrin and Pluronic® P85, an in vitro cell line and in vivo biodistribution studies on rat model. J. Drug Target. 2009, 17, 533–542. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhao, Y.; Wang, H.; Gong, J.; He, H.; Shin, M.C.; Yang, V.C.; Huang, Y. Low-molecular-weight protamine-modified PLGA nanoparticles for overcoming drug-resistant breast cancer. J. Control. Release 2014, 192, 47–56. [Google Scholar] [CrossRef]
- Lin, W.J.; Lee, W.C. Polysaccharide-modified nanoparticles with intelligent CD44 receptor targeting ability for gene delivery. Int. J. Nanomed. 2018, 13, 3989–4002. [Google Scholar] [CrossRef] [PubMed]
- Mehdizadeh, M.; Rouhani, H.; Sepehri, N.; Varshochian, R.; Ghahremani, M.H.; Amini, M.; Gharghabi, M.; Ostad, S.N.; Atyabi, F.; Baharian, A.; et al. Biotin decorated PLGA nanoparticles containing SN-38 designed for cancer therapy. Artif. Cells Nanomed. Biotechnol. 2017, 45, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Bao, W.; Liu, R.; Wang, Y.; Wang, F.; Xia, G.; Zhang, H.; Li, X.; Yin, H.; Chen, B. PLGA-PLL-PEG-Tf-based targeted nanoparticles drug delivery system enhance antitumor efficacy via intrinsic apoptosis pathway. Int. J. Nanomed. 2015, 10, 557–566. [Google Scholar] [Green Version]
- Venugopal, V.; Krishnan, S.; Palanimuthu, V.R.; Sankarankutty, S.; Kalaimani, J.K.; Karupiah, S.; Kit, N.S.; Hock, T.T. Anti-EGFR anchored paclitaxel loaded PLGA nanoparticles for the treatment of triple negative breast cancer. In-vitro and in-vivo anticancer activities. PLoS ONE 2018, 13, e0206109. [Google Scholar] [CrossRef] [PubMed]
- Babu, A.; Amreddy, N.; Muralidharan, R.; Pathuri, G.; Gali, H.; Chen, A.; Zhao, Y.D.; Munshi, A.; Ramesh, R. Chemodrug delivery using integrin-targeted PLGA-chitosan nanoparticle for lung cancer therapy. Sci. Rep. 2017, 7, 14674. [Google Scholar] [CrossRef] [PubMed]
- Ni, M.; Xiong, M.; Zhang, X.; Cai, G.; Chen, H.; Zeng, Q.; Yu, Z. Poly(lactic-co-glycolic acid) nanoparticles conjugated with CD133 aptamers for targeted salinomycin delivery to CD133+ osteosarcoma cancer stem cells. Int. J. Nanomed. 2015, 10, 2537–2554. [Google Scholar]
- Foroozandeh, P.; Aziz, A.A. Insight into cellular uptake and intracellular trafficking of nanoparticles. Nanoscale Res. Lett. 2018, 13, 339. [Google Scholar] [CrossRef] [PubMed]
- Kou, L.; Sun, J.; Zhai, Y.; He, Z. The endocytosis and intracellular fate of nanomedicines: Implication for rational design. Asian J. Pharm. Sci. 2013, 8, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Malinovskaya, Y.; Melnikov, P.; Baklaushev, V.; Gabashvili, A.; Osipova, N.; Mantrov, S.; Ermolenko, Y.; Maksimenko, O.; Gorshkova, M.; Balabanyan, V.; et al. Delivery of doxorubicin-loaded PLGA nanoparticles into U87 human glioblastoma cells. Int. J. Pharm. 2017, 524, 77–90. [Google Scholar] [CrossRef]
- Sahay, G.; Alakhova, D.Y.; Kabanov, A.V. Endocytosis of nanomedicines. J. Control. Release 2010, 145, 182–195. [Google Scholar] [CrossRef] [Green Version]
- Trindade, I.C.; Pound-Lana, G.; Pereira, D.G.S.; de Oliveira, L.A.M.; Andrade, M.S.; Vilela, J.M.C.; Postacchini, B.B.; Mosqueira, V.C.F. Mechanisms of interaction of biodegradable polyester nanocapsules with non-phagocytic cells. Eur. J. Pharm. Sci. 2018, 124, 89–104. [Google Scholar] [CrossRef]
- Alshaer, W.; Hillaireau, H.; Fattal, E. Aptamer-guided nanomedicines for anticancer drug delivery. Adv. Drug Deliv. Rev. 2018, 134, 122–137. [Google Scholar] [CrossRef] [PubMed]
- Salatin, S.; Yari Khosroushahi, A. Overviews on the cellular uptake mechanism of polysaccharide colloidal nanoparticles. J. Cell. Mol. Med. 2017, 21, 1668–1686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, C.H.; Lee, S.G.; Kang, M.J.; Lee, S.; Choi, Y.W. Surface modification of lipid-based nanocarriers for cancer cell-specific drug targeting. J. Pharm. Investig. 2017, 47, 203–227. [Google Scholar] [CrossRef]
- Ghosh, D.; Peng, X.; Leal, J.; Mohanty, R.P. Peptides as drug delivery vehicles across biological barriers. J. Pharm. Investig. 2018, 48, 89–111. [Google Scholar] [CrossRef] [PubMed]
- Kreuter, J. Drug delivery to the central nervous system by polymeric nanoparticles: What do we know? Adv. Drug Deliv. Rev. 2014, 71, 2–14. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, X.; Li, B.; Hou, Y.; Yang, J.; Yi, L. Development of a novel morphological paclitaxel-loaded PLGA microspheres for effective cancer therapy: In vitro and in vivo evaluations. Drug Deliv. 2018, 25, 166–177. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Zhang, Y.; Feng, J.; Gu, T.; Dong, Q.; Yang, X.; Sun, Y.; Wu, Y.; Chen, Y.; Kong, W. Preparation, characterization, and in vitro and in vivo investigation of chitosan-coated poly (d,l-lactide-co-glycolide) nanoparticles for intestinal delivery of exendin-4. Int. J. Nanomed. 2013, 8, 1141–1154. [Google Scholar]
- Alibolandi, M.; Ramezani, M.; Abnous, K.; Hadizadeh, F. AS1411 aptamer-decorated biodegradable polyethylene glycol-poly(lactic-co-glycolic acid) nanopolymersomes for the targeted delivery of gemcitabine to non-small cell lung cancer in vitro. J. Pharm. Sci. 2016, 105, 1741–1750. [Google Scholar] [CrossRef]
- Wei, J.; Sun, J.; Liu, Y. Enhanced targeting of prostate cancer-initiation cells by salinomycin-encapsulated lipid-PLGA nanoparticles linked with CD44 antibodies. Oncol. Lett. 2019, 17, 4024–4033. [Google Scholar]
- Kou, L.; Yao, Q.; Sivaprakasam, S.; Luo, Q.; Sun, Y.; Fu, Q.; He, Z.; Sun, J.; Ganapathy, V. Dual targeting of l-carnitine-conjugated nanoparticles to OCTN2 and ATB0,+ to deliver chemotherapeutic agents for colon cancer therapy. Drug Deliv. 2017, 24, 1338–1349. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Di, X.; Wu, M.; Sun, Z.; Zhong, L.; Wang, Y.; Fu, Q.; Kan, Q.; Sun, J.; He, Z. Targeting tumor highly-expressed LAT1 transporter with amino acid-modified nanoparticles: Toward a novel active targeting strategy in breast cancer therapy. Nanomedicine 2017, 13, 987–998. [Google Scholar] [CrossRef]
- Cespedes, A.; Perez, M.J.; Gomara, M.J.; Wandosell, F.; Haro, I. Atorvastatin in PLGA-PEG nanoparticles derivatized with the HIV-TAT peptide protects neuronal cultures in an oxygen-glucose deprivation (OGD) model. Am. J. Nanotechnol. Nanomed. 2018, 1, 55–63. [Google Scholar]
- Choi, D.H.; Park, Y.S. Arginine-rich peptide coated PLGA nanoparticles enhance polymeric delivery of antisense HIF1α-oligonucleotide to fully differentiated stiff adipocytes. Toxicol. Environ. Health Sci. 2019, 11, 1–10. [Google Scholar] [CrossRef]
- Nottelet, B.; Darcos, V.; Coudane, J. Aliphatic polyesters for medical imaging and theranostic applications. Eur. J. Pharm. Biopharm. 2015, 97, 350–370. [Google Scholar] [CrossRef]
- Reul, R.; Tsapis, N.; Hillaireau, H.; Sancey, L.; Mura, S.; Recher, M.; Nicolas, J.; Coll, J.L.; Fattal, E. Near infrared labeling of PLGA for in vivo imaging of nanoparticles. Polym. Chem. 2012, 3, 694–702. [Google Scholar] [CrossRef]
- Ratzinger, G.; Agrawal, P.; Körner, W.; Lonkai, J.; Sanders, H.M.; Terreno, E.; Wirth, M.; Strijkers, G.J.; Nicolay, K.; Gabor, F. Surface modification of PLGA nanospheres with Gd-DTPA and Gd-DOTA for high-relaxivity MRI contrast agents. Biomaterials 2010, 31, 8716–8723. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, S.; Pandey, U.; Gugulothu, D.; Patravale, V.; Samuel, G. Modification of PLGA nanoparticles for improved properties as a 99mTc-labeled agent in sentinel lymph node detection. Cancer Biother. Radiopharm. 2013, 28, 598–606. [Google Scholar] [CrossRef]
- Nomikou, N.; Curtis, K.; McEwan, C.; O’Hagan, B.M.G.; Callan, B.; Callan, J.F.; McHale, A.P. A versatile, stimulus-responsive nanoparticle-based platform for use in both sonodynamic and photodynamic cancer therapy. Acta Biomater. 2017, 49, 414–421. [Google Scholar] [CrossRef]
- Sivakumar, B.; Aswathy, R.G.; Romero-Aburto, R.; Mitcham, T.; Mitchel, K.A.; Nagaoka, Y.; Bouchard, R.R.; Ajayan, P.M.; Maekawa, T.; Sakthikumar, D.N. Highly versatile SPION encapsulated PLGA nanoparticles as photothermal ablators of cancer cells and as multimodal imaging agents. Biomater. Sci. 2017, 5, 432–443. [Google Scholar] [CrossRef]
- Li, Q.; Li, C.; Tong, W. Nile red loaded PLGA nanoparticles surface modified with Gd-DTPA for potential dual-modal imaging. J. Nanosci. Nanotechnol. 2016, 16, 5569–5576. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Vu-Quang, H.; Husum, D.M.U.; Tingskov, S.J.; Vinding, M.S.; Nielsen, T.; Song, P.; Nielsen, N.C.; Nørregaard, R.; Kjems, J. Theranostic poly(lactic-co-glycolic acid) nanoparticle for magnetic resonance/infrared fluorescence bimodal imaging and efficient siRNA delivery to macrophages and its evaluation in a kidney injury model. Nanomedicine 2017, 13, 2451–2462. [Google Scholar] [CrossRef] [PubMed]
- Xing, L.; Shi, Q.; Zheng, K.; Shen, M.; Ma, J.; Li, F.; Liu, Y.; Lin, L.; Tu, W.; Duan, Y.; et al. Ultrasound-mediated microbubble destruction (UMMD) facilitates the delivery of CA19-9 targeted and paclitaxel loaded mPEG-PLGA-PLL nanoparticles in pancreatic cancer. Theranostics 2016, 6, 1573–1587. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Li, X.; Hua, H.; Wang, A.; Liu, W.; Li, Y.; Fu, F.; Shi, Y.; Sun, K. Cyclic hexapeptide-conjugated nanoparticles enhance curcumin delivery to glioma tumor cells and tissue. Int. J. Nanomed. 2017, 12, 5717–5732. [Google Scholar] [CrossRef] [PubMed]
- Vats, M.; Mishra, S.K.; Baghini, M.S.; Chauhan, D.S.; Srivastava, R.; De, A. Near infrared fluorescence imaging in nano-therapeutics and photo-thermal evaluation. Int. J. Mol. Sci. 2017, 18, 924. [Google Scholar] [CrossRef] [PubMed]
- da Silva, C.L.; Del Ciampo, J.O.; Rossetti, F.C.; Bentley, M.V.; Pierre, M.B. Improved in vitro and in vivo cutaneous delivery of protoporphyrin IX from PLGA-based nanoparticles. Photochem. Photobiol. 2013, 89, 1176–1184. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Li, T.; Chen, Z.; Xie, X.; Zhang, H.; Feng, Y.; Li, S.; Qin, X.; Yang, H.; Wu, C.; et al. NIR-light-triggered anticancer strategy for dual-modality imaging-guided combination therapy via a bioinspired hybrid PLGA nanoplatform. Mol. Pharm. 2019, 16, 1367–1384. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Dong, Y.; Zhang, Y.; Ru, D.; Wu, Z.; Zhang, J.; Shen, M.; Duan, Y.; Sun, Y. GSH-sensitive Pt(IV) prodrug-loaded phase-transitional nanoparticles with a hybrid lipid-polymer shell for precise theranostics against ovarian cancer. Theranostics 2019, 9, 1047–1065. [Google Scholar] [CrossRef]
- Guo, Y.; Wang, X.Y.; Chen, Y.L.; Liu, F.Q.; Tan, M.X.; Ao, M.; Yu, J.H.; Ran, H.T.; Wang, Z.X. A light-controllable specific drug delivery nanoplatform for targeted bimodal imaging-guided photothermal/chemo synergistic cancer therapy. Acta Biomater. 2018, 80, 308–326. [Google Scholar] [CrossRef]
- Hao, Y.; Zhang, B.; Zheng, C.; Ji, R.; Ren, X.; Guo, F.; Sun, S.; Shi, J.; Zhang, H.; Zhang, Z.; et al. The tumor-targeting core-shell structured DTX-loaded PLGA@Au nanoparticles for chemo-photothermal therapy and X-ray imaging. J. Control. Release 2015, 220, 545–555. [Google Scholar] [CrossRef]
- Hahn, M.A.; Singh, A.K.; Sharma, P.; Brown, S.C.; Moudgil, B.M. Nanoparticles as contrast agents for in-vivo bioimaging: Current status and future perspectives. Anal. Bioanal. Chem. 2011, 399, 3–27. [Google Scholar] [CrossRef] [PubMed]
- Sah, H.; Thoma, L.A.; Desu, H.R.; Sah, E.; Wood, G.C. Concepts and practices used to develop functional PLGA-based nanoparticulate systems. Int. J. Nanomed. 2013, 8, 747–765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, H.; Guo, Y.; Peng, L.; Fang, H.; Wang, Z.; Zheng, Y.; Ran, H.; Chen, Y. In vivo targeted, responsive, and synergistic cancer nanotheranostics by magnetic resonance imaging-guided synergistic high-intensity focused ultrasound ablation and chemotherapy. ACS Appl. Mater. Interfaces 2018, 10, 15428–15441. [Google Scholar] [CrossRef] [PubMed]
- Mosafer, J.; Abnous, K.; Tafaghodi, M.; Mokhtarzadeh, A.; Ramezani, M. In vitro and in vivo evaluation of anti-nucleolin-targeted magnetic PLGA nanoparticles loaded with doxorubicin as a theranostic agent for enhanced targeted cancer imaging and therapy. Eur. J. Pharm. Biopharm. 2017, 113, 60–74. [Google Scholar] [CrossRef] [PubMed]
- Singh, Y.; Durga Rao Viswanadham, K.K.; Kumar Jajoriya, A.; Meher, J.G.; Raval, K.; Jaiswal, S.; Dewangan, J.; Bora, H.K.; Rath, S.K.; Lal, J.; et al. Click biotinylation of PLGA template for biotin receptor oriented delivery of doxorubicin hydrochloride in 4T1 cell-induced breast cancer. Mol. Pharm. 2017, 14, 2749–2765. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Zhang, M.; Zeng, F.; Jin, H.; Xu, Q.; Huang, Y. Dual-targeting magnetic PLGA nanoparticles for codelivery of paclitaxel and curcumin for brain tumor therapy. ACS Appl. Mater. Interfaces 2016, 8, 32159–32169. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.L.; Liu, F.Q.; Guo, Y.; Cheng, J.; Yang, L.; Lu, M.; Li, P.; Xu, J.; Yu, T.; Wang, Z.G.; et al. PA/US dual-modality imaging to guide VEGFR-2 targeted photothermal therapy using ZnPc-/PFH-loaded polymeric nanoparticles. Biomater. Sci. 2018, 6, 2130–2143. [Google Scholar] [CrossRef] [PubMed]
- Euhus, D.M.; Hudd, C.; Laregina, M.C.; Johnson, F.E. Tumor measurement in the nude mouse. J. Surg. Oncol. 1986, 31, 229–234. [Google Scholar] [CrossRef] [PubMed]
- Tomayko, M.M.; Reynolds, C.P. Determination of subcutaneous tumor size in athymic (nude) mice. Cancer Chemother. Pharmacol. 1989, 24, 148–154. [Google Scholar] [CrossRef]
- Guo, J.; Gao, X.; Su, L.; Xia, H.; Gu, G.; Pang, Z.; Jiang, X.; Yao, L.; Chen, J.; Chen, H. Aptamer-functionalized PEG–PLGA nanoparticles for enhanced anti-glioma drug delivery. Biomaterials 2011, 32, 8010–8020. [Google Scholar] [CrossRef] [PubMed]
- Wohlfart, S.; Khalansky, A.S.; Gelperina, S.; Maksimenko, O.; Bernreuther, C.; Glatzel, M.; Kreuter, J. Efficient chemotherapy of rat glioblastoma using doxorubicin-loaded PLGA nanoparticles with different stabilizers. PLoS ONE 2011, 6, e19121. [Google Scholar] [CrossRef] [PubMed]
- Cook, R.L.; Householder, K.T.; Chung, E.P.; Prakapenka, A.V.; DiPerna, D.M.; Sirianni, R.W. A critical evaluation of drug delivery from ligand modified nanoparticles: Confounding small molecule distribution and efficacy in the central nervous system. J. Control. Release 2015, 220, 89–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Deng, C.; Meng, F.; Zhang, J.; Sun, H.; Zhong, Z. Hyaluronic acid coated PLGA nanoparticulate docetaxel effectively targets and suppresses orthotopic human lung cancer. J. Control. Release 2017, 259, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Moreno, D.; Zalba, S.; Navarro, Í.; De Ilarduya, C.T.; Garrido, M.J. Pharmacodynamics of cisplatin-loaded PLGA nanoparticles administered to tumor-bearing mice. Eur. J. Pharm. Biopharm. 2010, 74, 265–274. [Google Scholar] [CrossRef] [PubMed]
- Mattheolabakis, G.; Taoufik, E.; Haralambous, S.; Roberts, M.L.; Avgoustakis, K. In vivo investigation of tolerance and antitumor activity of cisplatin-loaded PLGA-mPEG nanoparticles. Eur. J. Pharm. Biopharm. 2009, 71, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Saneja, A.; Kumar, R.; Mintoo, M.J.; Dubey, R.D.; Sangwan, P.L.; Mondhe, D.M.; Panda, A.K.; Gupta, P.N. Gemcitabine and betulinic acid co-encapsulated PLGA−PEG polymer nanoparticles for improved efficacy of cancer chemotherapy. Mater. Sci. Eng. C 2019, 98, 764–771. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; You, J.; Shao, H.; Yan, C. Effects of surface modification of As2O3-loaded PLGA nanoparticles on its anti-liver cancer ability: An in vitro and in vivo study. Colloids Surf. B Biointerfaces 2018, 169, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.; Yang, Y.; Ling, Y.; Huang, Y.; Li, T.; Li, X. Improved therapeutic effect of folate-decorated PLGA–PEG nanoparticles for endometrial carcinoma. Bioorg. Med. Chem. 2011, 19, 4057–4066. [Google Scholar] [CrossRef] [PubMed]
- Kang, T.; Gao, X.; Hu, Q.; Jiang, D.; Feng, X.; Zhang, X.; Song, Q.; Yao, L.; Huang, M.; Jiang, X.; et al. iNGR-modified PEG-PLGA nanoparticles that recognize tumor vasculature and penetrate gliomas. Biomaterials 2014, 35, 4319–4332. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Hao, W.; Xu, L.; Gao, Y.; Wang, X.; Zhu, D.; Chen, Z.; Zhang, X.; Chen, H.; Mei, L. A pH-sensitive methenamine mandelate-loaded nanoparticle induces DNA damage and apoptosis of cancer cells. Acta Biomater. 2017, 62, 246–256. [Google Scholar] [CrossRef]
- Deng, K.; Li, C.; Huang, S.; Xing, B.; Jin, D.; Zeng, Q.; Hou, Z.; Lin, J. Recent progress in near infrared light triggered photodynamic therapy. Small 2017, 13, 1702299. [Google Scholar] [CrossRef]
- Li, T.; Yan, L. Functional polymer nanocarriers for photodynamic therapy. Pharmaceuticals 2018, 11, 133. [Google Scholar] [CrossRef] [PubMed]
- Bœuf-Muraille, G.; Rigaux, G.; Callewaert, M.; Zambrano, N.; Van Gulick, L.; Roullin, V.G.; Terryn, C.; Andry, M.C.; Chuburu, F.; Dukic, S.; et al. Evaluation of mTHPC-loaded PLGA nanoparticles for in vitro photodynamic therapy on C6 glioma cell line. Photodiagn. Photodyn. Ther. 2019, 25, 448–455. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Yan, S.Z.; Qi, S.S.; Xu, Q.; Han, S.S.; Guo, L.Y.; Zhao, N.; Chen, S.L.; Yu, S.Q. Transferrin-modified nanoparticles for photodynamic therapy enhance the antitumor efficacy of hypocrellin A. Front. Pharmacol. 2017, 8, 815. [Google Scholar] [CrossRef] [PubMed]
- Son, J.; Yang, S.M.; Yi, G.; Roh, Y.J.; Park, H.; Park, J.M.; Choi, M.G.; Koo, H. Folate-modified PLGA nanoparticles for tumor-targeted delivery of pheophorbide a in vivo. Biochem. Biophys. Res. Commun. 2018, 498, 523–528. [Google Scholar] [CrossRef] [PubMed]
- Xin, Y.; Liu, T.; Yang, C. Development of PLGA-lipid nanoparticles with covalently conjugated indocyanine green as a versatile nanoplatform for tumor-targeted imaging and drug delivery. Int. J. Nanomed. 2016, 11, 5807–5821. [Google Scholar] [CrossRef]
- Vivek, R.; Varukattu, N.; Chandrababu, R.; Alok, S.; Thondhi, P.; Alagarsamy, V.; Kannan, S. Multifunctional nanoparticles for trimodal photodynamic therapy-mediated photothermal and chemotherapeutic effects. Photodiagn. Photodyn. Ther. 2018, 23, 244–253. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Shi, L.; Tu, Q.; Wang, H.; Zhang, H.; Wang, P.; Zhang, L.; Huang, Z.; Zhao, F.; Luan, H.; et al. Treating cutaneous squamous cell carcinoma using 5-aminolevulinic acid polylactic-co-glycolic acid nanoparticle-mediated photodynamic therapy in a mouse model. Int. J. Nanomed. 2015, 10, 347–355. [Google Scholar]
- Peng, Y.; Nie, J.; Cheng, W.; Liu, G.; Zhu, D.; Zhang, L.; Liang, C.; Mei, L.; Huang, L.; Zeng, X. A multifunctional nanoplatform for cancer chemo-photothermal synergistic therapy and overcoming multidrug resistance. Biomater. Sci. 2018, 6, 1084–1098. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.T.; Dai Phung, C.; Thapa, R.K.; Pham, T.T.; Tran, T.H.; Jeong, J.-H.; Ku, S.K.; Choi, H.-G.; Yong, C.S.; Kim, J.O. Multifunctional nanoparticles as somatostatin receptor-targeting delivery system of polyaniline and methotrexate for combined chemo–photothermal therapy. Acta Biomater. 2018, 68, 154–167. [Google Scholar] [CrossRef] [PubMed]
- Senthilkumar, M.; Mishra, P.; Jain, N.K. Long circulating PEGylated poly(d,l-lactide-co-glycolide) nanoparticulate delivery of Docetaxel to solid tumors. J. Drug Target. 2008, 16, 424–435. [Google Scholar] [CrossRef] [PubMed]
- Moghimi, S.M.; Muir, I.S.; Illum, L.; Davis, S.S.; Kolb-Bachofen, V. Coating particles with a block co-polymer (poloxamine-908) suppresses opsonization but permits the activity of dysopsonins in the serum. Biochim. Biophys. Acta 1993, 1179, 157–165. [Google Scholar] [CrossRef]
- Milane, L.; Duan, Z.F.; Amiji, M. Pharmacokinetics and biodistribution of lonidamine/paclitaxel loaded, EGFR-targeted nanoparticles in an orthotopic animal model of multi-drug resistant breast cancer. Nanomedicine 2011, 7, 435–444. [Google Scholar] [CrossRef] [Green Version]
- Dhar, S.; Kolishetti, N.; Lippard, S.J.; Farokhzad, O.C. Targeted delivery of a cisplatin prodrug for safer and more effective prostate cancer therapy in vivo. Proc. Natl. Acad. Sci. USA 2011, 108, 1850–1855. [Google Scholar] [CrossRef] [Green Version]
- Avgoustakis, K.; Beletsi, A.; Panagi, Z.; Klepetsanis, P.; Karydas, A.G.; Ithakissios, D.S. PLGA-mPEG nanoparticles of cisplatin: In vitro nanoparticle degradation, in vitro drug release and in vivo drug residence in blood properties. J. Control. Release 2002, 79, 123–135. [Google Scholar] [CrossRef]
- Ma, Y.; Sadoqi, M.; Shao, J. Biodistribution of indocyanine green-loaded nanoparticles with surface modifications of PEG and folic acid. Int. J. Pharm. 2012, 436, 25–31. [Google Scholar] [CrossRef]
- Yin, P.; Wang, Y.; Qiu, Y.; Hou, L.; Liu, X.; Qin, J.; Duan, Y.; Liu, P.; Qiu, M.; Li, Q. Bufalin-loaded mPEG-PLGA-PLL-cRGD nanoparticles: Preparation, cellular uptake, tissue distribution, and anticancer activity. Int. J. Nanomed. 2012, 7, 3961–3969. [Google Scholar] [CrossRef]
- Huang, J.; Zhang, H.; Yu, Y.; Chen, Y.; Wang, D.; Zhang, G.; Zhou, G.; Liu, J.; Sun, Z.; Sun, D.; et al. Biodegradable self-assembled nanoparticles of poly(d,l-lactide-co-glycolide)/hyaluronic acid block copolymers for target delivery of docetaxel to breast cancer. Biomaterials 2014, 35, 550–566. [Google Scholar] [CrossRef]
- Chen, Z.; Tai, Z.; Gu, F.; Hu, C.; Zhu, Q.; Gao, S. Aptamer-mediated delivery of docetaxel to prostate cancer through polymeric nanoparticles for enhancement of antitumor efficacy. Eur. J. Pharm. Biopharm. 2016, 107, 130–141. [Google Scholar] [CrossRef]
- Choi, S.J.; Choy, J.H. Layered double hydroxide nanoparticles as target-specific delivery carriers: Uptake mechanism and toxicity. Nanomedicine 2011, 6, 803–814. [Google Scholar] [CrossRef]
- Jin, H.; Pi, J.; Zhao, Y.; Jiang, J.; Li, T.; Zeng, X.; Yang, P.; Evans, C.E.; Cai, J. EGFR-targeting PLGA-PEG nanoparticles as a curcumin delivery system for breast cancer therapy. Nanoscale 2017, 9, 16365–16374. [Google Scholar] [CrossRef] [PubMed]
- Paolini, M.; Poul, L.; Berjaud, C.; Germain, M.; Darmon, A.; Bergere, M.; Pottier, A.; Levy, L.; Vibert, E. Nano-sized cytochrome P450 3A4 inhibitors to block hepatic metabolism of docetaxel. Int. J. Nanomed. 2017, 12, 5537–5556. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, N.; Alam, M.A.; Ahmad, R.; Naqvi, A.A.; Ahmad, F.J. Preparation and characterization of surface-modified PLGA-polymeric nanoparticles used to target treatment of intestinal cancer. Artif. Cells Nanomed. Biotechnol. 2018, 46, 432–446. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Parmar, A.; Kori, S.; Sandhir, R. PLGA-based nanoparticles: A new paradigm in biomedical applications. Trends Anal. Chem. 2016, 80, 30–40. [Google Scholar] [CrossRef]
- Navarro, S.M.; Morgan, T.W.; Astete, C.E.; Stout, R.W.; Coulon, D.; Mottram, P.; Sabliov, C.M. Biodistribution and toxicity of orally administered poly(lactic-co-glycolic) acid nanoparticles to F344 rats for 21 days. Nanomedicine 2016, 11, 1653–1669. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulation | Anticancer Agent | Target (Cell Line) | Surface Modification Method | Ref. |
|---|---|---|---|---|
| mPEG-PLGA NPs | Mitramycin | Pancreatic carcinoma (BxPC-3 and MIA Paca-2 cells) | Mitramycin was loaded onto mPEG-PLGA NPs by the single-emulsion solvent evaporation method using poloxamer 188 as a stabilizer | [87] |
| CS-modified PLGA NPs | PTX | Breast cancer (MDA-MB- 231 cells) | PTX-loaded PLGA NPs were prepared by the nanoprecipitation method using a high-gravity rotating packed bed. Then, PLGA NPs were modified with CS through electrostatic adherence | [88] |
| Pluronic® P85 or Tf-modified PLGA NPs | PTX | Glioma (C6 cells) | PTX-loaded PLGA NPs were prepared by the nanoprecipitation method. PLGA NPs were coated with Pluronic® P85 or conjugated with Tf | [89] |
| C24-LMWP peptide-modified PLGA NPs | DOX | Drug-resistant lung cancer (A549/T cells) and drug-resistant breast cancer (MCF-7/ADR cells) | Desalted DOX-loaded PLGA NPs were prepared by the nanoprecipitation method. Then, a C24-LMWP hybrid peptide was introduced to PLGA NPs by electrostatic interaction | [90] |
| HA-PEG- PLGA or CD-PEG-PLGA NPs | pDNA lipoplex | Glioblastoma (U87 cells) | HA or CD was conjugated to PLGA-PEG-NH2 using a reducing agent and a catalyst. HA-PEG-PLGA or CD-PEG-PLGA NPs were prepared by the dialysis method | [91] |
| PLGA-PEG-biotin NPs | SN-38 (active metabolite of irinotecan) | Breast cancer (4T1 cells) | NHS-Biotin and H2N-PEG-NH2 were conjugated. Then, PEG-biotin was conjugated to PLGA-NHS to synthesize PLGA-PEG-biotin. PLGA-PEG-biotin NPs were prepared by the modified emulsification solvent evaporation method | [92] |
| PLGA-PLL-PEG-Tf NPs | DNR | Leukemia (K562 cells) | DNR-loaded PLGA-PLL-PEG NPs were prepared by the modified double-emulsion solvent evaporation/diffusion method. Tf was conjugated to the surface of NPs with CDI | [93] |
| Anti-EGFR mAb-PLGA-PEG NPs | PTX | Triple-negative breast cancer (MDA-MB-468 cells) | PTX-loaded PLGA-PEG NPs were prepared by the nanoprecipitation method. The anti-EGFR mAb was anchored on the surface of NPs by crosslinking with MBS | [94] |
| CS-RGD-modified PLGA NPs | PTX or CDDP | Lung cancer (H1299 and A549 cells) | Drug-loaded PLGA NPs were prepared by the emulsification solvent evaporation (PTX) or double emulsion (CDDP) method. CS-RGD was synthesized by conjugating the GRGDSP peptide to chitosan via maleimide-PEG-NHS. CS-RGD was physically adsorbed onto the PLGA NPs | [95] |
| Glucose-PLGA | DTX | Human laryngeal carcinoma (Hep-2 cells) | DTX-loaded glucose-PLGA NPs were prepared by the single-emulsion solvent evaporation method. | [70] |
| CD133 aptamer- PEG-PLGA | Salinomycin | CD133-positive osteosarcoma (Saos-2 cells) and cancer stem cells | Salinomycin was loaded into PLGA-PEG-COOH NPs by the single-emulsion solvent evaporation method. CD133 aptamers were conjugated to PLGA-PEG-COOH NPs by EDC/NHS coupling. | [96] |
| Surface Functionalities | Mean Diameter (nm) | Type of Endocytosis | Ref. |
|---|---|---|---|
| AS1411 aptamer | 128 | Receptor (nucleotin)-mediated endocytosis | [109] |
| CD44 antibodies | 140 | Receptor (CD44)-mediated endocytosis | [110] |
| l-carnitine | 189 | Carrier (OCTN2)-mediated endocytosis | [111] |
| Glutamate- polyoxyethylene stearate | 152–181 | Carrier (LAT1)-mediated endocytosis | [112] |
| CS | 140–173 | Adsorption-mediated endocytosis | [88] |
| PEG-HIV-TAT | 97–176 | Adsorption-mediated endocytosis | [113] |
| Arginine-rich peptide | 156 | Adsorption-mediated endocytosis | [114] |
| Polymer | Drug | Targeting Material | Target Receptor (Cell Line) | Imaging Technique | Imaging Modality | Ref. |
|---|---|---|---|---|---|---|
| PLGA-mPEG | Platinum(II) prodrug | cRGD | Integrin (SKOV-3 cells) | US and NIRF | PFH and Cy7 | [128] |
| PLGA-PEG-COOH | PTX | Herceptin | HER2 receptor (SKBR-3 cells) | PA and US | PFH and SPIO | [129] |
| PLGA-PEG-COOH | DOX | AS1411 aptamer | Nucleolin receptor (C26 cells) | MR | SPIO | [134] |
| PLGA-PEG | DOX | Biotin | Biotin receptor (4T1 cells) | FL | DOX | [135] |
| PLGA-PEG-COOH | DOX | FA | Folate receptor (Bel-7402 cells) | US and MR | PFH and Fe3O4 | [133] |
| mPEG-PLGA-PLL | PTX | Anti-CA19-9 Ab | CA19-9 (capan-1 cells) | FL | DiR | [123] |
| Maleimide-PEG-PLGA | Curcumin | c(RGDf(N-me)V) | αvβ3, αvβ5, and α5β1 integrins (C6 cells) | FL | DiR | [124] |
| PLGA | DTX | Angiopep-2 | LRP-1 (U87-MG cells) | NIRF and X-ray | IR780 and gold nanoshell | [130] |
| PLGA-Glc | DTX | Glucose | Glucose transporter (Hep-2 cells) | NIRF | Cy5.5 | [70] |
| PLGA | DTX | HA | CD44 receptor (MDA-MB-231 cells) | NIRF | Cy5.5 | [65] |
| PLGA-PEG | Curcumin and PTX | T7 (HAIYPRH) peptide | Tf receptor (U87 cells) | FL, X-ray, and MR | DiR and MNP | [136] |
| PLGA | ZnPc | Anti-VEGFR-2 Ab | VEGFR-2 (MDA-MB-231 cells) | PA and US | ZnPc and PFH | [137] |
| Drug@Formulation | Target (Cell Line) | Functions | Therapeutic Benefits | Ref. |
|---|---|---|---|---|
| As2O3@PLGA-PEG/LA NPs | Liver cancer (HepG2 cells) | EPR effect; controlled release of As2O3 | 1.49- and 1.09-fold reduction in tumor volume compared with saline and free As2O3, respectively, in HepG2 tumor-bearing mice | [147] |
| CBP/ICG@FA-PEG-PLGA NPs | Breast cancer (MCF-7 cells) | EPR effect; targeted delivery via the folate receptor; combination of chemo, photodynamic, and photothermal therapy | The strongest tumor growth suppression potentials in NPs with NIR laser irradiation group rather than the other group | [157] |
| CPT@RVG-PLGA NPs | Glioblastoma (GL261-Luc2 cells) | Brain-specific delivery of CPT | Prolonged tumor doubling time and increased median survival (3.15/23 days) compared with either saline (2.46/16.5 days) or blank RVG-PLGA (2.50/19 days) in mice bearing intracranial GL261-Luc2 gliomas | [142] |
| CP@PLGA NPs | Colon cancer (DHD/K12PROb cells) | Higher activation of caspase-3-mediated apoptosis | Comparable reduction in tumor volume with free CP in DHD/K12PROb tumor-xenografted mice | [144] |
| CP@mPEG-PLGA NPs | Colorectal cancer (HT 29 cells) | EPR effect; prolonged CP residence in the systemic circulation | Increased survival rate of HT 29 tumor-bearing mice compared with saline, free CP, or blank NPs | [145] |
| DOX@lecithin-PLGA NPs | Glioblastoma (GB 101/8 cells) | Adsorption of apolipoprotein A-1 on the surface of the NPs and subsequent improvement of endocytosis into vascular endothelial cells via lipoprotein receptors | Reduced mean tumor area (9.6 ± 10.7 mm2) compared with vehicle (32.1 ± 3.8 mm2) and free DOX (21.7 ± 13.4 mm2) in rats with orthotopic glioblastoma | [141] |
| DOX@LMWP/PLGA NPs | Breast cancer (MCF-7/ADR cells) | Targeted nuclear delivery of DOX; tumor penetration by breaking down the diffusion barriers caused by interstitial fluid pressure | Near-complete tumor growth arrest in MCF-7/ADR tumor-bearing mice compared with vehicle, free DOX, or DOX-loaded PLGA NPs | [90] |
| DTX@HPLGA NPs | Lung cancer (A549-Luc cells) | Enhanced colloidal stability; superior tumor selectivity in vivo | Improved median survival (46 days) compared with vehicle (20 days), free DTX (34 days), and blank HPLGA NPs (24 days) in orthotopic A549-Luc lung xenografts | [143] |
| DTX@PLGA-PDA-TPGS NPs + NIR | Drug-resistant breast cancer (MCF-7/ADR cells) | Improved photothermal conservation by PDA; inhibition of P-glycoprotein by TPGS | Approximate 10-fold reduction in tumor size and weight compared with Taxotere® | [159] |
| DTX@ANG/GS/PLGA NPs + NIR | Glioblastoma (U87-MG cells) | DTX accumulation in the tumor; heat-induced tumor cell damage | The greatest tumor inhibition rate among all groups comprising saline, 808 nm irradiation, free DTX, GS/PLGA/DTX NPs, and ANG/GS/PLGA/DTX NPs. | [130] |
| GEM/BA@mPEG-PLGA NPs | Ehrlich ascites carcinoma (EAC cells) | Combination drug delivery; improved pharmacokinetic properties | Reduced mean tumor volume (195.5 mm3) compared with saline (1236.5 mm3), GEM solution (553.1 mm3), GEM NPs (367.8 mm3), or GEM + BA solution (213.5 mm3) in mice bearing Ehrlich tumors | [146] |
| MM@NaHCO3/PLGA NPs | Breast cancer (MCF-7 cells) | EPR effect; pH-responsive degradation of NPs due to CO2 bubbles generated from NaHCO3 and subsequent rapid release of MM in lysosomes | Highest tumor growth inhibition compared with vehicle, blank NPs, or MM–loaded NPs in MCF-7 tumor-xenografted mice | [150] |
| MTX@PANI-LT-PLGA NPs + NIR | Breast cancer (MDA-MB-231 cells) | Targeting somatostatin receptors by LT modification; hyperthermia effect | Higher tumor suppression compared with saline, free MTX, PANI PLGA NPs, MTX/PANI PLGA NPs, or MTX/PANI LT-PLGA NPs in mice | [160] |
| PTX@FA-PEG-PLGA NPs | Endometrial carcinoma (HEC-1A cells) | EPR effect; targeted delivery via the folate receptor | Higher anti-tumor efficacy than free PTX and non-targeted NPs in mice | [148] |
| PTX@AS1411-PEG-PLGA NPs | Glioma (C6 cells) | Targeted delivery to the tumor and angiogenic blood vessels by AS1411 aptamer | The highest average tumor inhibition based on tumor volume and weight (81.68 and 79.93%), compared with non-targeted NPs (66.95 and 68.69%) and Taxol® (68.69 and 46.75%) | [140] |
| PTX@iNGR-PEG-PLGA NPs | Glioblastoma (U87-MG cells) | Targeted delivery to angiogenic blood vessels and tumor penetration by iNGR | Prolonged median survival (42.5 days) compared with saline (17.5 days), PTX-loaded PEG-PLGA NPs (27 days), Taxol® (21.5 days), and PTX-loaded cNGR-PEG-PLGA NPs (34 days) in mice bearing intracranial U87-MG glioblastoma | [149] |
| Drug@Formulation | Animal Model | Pharmacokinetic Alterations | Ref. |
|---|---|---|---|
| DTX@PEG-PLGA NPs | Balb/C mice bearing C26 tumors | ▪ Plasma CL (mL/h/kg): 407.1 in solution; 148.4 in DTX-PEG-PLGA NPs | [161] |
| PTX@P85/Tf-PLGA NPs | SD rats bearing C6 glioma | ▪ Plasma AUC (μg·h/mL): 108.23 in solution; 362.52 in PLGA-NPs; 391.54 in P85-PLGA-NPs; 551.83 in Tf-PLGA-NPs ▪ Plasma t1/2 (h): 2.1 in solution; 3.96 in PLGA-NPs; 4.33 in P85-PLGA-NPs; 5.43 in Tf-PLGA-NPs | [162] |
| Lonidamine@PLGA-PEG-EGFR peptide NPs | Female nude mice xenografted with MDA-MB-231 tumors | ● Plasma ▪ AUC (μg·h/mL): 768.8 in solution; 992.44 in PLGA-PEG NPs; 1316.01 in PLGA-PEG-EGFR NPs ▪ MRT (h): 2.36 in solution; 3.77 in PLGA-PEG NPs; 4.38 in PLGA-PEG-EGFR NPs ▪ t1/2 (h): 1.67 in solution; 3.92 in PLGA-PEG NPs; 4.16 in PLGA-PEG-EGFR NPs ● Tumor ▪ AUC (μg·h/mL): 4.58 in solution; 32.09 in PLGA-PEG NPs; 49.96 in PLGA-PEG-EGFR NPs ▪ MRT (h): 1.94 in solution; 4.46 in PLGA-PEG NPs; 7.25 in PLGA-PEG-EGFR NPs ▪ λz (h): 0.63 in solution; 0.26 in PLGA-PEG NPs; 0.13 in PLGA-PEG-EGFR NPs | [163] |
| Pt@PLGA-PEG NPs | SD rats | ● Plasma ▪ AUC0–24 h (μg·h/mL): 14.7 in Pt prodrug; 48.9 in PLGA-PEG NPs ▪ Vd (mL/kg): 210.9 in Pt prodrug; 43.2 in PLGA-PEG NPs ● Blood ▪ AUC0–24 h (μg·h/mL): 3.8 in Pt prodrug; 12.7 in PLGA-PEG NPs ▪ Vd (mL/kg): 678.7 in Pt prodrug; 81.9 in PLGA-PEG NPs | [164] |
| ICG@PEG/FA-PLGA NPs | Female NCr mice xenografted with MDA-MB-231 tumors | ▪ AUC0–12 h ratio (PEG/FA-PLGA NPs: PLGA NPs): 3.45 in plasma;2.94 in tumor; 0.87 in liver | [166] |
| Bufalin@PEG-PLGA-PLL-RGD NPs | Mice bearing colon cancer | ▪ t1/2 (h): 3.35 in solution; 7.17 in PEG-PLGA-PLL-RGD NPs ▪ MRT (h): 3.45 in solution; 7.63 in PEG-PLGA-PLL-RGD NPs ▪ Vd (L/kg): 63.37 in solution; 187.83 in PEG-PLGA-PLL-RGD NPs | [167] |
| DTX@PLGA/HA NPs | SD rats | ▪ AUC (μg·h/L): 6110 in solution; 9394 in PLGA NPs; 23,175 in PLGA/HA NPs ▪ Vd (L/kg): 6.94 in solution; 7.52 in PLGA NPs; 3.16 in PLGA/HA NPs | [168] |
| DTX@PLGA-PEG-Apt NPs | SD rats | ▪ AUC (ng·h/mL): 1393.6 in solution; 3996.9 in PLGA-PEG NPS; 3807.4 in PLGA-PEG-Apt NPs ▪ CL (L/h): 3.588 in solution; 1.251 in PLGA-PEG NPS; 1.313 in PLGA-PEG-Apt NPs ▪ Vd (L): 0.501 in solution; 0.165 in PLGA-PEG NPS; 0.166 in PLGA-PEG-Apt NPs | [169] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, K.-T.; Lee, J.-Y.; Kim, D.-D.; Yoon, I.-S.; Cho, H.-J. Recent Progress in the Development of Poly(lactic-co-glycolic acid)-Based Nanostructures for Cancer Imaging and Therapy. Pharmaceutics 2019, 11, 280. https://doi.org/10.3390/pharmaceutics11060280
Kim K-T, Lee J-Y, Kim D-D, Yoon I-S, Cho H-J. Recent Progress in the Development of Poly(lactic-co-glycolic acid)-Based Nanostructures for Cancer Imaging and Therapy. Pharmaceutics. 2019; 11(6):280. https://doi.org/10.3390/pharmaceutics11060280
Chicago/Turabian StyleKim, Ki-Taek, Jae-Young Lee, Dae-Duk Kim, In-Soo Yoon, and Hyun-Jong Cho. 2019. "Recent Progress in the Development of Poly(lactic-co-glycolic acid)-Based Nanostructures for Cancer Imaging and Therapy" Pharmaceutics 11, no. 6: 280. https://doi.org/10.3390/pharmaceutics11060280