3.1. Production of Amorphous Solid Dispersions/Amorphous Polymeric Salts
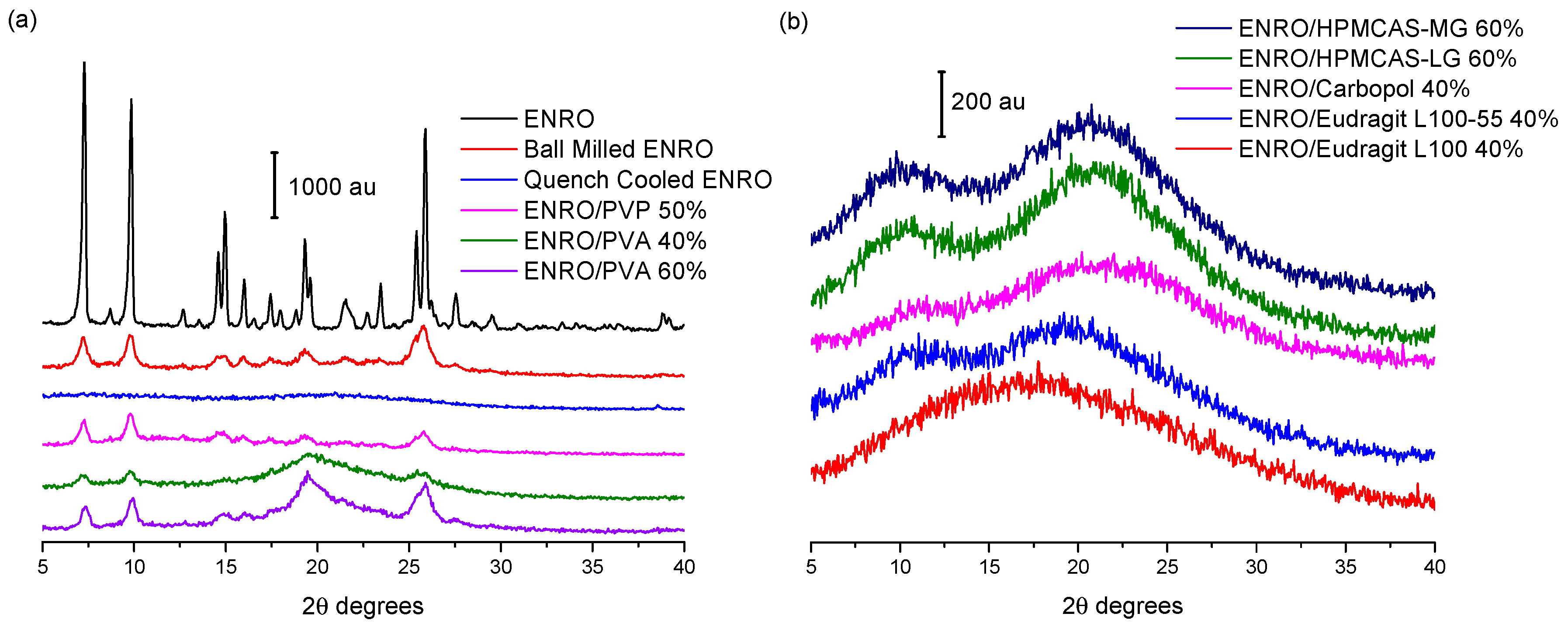
Ball milling was first carried out on crystalline “as received” ENRO to determine whether it is possible to amorphize the drug in this manner. However, following four hours of milling at room temperature, a disordered, semi-crystalline solid was obtained (
Figure 2a). This was also the case with CIP [
1]. The most intense peaks in the X-ray diffractogram of the unprocessed ENRO powder are visible at 7.4, 9.8, 14.9, and 25.8 2θ degrees. These peaks are also present in the diffractogram of ball milled ENRO; however, their intensity is reduced. Quench cooling ENRO, on the other hand, resulted in an X-ray amorphous material (
Figure 2a).
In previous studies with CIP, X-ray amorphous solid dispersions were obtained when the drug was ball milled with Eudragit L100, Eudragit L100-55, Carbopol, HPMCAS-LG, and HPMCAS-MG. All of these polymers are acidic, and FTIR analysis confirmed the presence of an ionic bond between the positively charged piperazine amino group of CIP and the carboxylate groups of the polymers in the ASDs [
9]. These acidic polymers also proved to be suitable co-formers for ENRO, with each resulting in an X-ray amorphous formulation (
Figure 2b). As was the case with CIP, a polymer concentration of 60% (
w/
w) was required to fully amorphize mixtures of CIP and HPMCAS, whereas 40% (
w/
w) was adequate for Eudragit L100, Eudragit L100-55, and Carbopol. Although the product obtained with 40% (
w/
w) HPMCAS-LG was almost X-ray amorphous following 4 h of milling, very small peaks could still be detected by PXRD at 9.8 and 25.8 2θ degrees, corresponding to the most prominent peaks of anhydrous ENRO (
Figure S1). A slightly more crystalline product was obtained with HPMCAS-MG under the same conditions, which decreased in intensity following a further 2 h of milling but did not disappear entirely. In contrast to CIP, which required a total of 6 h of milling and a reduced temperature of 2–5 °C to form ASDs with 60% (
w/
w) HPMCAS [
9], 4 h of milling at room temperature was adequate for the corresponding ENRO ASDs (
Figure S1). This indicates that the polymers may interact more readily with ENRO than CIP possibly due to the weaker crystal lattice of ENRO, which would facilitate its amorphization. These results show that the presence of an extra ethyl group in the structure of ENRO does not appear to negatively affect its ability to interact with these acidic polymers. To enable closer comparison with the equivalent CIP ASDs, the ENRO/HPMCAS ASDs containing 60% (
w/
w) polymer that were milled for 6 h were used for further studies.
In contrast to the acidic excipients, when CIP was milled with neutral polymers such as PVP and PVA at a concentration of 40–60% (
w/
w), a semi-crystalline product was obtained [
9]. This was also the case with ENRO (
Figure 2a). The fact that X-ray amorphous solid dispersions were only formed when ENRO was milled with acidic polymers containing carboxylic acid groups suggests that the drug is interacting with these substances via ionic bonds, as was the case with CIP. Likewise, in all of the ENRO salts produced by Karanam et al. containing an acidic counterion, proton transfer from the acid to the piperazine tertiary amine (N3) of the drug occurred, resulting in an ionic interaction between the two moieties [
2]. A similar reaction may take place between the N3 of ENRO and the polymers in these ASDs.
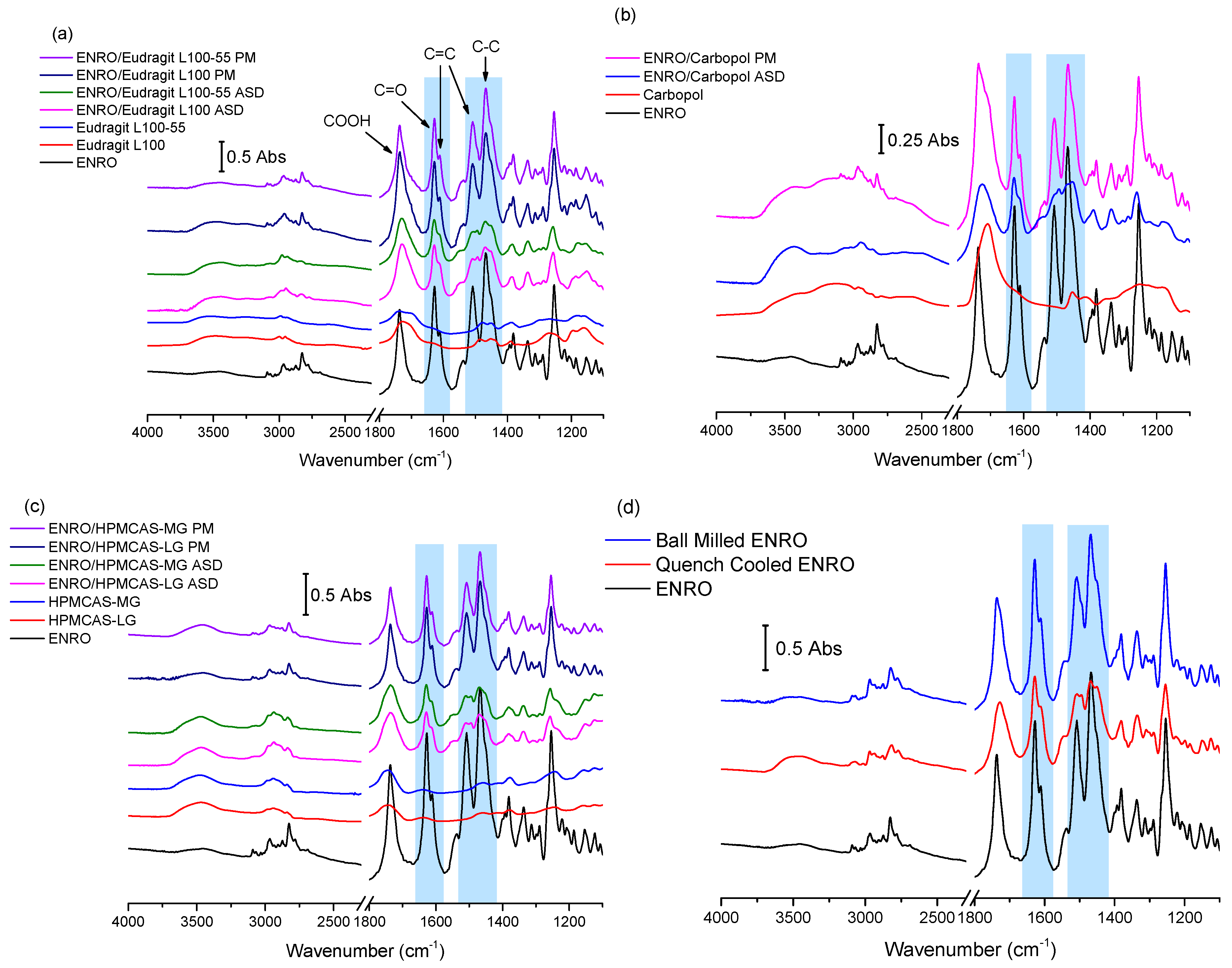
3.2. Solid-State Fourier Transform Infrared Spectroscopy
The results of FTIR analysis of the ASDs, PMs, and starting materials are shown in
Figure 3a–d. A sharp peak is located at 1737 cm
−1 in the spectrum of crystalline ENRO due to the carbonyl stretch of its unionized carboxylic acid group. While the process of ball milling introduced some disorder to the crystal lattice of ENRO, the FTIR spectrum of the ball milled drug was almost identical to the crystalline ENRO starting material. The greater molecular disorder of quench cooled ENRO, on the other hand, is evident in the broader and less intense peaks of its spectrum (
Figure 3d). Slight peak shifts were also seen with this sample, in particular, the carboxylic acid C=O stretch, which shifted to 1728 cm
−1. This can be attributed to changes in the drug’s intermolecular interactions upon amorphization, such as hydrogen bonding [
25]. Interestingly, the COOH carbonyl stretch of the drug also shifted to lower wavenumbers in the spectrum of the crystalline ENRO saccharinate salt, in which the piperazine N3 amino group of the drug is positively charged [
6]. This carbonyl peak underwent a similar shift with all of the ASDs. Therefore, while the carboxylic acid of ENRO remains unionized in the ASDs, changes in the hydrogen bonding of this group clearly occur upon amorphization. This shift may also be related to changes in the ionization state of the drug.
The main differences between the spectra of ENRO and the ASDs may be seen in the 1650–1450 cm−1 region. In the case of crystalline ENRO, the carbonyl stretch of its ketone group appears as a sharp, strongly absorbing peak at 1628 cm−1. The medium intensity shoulder at 1611 cm−1 may be assigned to C=C stretching vibrations of the drug’s aromatic ring. While these peaks are not significantly shifted in the spectra of the ASDs, differences in their relative absorbance were observed. In crystalline ENRO, the absorbance of the ketone peak is approximately 1.8 times greater than that of the aromatic peak. This ratio decreases to 1.5–1.7 for each of the ASDs. However, a similar decrease in the relative absorbance of these peaks was also seen with quench cooled ENRO and is therefore likely due to changes in the interactions of these groups upon amorphization.
The peaks at 1508 and 1469 cm
−1 in the spectrum of ENRO may be attributed to C=C stretching of the aromatic ring, and C–C stretching of the drug’s piperazine group, respectively [
26]. The shape of these peaks was altered notably in the ASDs, and the presence of multiple overlapping peaks became evident. In order to separate the individual peaks in this region and to quantify their relative absorbance, deconvolution of the spectra, with Gaussian peak fitting, was carried out. The resulting spectra are shown in
Figure S2. Deconvolution allowed the detection of a further peak at approximately 1453 cm
−1 in the spectrum of ENRO, which may be tentatively assigned to the C–H bending vibrations of the ethyl group. This peak is also present in the spectra of ball milled and quench cooled ENRO, and all of the ASDs, along with an additional peak at approximately 1494 cm
−1. Although a slight broadening is visible at this wavenumber in the spectrum of crystalline ENRO, it is not as distinct as with the other samples. Clear differences in the relative absorbance of these peaks may also be seen between the pure drug and ASDs. For instance, in crystalline ENRO, the area of the peak at 1508 cm
−1 is approximately two times smaller than the combined area of the peaks at 1469–1453 cm
−1. While a similar ratio was obtained with the equivalent peaks in ball milled ENRO, with the quench cooled form of the drug, it decreased to 1.9. With the ASDs, on the other hand, this ratio decreased further to 1.3–1.55. Similarly, in the spectra of crystalline and ball milled ENRO, the absorbance of the peak at 1469 cm
−1 is clearly greater than that at 1453 cm
−1. By contrast, in each of the ASDs, as well as quench cooled ENRO, the maximum absorbance of these peaks did not differ greatly. Similar changes in this region were seen in the spectra of the partially crystalline ENRO/PVA solid dispersion, whereas the less disordered ENRO/PVP more closely resembled the crystalline ENRO starting material (
Figure S3).
As previously mentioned, the terminal tertiary amine of ENRO (N3) may be protonated in these ASDs, forming ionic bonds with the carboxylate groups of the polymers. If this is the case, the main differences in the FTIR spectra of the ASDs compared to the starting materials or PMs can be attributed to the change in ionization state of the drug, and the presence of an additional
+N–H bond. Unfortunately, the
+N–H stretch is difficult to assign with certainty, as it will produce a weak band in the 3000–2600 cm
−1 region that possibly overlaps with others, such as that of the C–H stretch of the neighbouring aliphatic group [
26]. Similarly, the
+N–H bend of a tertiary amine salt generally appears as a very weak band in the 1610–1500 cm
−1 region [
27] and therefore is likely to be obscured by more intense peaks in the spectra of the ASDs. However, as described above, a number of differences in the 1450–1550 cm
−1 region of the spectra of ENRO and the ASDs were observed. Therefore, it is possible that the presence of the peak corresponding to the
+N–H bend contributed to the variations in this area of the spectra. In addition, as the peaks in this region correspond to groups surrounding the terminal amino group of ENRO, it is likely that they would be altered upon the protonation of N3.
The hypothesis that ENRO is protonated in these ASDs is supported by the FTIR analysis of ENRO salts conducted by Karanam et al. [
2]. In the spectra of each of the salts containing an acidic counterion, a decrease in the absorbance of the peak around 1469 cm
−1 relative to that at 1508 cm
−1 can be seen, in common with the ENRO ASDs. Single crystal X-ray diffraction confirmed that the N3 of the drug was protonated in these salts and formed an ionic bond with the carboxylate groups of the acids. Therefore, it is likely that ENRO is in the same cationic state in these ASDs and interacts with the acidic groups of the polymers to form amorphous polymeric salts (APSs). The fact that the spectrum of quench cooled ENRO is similar to that of the ASDs may be due to the partial conversion of the drug to the zwitterion.
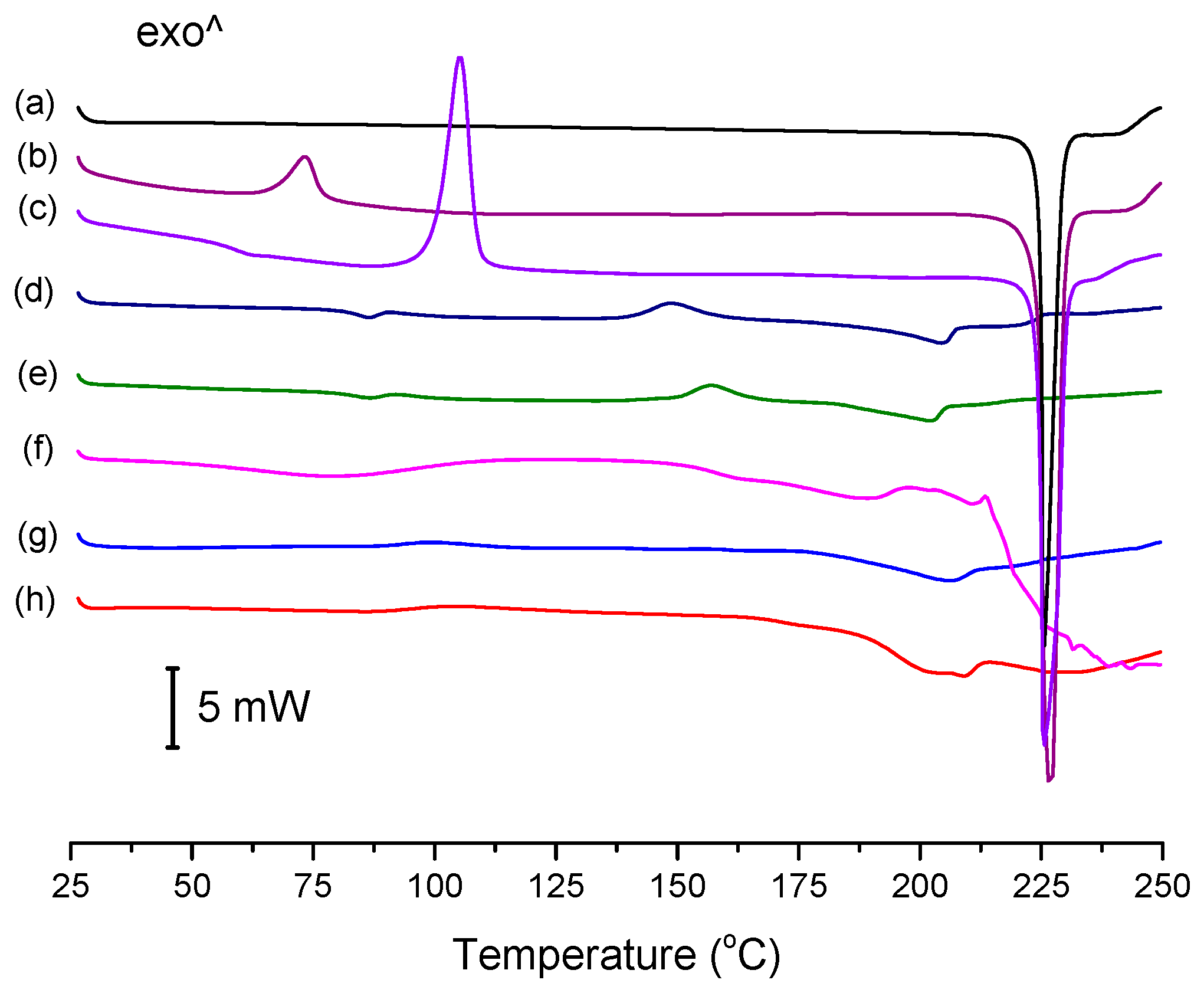
3.3. Thermal Analysis
The conventional DSC thermograms of ENRO and the ASDs are shown in
Figure 4. The melting point onset of crystalline ENRO, as well as the ball milled and quench cooled drug, was approximately 225 °C. Its lower melting point in comparison to CIP (approximately 272 °C) [
1] can be explained by the less extensive intermolecular bonds in ENRO. In contrast to the pure drug, the thermograms of the ASDs were missing a clear melting point. Similarly, the ASDs did not show distinct crystallization exotherms during DSC analysis, although the small, broad peaks visible at approximately 157 °C and 148 °C in the thermograms of ENRO/HPMCAS-LG and ENRO/HPMCAS-MG, respectively, may be due to some crystallization. The indistinct nature of the thermograms can be attributed to the amorphous nature of these formulations and their stability upon heating [
28]. By contrast, ball milled and quench cooled ENRO had clear crystallization peaks at approximately 73 °C and 106 °C, respectively, confirming their lower resistance to crystallization. The particularly low crystallization temperature of ball milled ENRO is to be expected, as the residual crystallinity present in this sample would enable crystal growth to occur more quickly upon heating.
The
Tg of quench cooled ENRO was detected at 58.9 °C, which is significantly lower than that of CIP (86.7 °C). Again, this may be attributed to the weaker intermolecular interactions present in ENRO. As a distinct
Tg could not be found for all of the ENRO ASDs using conventional DSC; they were therefore analyzed by MTDSC. The resultant
Tgs are listed in
Table 1. In each case, a single
Tg was detected. This suggests that the drug is miscible with each of these polymers, and that they form a single homogeneous phase [
29]. Due to its low amorphous content and high crystallization rate, no
Tg could be determined for ball milled ENRO with either DSC technique.
The Gordon–Taylor (G–T) equation was used to calculate the expected
Tgs of the ASDs, given their weight percentage of drug and polymer. From
Table 1, it can be seen that the experimental
Tgs of the ASDs containing Carbopol, Eudragit L100, and Eudragit L100-55 were substantially higher than the theoretically derived values. Such large positive deviations from the predicted
Tgs suggest that strong interactions exist between the components and are particularly indicative of polymeric salt formation [
30]. By contrast, the experimental and G–T
Tgs of the HPMCAS-containing ASDs differed by only a few degrees. This suggests that these polymers are fully miscible with ENRO but do not form strong interactions with the drug or that the heteromolecular drug–polymer interactions may be of a similar strength to the homomolecular interactions present in the individual raw materials [
31].
Similar results have been obtained with the equivalent CIP ASDs, whereby the experimental
Tgs of those containing Eudragit L100 and L100-55 deviated from the predicted values by a much greater degree than those containing either grade of HPMCAS. The apparently weaker interactions present in the latter ASDs were attributed to the lower proportion of carboxylic acid groups present in HPMCAS compared to the other polymers [
9]. This would explain why a polymer concentration of 60% (
w/
w) was required to produce X-ray amorphous solid dispersions with these polymers, whereas a concentration of 40% (
w/
w) was sufficient with the others.
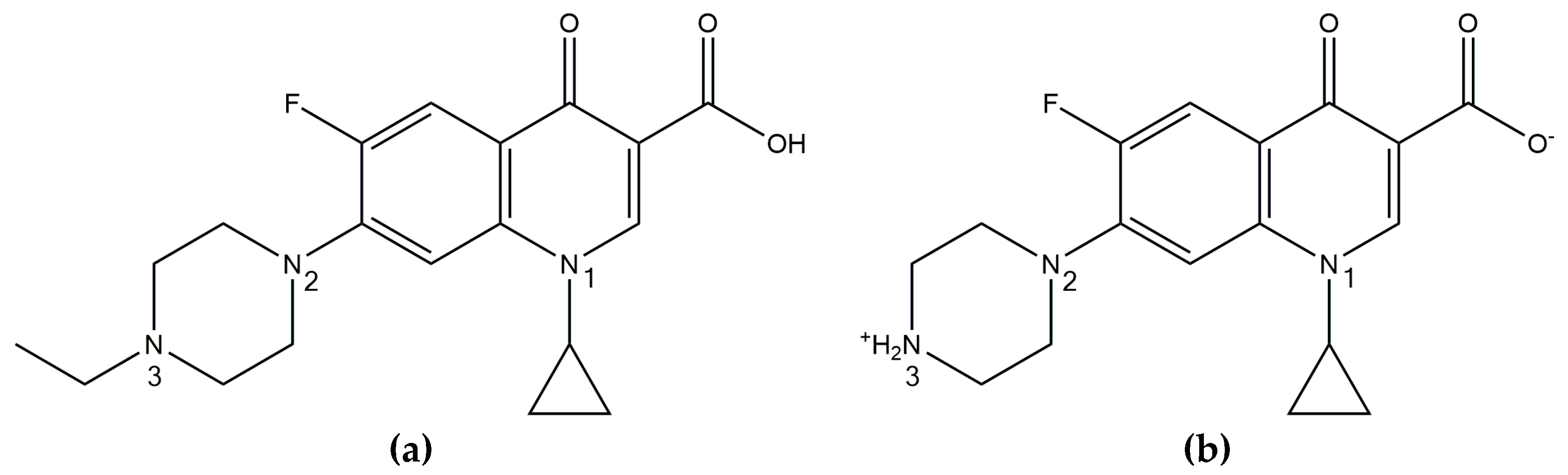
As previously mentioned, CIP exists as a zwitterion in the solid state, with a positively charged piperazine amino group and negatively charged carboxylate group. However, it has been shown to convert to the unionized form upon melting, due to intramolecular proton transfer. This was visualized as a small endothermic peak in the DSC thermogram of the drug, just prior to the melting endotherm. However, this low energy event was only visible when CIP was heated at 500 °C/min [
1]. HSDSC analysis was therefore carried out on crystalline ENRO in order to determine if it also undergoes proton transfer at high temperatures, in this case from the unionized form to the zwitterion. However, even when heated at the maximum heating rate of 500 °C/min, the drug did not show any evidence of solid-state transformation (
Figure S4). After heating ENRO to the endset of melting and allowing it to cool slowly, PXRD and FTIR analysis confirmed that the drug remained in the unionized anhydrous state; thus, the ethyl moiety attached to N3 prevented the proton transfer.
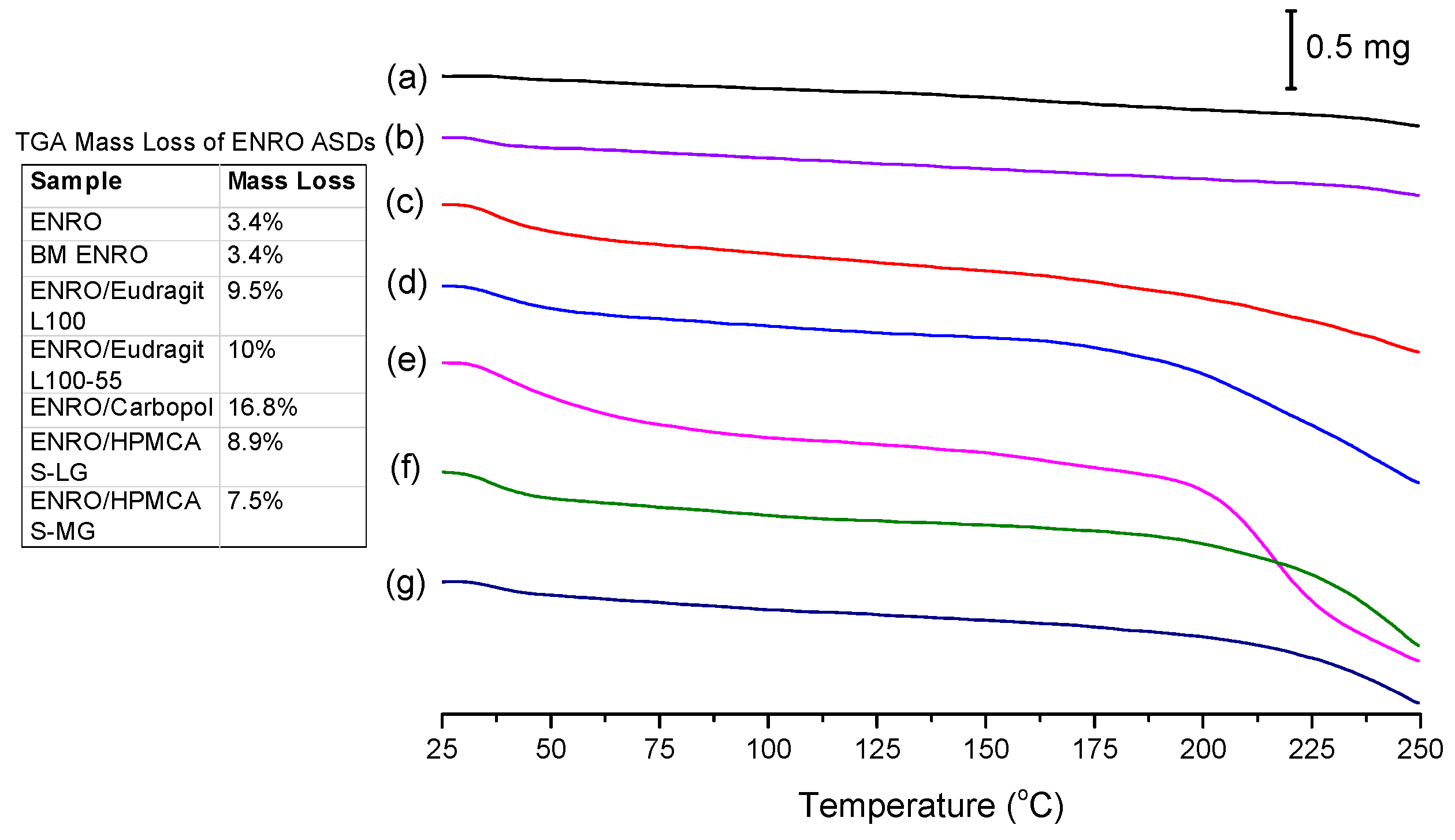
While crystalline ENRO is pale yellow, quench cooled ENRO is a more vibrant golden color, and when heated to 250 °C, the drug becomes dark orange/rusty. CIP also turns from off-white to a yellow color prior to melting; however, when heated past its melting point, it becomes brown due to substantial degradation. From the TGA curves obtained with ENRO and the ASDs (
Figure 5), crystalline and ball milled ENRO do not appear to undergo substantial thermal degradation, decreasing in mass by only 3.4% over the course of the TGA analysis. CIP, on the other hand, is much more prone to thermal degradation, with a mass loss of 12.8% and 17.3% being obtained with the crystalline and ball milled forms of the drug, respectively [
1]. An initial mass loss was observed below 70 °C with all of the amorphous formulations due to water evaporation. This is to be expected with ASDs, as the hygroscopic nature of amorphous drugs and polymers results in the absorption of atmospheric water vapor. The amorphous samples also degraded to a greater degree than the pure drug, in particular, the Carbopol ASD. Amorphous solids are typically more reactive than their crystalline counterparts, as their higher molecular mobility can enable such degradation reactions to occur [
32]. Alternatively, this mass loss may simply be due to degradation of the polymers.
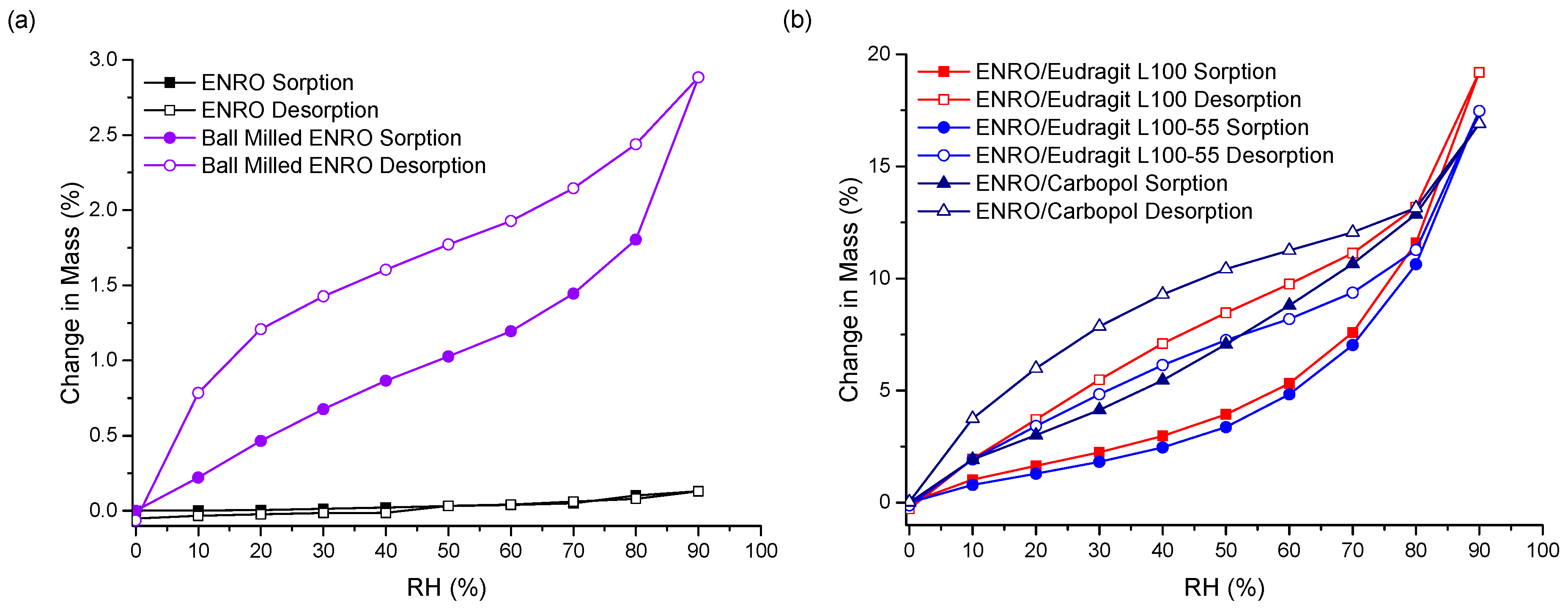
3.4. Water Sorption Studies
The stability of the ASDs when exposed to various humidity levels was examined by DVS. At the end of the sorption cycle, at 90% RH, ENRO absorbed only 0.13% (
w/
w) water. This increased to 2.9% for the ball milled drug, due to the increase in disordered material (
Figure 6a). CIP also absorbed low levels of water during DVS analysis, increasing in mass by only 0.6% (
w/
w) [
33]. PXRD analysis of the drugs at the end of the sorption studies revealed that both ENRO remained in the same solid state, with PXRD patterns matching those of the starting materials (
Figure S5) [
33]. In contrast to the crystalline drug, the ENRO ASDs were far more hygroscopic, absorbing 16–19% of their mass in water. Very similar levels of water uptake were observed with the CIP ASDs [
9]. The higher hygroscopicity of the amorphous formulations can be explained by the random orientation of their molecules. This leads to a larger free volume and enables the penetration of water into the samples [
34]. In addition, polymers are often more hygroscopic than the amorphous form of a drug, which increases the tendency of an ASD to take up moisture [
35].
As can be seen in
Figure 6b, the isotherms obtained with the ASDs containing Eudragit L100, Eudragit L100-55 and Carbopol were very similar in shape, with significant hysteresis. Hysteresis is commonly encountered with amorphous or porous solids, as water can absorb into the interior of the material [
36]. If water diffuses into the sample bulk more quickly than it can return to the surface, then, at the same RH level, a greater amount of moisture will be present during desorption than sorption, resulting in the appearance of hysteresis.
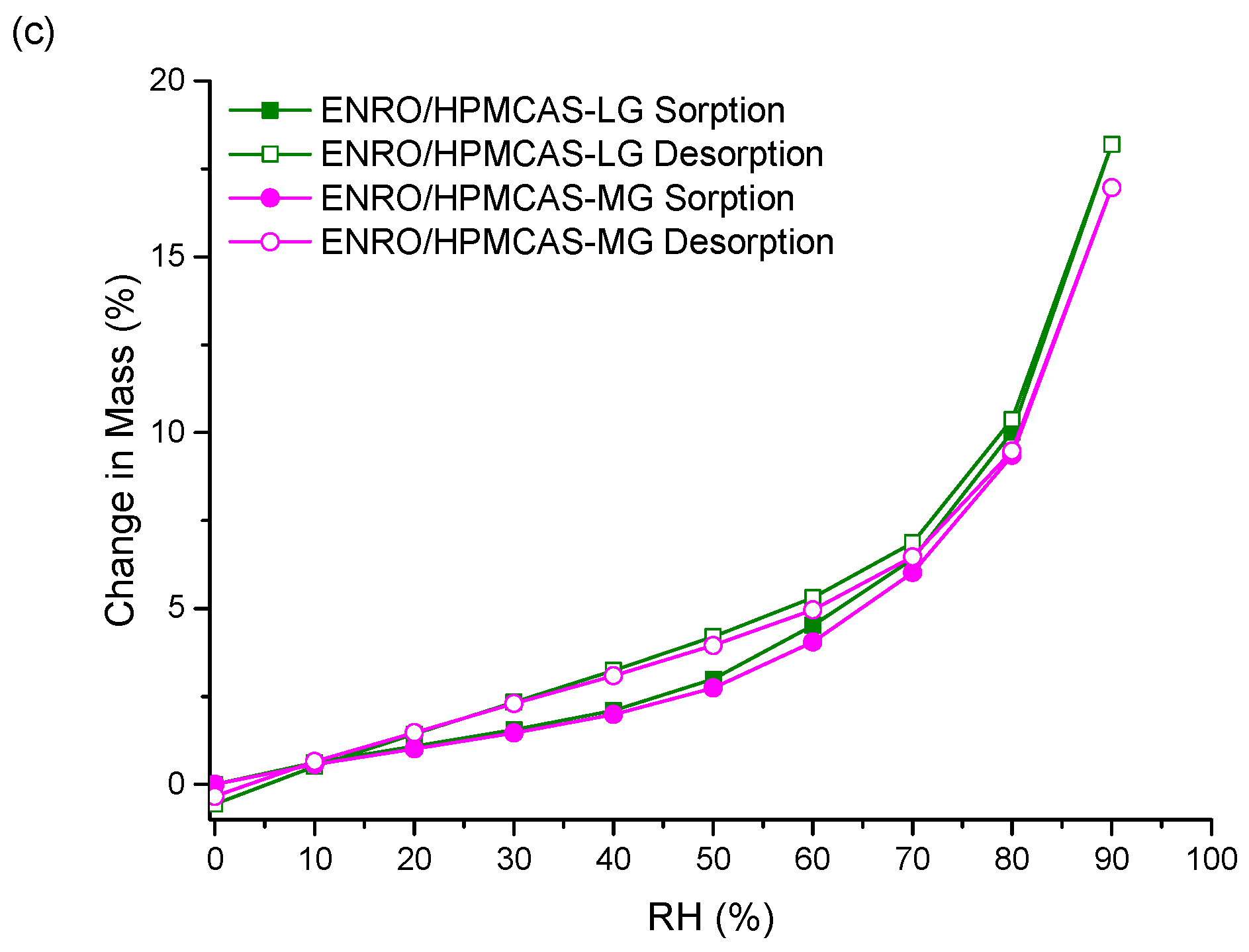
Unlike the other ASDs, the isotherms of both ENRO/HPMCAS ASDs were convex in shape with a small amount of hysteresis, suggesting that water was mainly adsorbed to the outer surfaces of these samples (
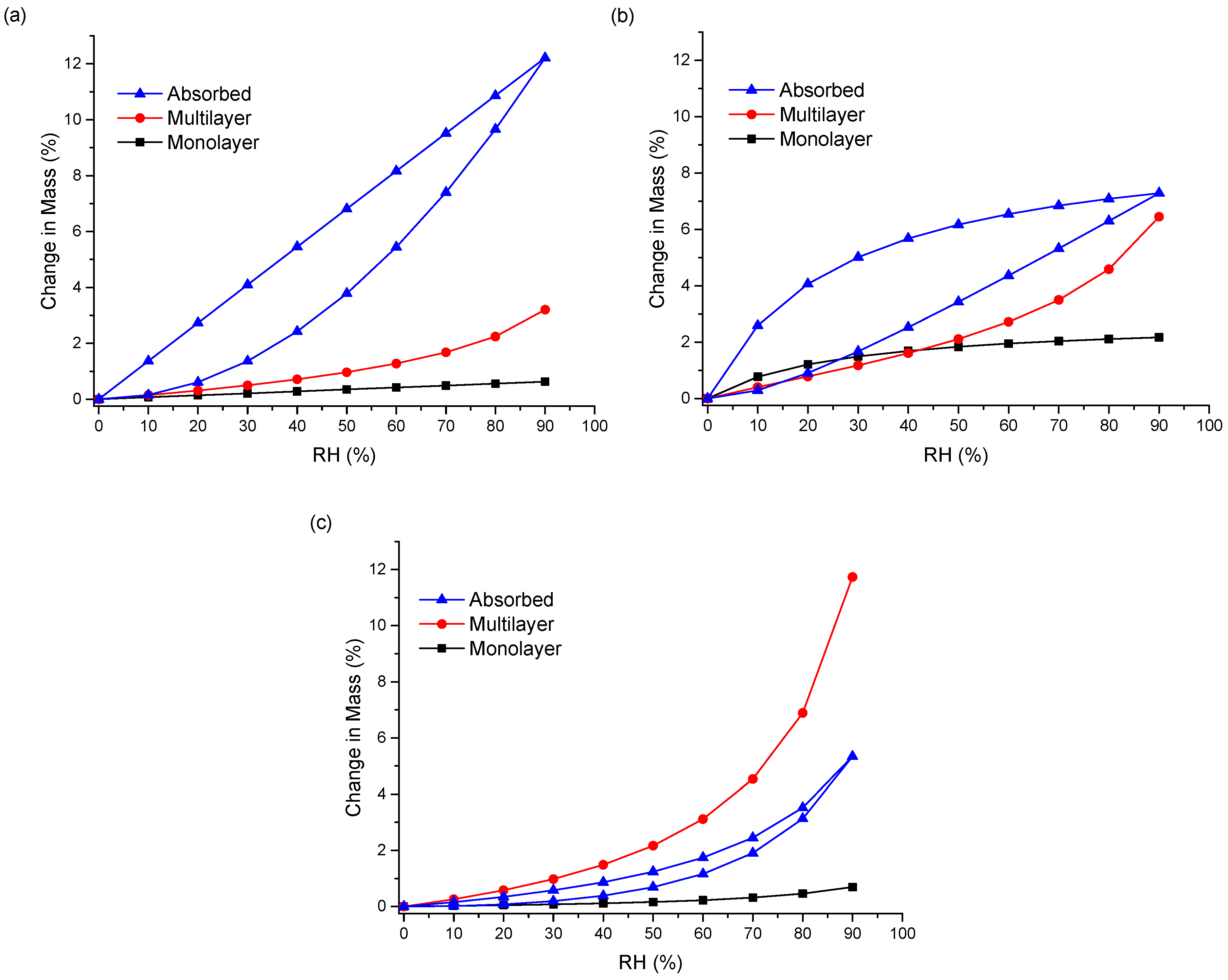
Figure 6c). Therefore, the water uptake behavior of the ENRO ASDs differs depending on the polymer used. This was further examined by fitting the sorption and desorption data to the Young–Nelson equations. According to the Young–Nelson model, water can be taken up by a sample in three different ways: adsorbed as a monomolecular layer, adsorbed as a multilayer, or absorbed into the interior of the solid [
22]. The parameters calculated using the Young–Nelson equations are listed in
Table S1, and the isotherms obtained using this approach are shown in
Figure 7 and
Figure S6. The corresponding CIP ASDs were also examined for comparison (
Figure S7).
As predicted from the DVS isotherms, the major water uptake mechanism of the ENRO ASDs containing Eudragit L100, Carbopol, and Eudragit L100-55 was water absorption (
Figure 7a,b and
Figure S6a). The small degree of absorption that occurred with the ENRO/HPMCAS ASDs confirms that they are somewhat porous, but less so than the other ASDs, as suggested by the minor hysteresis in their DVS isotherms. Unlike the other samples, the majority of water taken up by ENRO/HPMCAS ASDs was bound to their exterior surfaces as a multilayer. Multilayer formation begins at low RH levels and appears to occur simultaneously with monolayer adsorption (
Figure 7c and
Figure S6b). By contrast, the major water uptake mechanism for the CIP ASDs containing HPMCAS was absorption (
Figure S7). This suggests that the CIP/HPMCAS ASDs are more porous than the corresponding ENRO ASDs, or the polymers may be capable of swelling to a greater degree in the former formulations. As with the ENRO ASDs, water is primarily absorbed into the interior of the CIP ASDs containing Eudragit L100, Eudragit L100-55, and Carbopol. However, the water distribution patterns obtained with the ENRO and CIP ASDs containing Carbopol differed somewhat from the others. The monolayer adsorption of these samples increased more gradually over the course of the study and was also more extensive. This may be due to the presence of more hydrophilic groups on the surface of these ASDs, which can interact with water molecules [
23].
With both sets of ASDs, the highest value of E was obtained with those containing HPMCAS-LG, followed by HPMCAS-MG (
Table S1). However, this constant was more than 10 times larger for the ENRO/HPMCAS samples than those containing CIP. This indicates that water molecules form much stronger and extensive interactions with the surface of these samples [
37] and explains why water appears to be mainly adsorbed to the surface of these ASDs in a multilayer. The value of the regression coefficient, r, was ≥0.98 for all of the ASDs, showing that there was a good fit between the experimental and estimated values of the different parameters (
Table S1). Therefore, application of the Young–Nelson model is a suitable approach for comparing the water uptake of these samples.
The permeation of water molecules into the interior of an amorphous solid can increase its free volume, resulting in a decrease in
Tg [
38]. Water sorption is also known to increase the molecular mobility and thus crystallization rate of amorphous substances, and to decrease the crystallization onset temperature [
39]. However, despite the plasticizing effects of sorbed water, all five of the ENRO ASDs remained X-ray amorphous following DVS analysis (
Figure S5). This was also the case for the corresponding CIP polymeric ASDs [
9]. The high stability of these ASDs may be due to stabilizing drug–polymer interactions, the presence of which was suggested by the results of FTIR and DSC analysis. Polymers are also known to have anti-plasticizing effects and to reduce the molecular mobility of amorphous formulations, while steric hindrance from polymer chains can prevent the nucleation and crystal growth of drug molecules [
8,
40]. In contrast to the polymeric ASDs, amorphous CIP salts containing succinic acid or amino acids as counterions were unstable in humid environments and crystallized during DVS studies [
33].
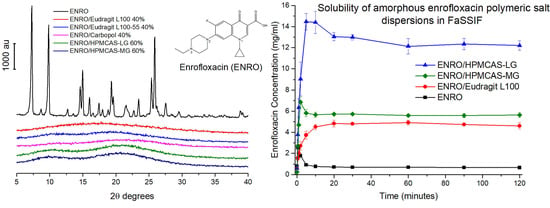
3.5. Solubility and Dissolution Studies
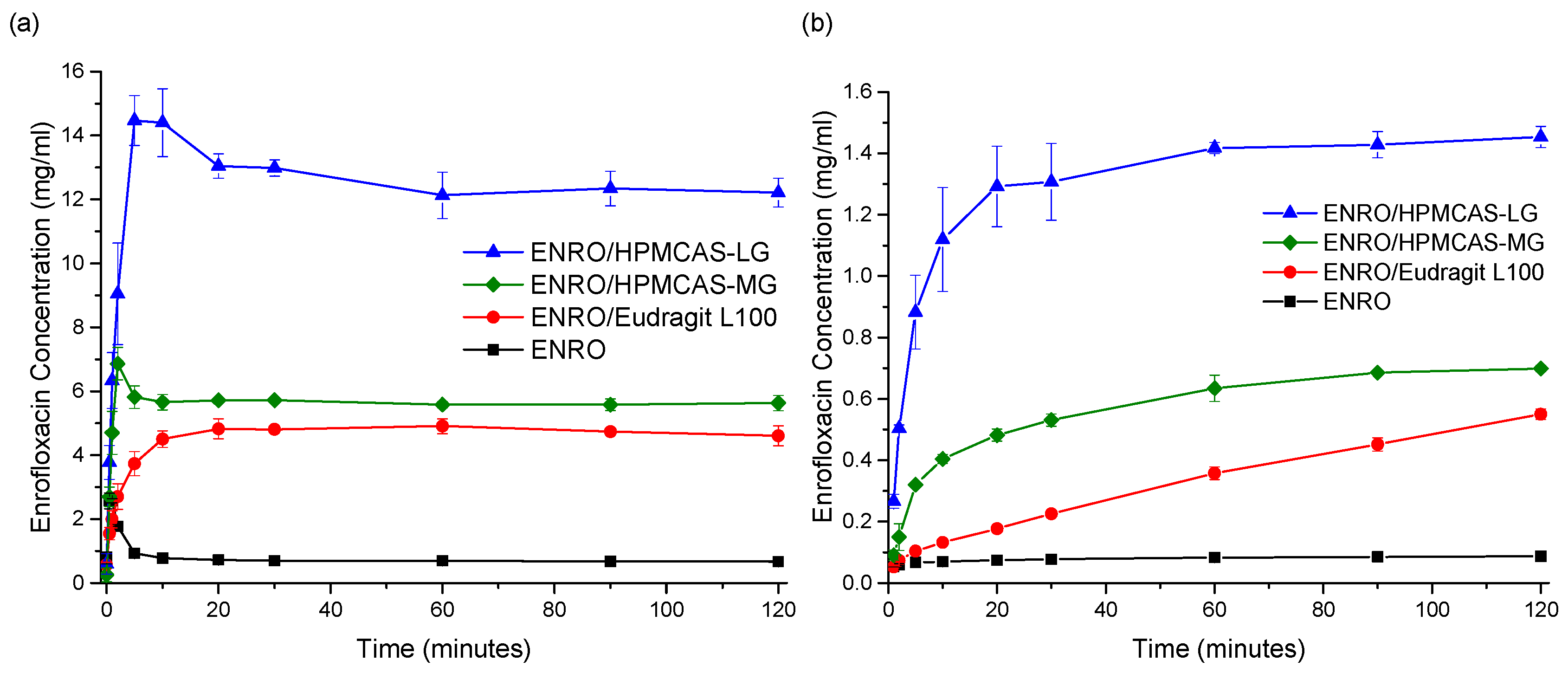
Due to issues with clumping and viscosity, solubility studies could not be carried out accurately on the ASDs containing Eudragit L100-55 and Carbopol, and these samples were therefore excluded from further studies. The superior solubility of the remaining ASDs in FaSSIF in comparison to crystalline ENRO is clear from
Figure 8a. With the pure drug, a peak in concentration was seen at 30–60 s, which then quickly fell to a constant level of approximately 0.7 mg/mL. A steep initial increase in drug concentration was also seen with the ASDs containing HPMCAS-LG and HPMCAS-MG, which peaked after 5 and 2 min, respectively. While this supersaturation then fell after 10–15 min, the concentration was still significantly higher than that obtained with crystalline ENRO. This solubility enhancement was sustained for the remainder of the study, with final concentrations of 12.2 mg/mL and 5.6 mg/mL being obtained with ENRO/HPMCAS-LG and ENRO/HPMCAS-MG, respectively. In contrast to the other samples, a more gradual increase in drug concentration was seen with ENRO/Eudragit L100, followed by a plateau after 20 min. This sample was also less soluble than those containing HPMCAS, reaching a concentration of 4.6 mg/mL after 2 h.
The dissolution behavior observed with these ASDs is similar to that described by the “spring” and “parachute” model [
41]. In this model, ASDs are described as “springs”, as their high energy and lack of a crystal lattice results in rapid drug dissolution and supersaturation. However, this supersaturated state is thermodynamically unstable, and crystallization of a lower energy, less soluble form of the drug soon follows. Fortunately, excipients such as polymers may be used to inhibit or delay the precipitation of dissolved drug and thus act as “parachutes” [
41]. Polymers can prevent nucleation and crystal growth via interactions with the drug, steric hindrance, and increased viscosity [
42]. Although the concentration obtained with the ENRO/HPMCAS ASDs did decrease somewhat over the course of the study, the polymers present in these ASDs most likely prevented extensive crystallization of the drug in solution, enabling supersaturation to be maintained for at least 2 h. By avoiding the rapid generation of supersaturation, less nucleation and crystallization would be expected to occur with the ENRO/Eudragit L100 ASD. This was confirmed by PXRD analysis of the excess solid recovered at the end of the solubility studies. In each case, enrofloxacin hexahydrate [
2] was detected; however, with ENRO/Eudragit L100, the sample was far less crystalline (
Figure S8).
Similarly enhanced concentrations were obtained with the ENRO ASDs in dissolution studies in comparison to crystalline ENRO. Following 2 h, the highest concentration was achieved with ENRO/HPMCAS-LG, at 1.45 ± 0.03 mg/mL (44.8 ± 1.2% of ENRO released), followed by ENRO/HPMCAS-MG (0.70 ± 0.01 mg/mL, 40.5 ± 0.6% of ENRO released) and ENRO/Eudragit L100 (0.55 ± 0.02 mg/mL, 57.4 ± 1.8% of ENRO released). Crystalline ENRO, on the other hand, only attained 0.09 ± 0.00 mg/mL (104.7 ± 4.5% of ENRO released) over the course of the study (
Figure 8b). Apart from concentration, the ASDs also differed in the shape of their dissolution profiles. With ENRO/HPMCAS-LG, the drug concentration increased quite rapidly at the start of the study and then remained fairly constant for the remainder. While a similar profile was obtained with ENRO/HPMCAS-MG, the initial drug release was more gradual than with the LG grade of polymer. As was the case in the solubility study, the final concentration obtained with ENRO/HPMCAS-MG was approximately half that of ENRO/HPMCAS-LG. However, ASDs containing different grades of HPMCAS are known to demonstrate different rates and extents of drug release, due to differences in their succinoyl and acetyl content [
43]. This may affect the pH of the diffusion layer surrounding the ASD particles, or the strength of drug–polymer interactions.
A steady, linear increase in drug concentration was observed with ENRO/Eudragit L100. As no leveling off occurred during the study, it is possible that the drug concentration would continue to rise during longer-term studies, similar to an extended release formulation. The gradual dissolution of ENRO from this ASD may be due to strong drug–polymer interactions, which could delay the dissociation and dissolution of the drug [
44]. Such interactions would also explain the higher than predicted
Tg of this formulation and the absence of crystallization during DSC analysis, unlike the ASDs containing HPMCAS. Alternatively, this polymer may be less soluble than HPMCAS, which would reduce the diffusion of water into the ASD and, thus, its dissolution rate.
Visible differences in the behavior of the ENRO ASD powders were also evident during dissolution studies. Both ENRO/Eudragit L100 and ENRO/HPMCAS-MG formed clumps when added to the dissolution vessels. While these eventually dissolved in the case of ENRO/HPMCAS-MG, with ENRO/Eudragit L100, they remained largely intact for the duration of the study. This would have hindered the release of the drug and reduced the surface area exposed to the dissolution medium. By contrast, no clumping was observed with ENRO/HPMCAS-LG, which enabled faster dissolution and higher concentrations of ENRO to be achieved.
From the results of this study and that of a previous investigation involving CIP, it can be concluded that ENRO is the more soluble of the two fluoroquinolones in FaSSIF. CIP was found to have a solubility of only 0.14 mg/mL in this medium [
9], which is five times lower than that of ENRO. Higher drug concentrations were also obtained with the ENRO ASDs than the equivalent CIP ASDs. Similarly, ENRO has been reported to be more soluble than CIP in pH 7.4 phosphate buffer [
5]. As previously mentioned, the greater solubility of ENRO may be explained by its weaker crystal lattice, which would facilitate the release of drug molecules into solution.
3.6. Bacterial Studies
The minimum inhibitory concentrations (MICs) and minimum bactericidal concentrations (MBCs) of ENRO and the ASDs are listed in
Table 2. Values that differ significantly from those of ENRO are shown in bold. In each case, the MBC should be greater than the MIC, as a larger quantity of drug is required to bring about bacterial death rather than growth inhibition. If the ratio of MBC to MIC is ≤4, this indicates that a drug is bactericidal [
45], which was the case for ENRO and the ASDs in all species of bacteria in this study. A MIC of ≤0.5 µg/mL may be considered as susceptible to ENRO, while ≥2 µg/mL indicates bacterial resistance, and 1 µg/mL is intermediate [
46]. Therefore, from the results of this study, it can be concluded that
E. coli,
S. aureus, and
K. pneumoniae are susceptible to ENRO, while
P. aeruginosa is not. As was the case with CIP,
E. coli was found to be the most susceptible of these bacteria to ENRO, having a MIC of 0.004–0.0016 µg/mL. Quite low MIC levels were also obtained in
K. pneumoniae (0.032–0.125 µg/mL), followed by
S. aureus (MIC 0.125–0.25 µg/mL). By contrast, much higher MIC and MBC values of 4–8 µg/mL were obtained with
P. aeruginosa. However, the outer membrane of this bacteria is known to be far less permeable than that of
E. coli, while fluoroquinolones are also believed to be substrates for an active efflux system within
P. aeruginosa [
47]. In each case, the MIC values obtained for ENRO in these four species agree well with those reported previously [
46].
As can be seen from
Table 2, the formulation of ENRO as an ASD did not significantly affect its antibacterial activity in any species of bacteria, while the MIC and MBC obtained with ENRO/HPMCAS-MG was significantly lower in
E. coli and
K. pneumoniae than in the pure drug. Similar results were previously obtained with CIP ASDs, whereby the MIC and MBC of CIP/HPMCAS-MG was significantly lower than crystalline CIP in all four of these species, while the MIC of CIP/HPMCAS-LG was also significantly reduced in
E. coli, and its MBC was lower in both
E. coli and
S. aureus. These ASDs were also found to increase the passive transmembrane permeability of CIP [
9]. Therefore, it is possible that the formulation of ENRO as an ASD with HPMCAS-MG also improved its permeability, enabling more of the drug to be transported through the bacterial cell membranes via passive diffusion.













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}