Design and Characterization of Inulin Conjugate for Improved Intracellular and Targeted Delivery of Pyrazinoic Acid to Monocytes

 and
and 
Abstract
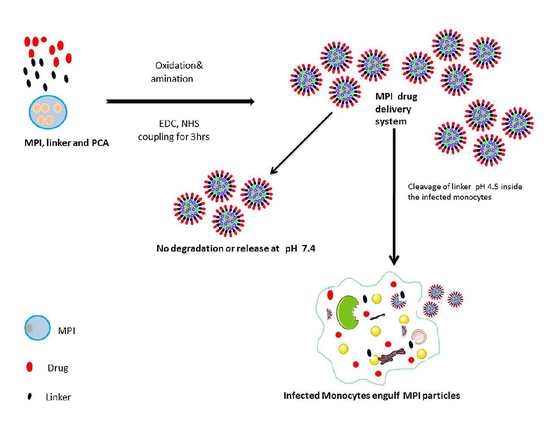
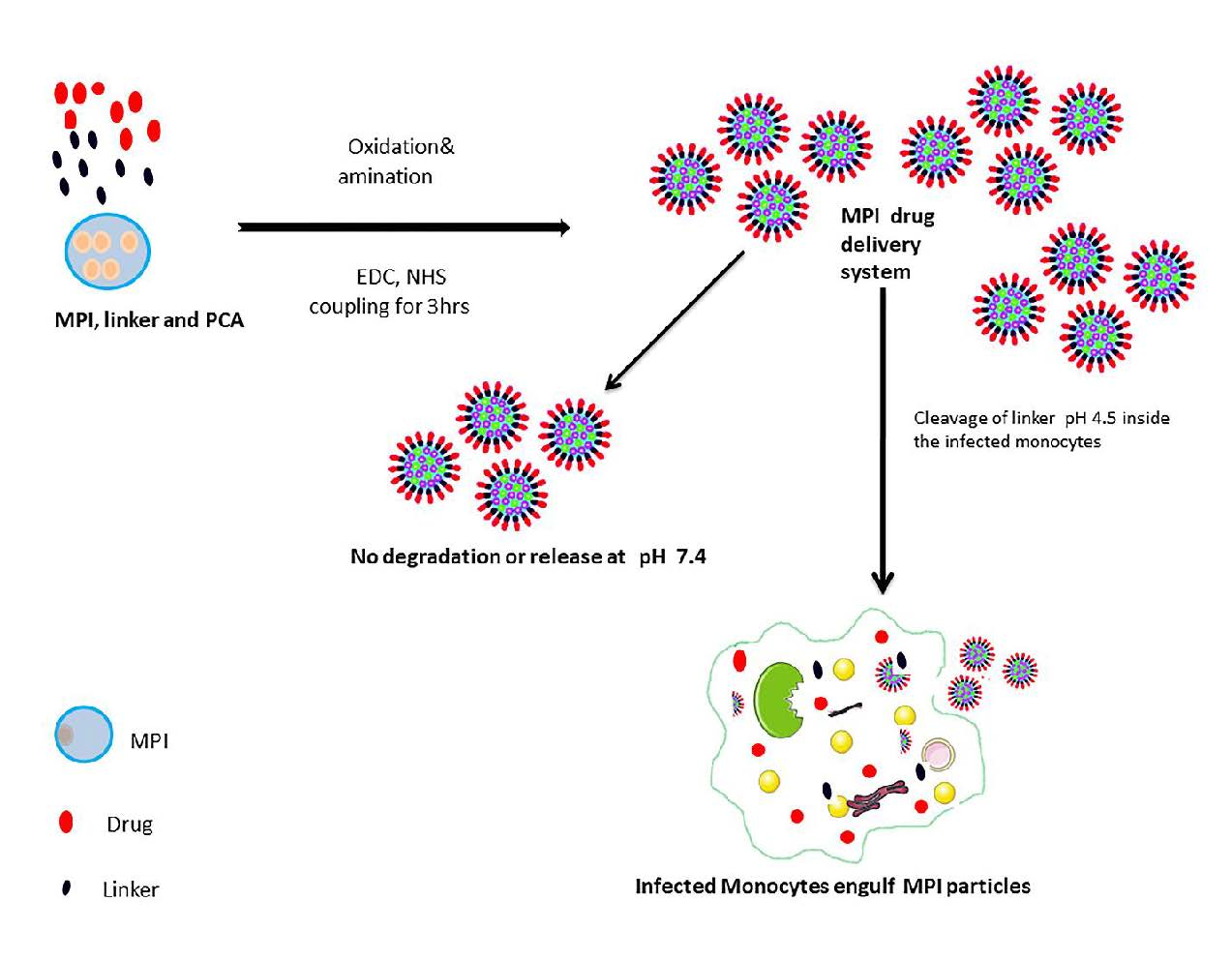
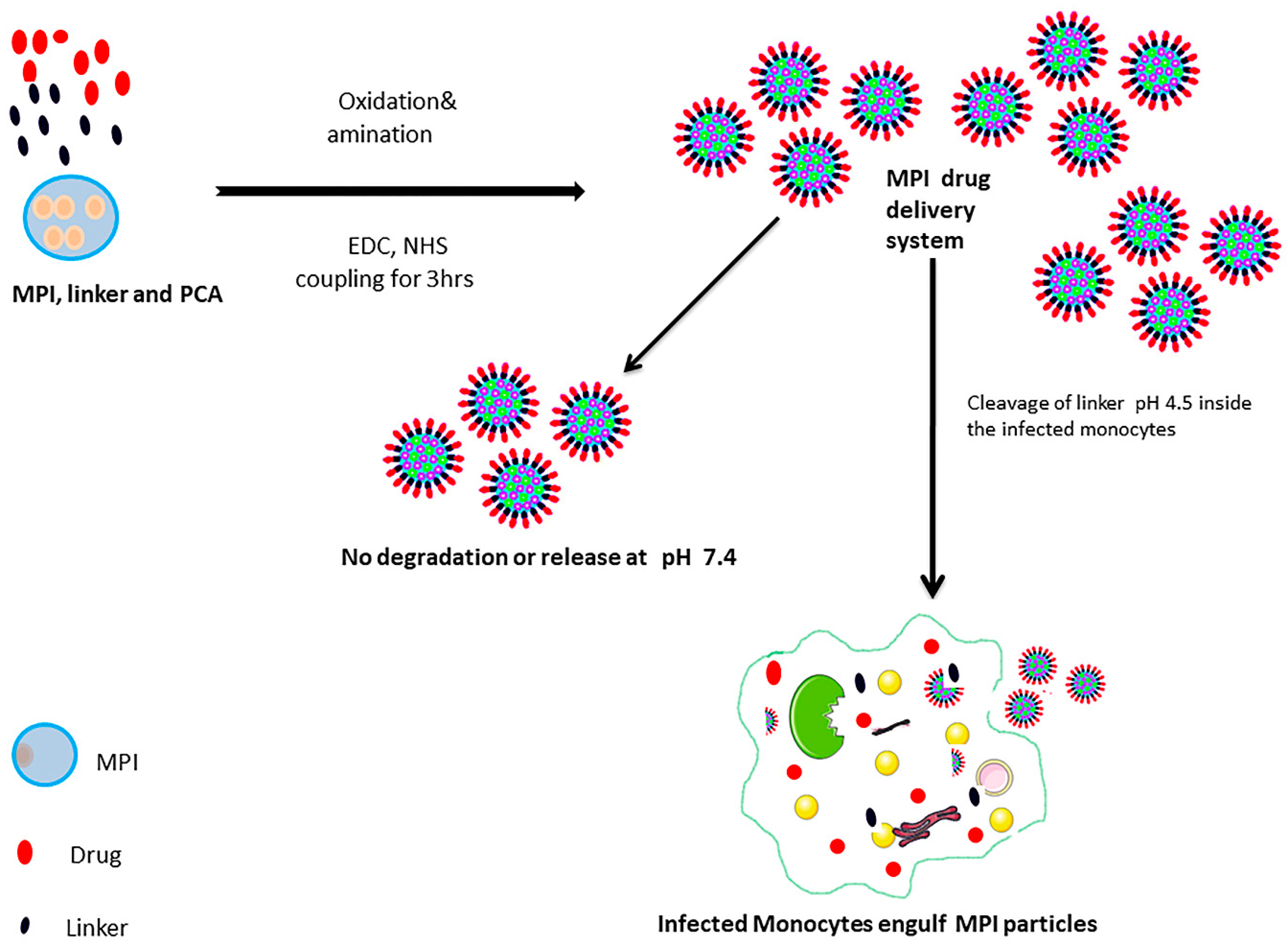
:
{kind=link}
{kind=link}
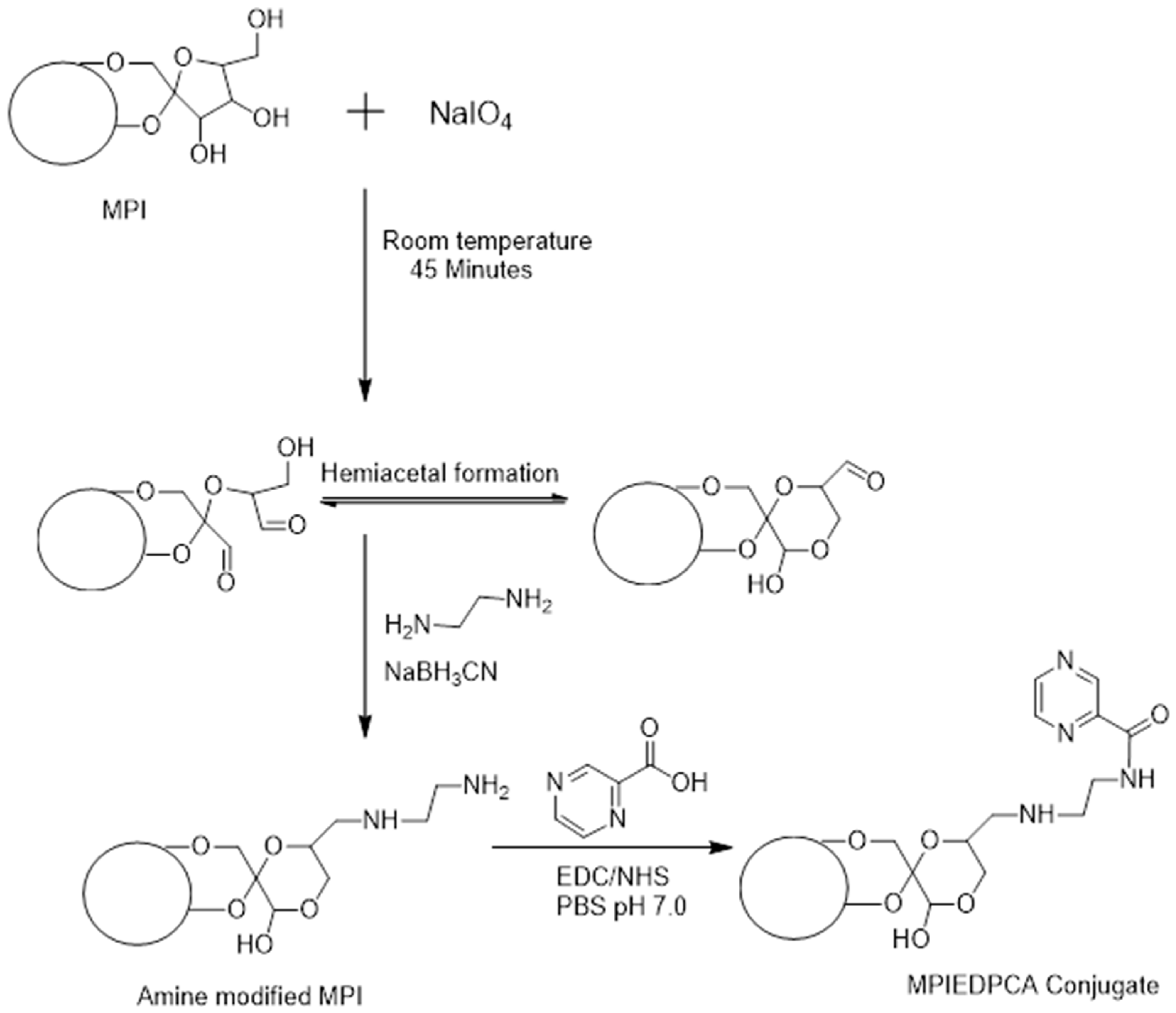{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Experimental/Methods
Oxidation and Diamine Coupling of Inulin Particles
2.3. Diamine Coupling of Oxidized MPI Particles
2.4. PCA Loading onto Amine Modified MPI Particles
2.5. Characterization of MPI-ED-PCA
2.5.1. Fourier Transform Infrared Spectroscopy (FTIR)
2.5.2. Proton Nuclear Magnetic Resonance (1H NMR) Spectroscopy NMR
2.5.3. Zeta Potential Measurements
2.5.4. Thermogravimetric Analysis (TGA)
2.5.5. Differential Scanning Calorimetry (DSC)
2.5.6. Drug Loading
2.5.7. In-Vitro Cleavage
2.5.8. Efficient Uptake of FITC Labelled MPI Particles by RAW 264.7 Macrophage Cells
2.5.9. Flow Cytometry Measurements (FACS)
3. Results/Discussion
3.1. Synthesis and Physicochemical Characterization of MPIEDPCA
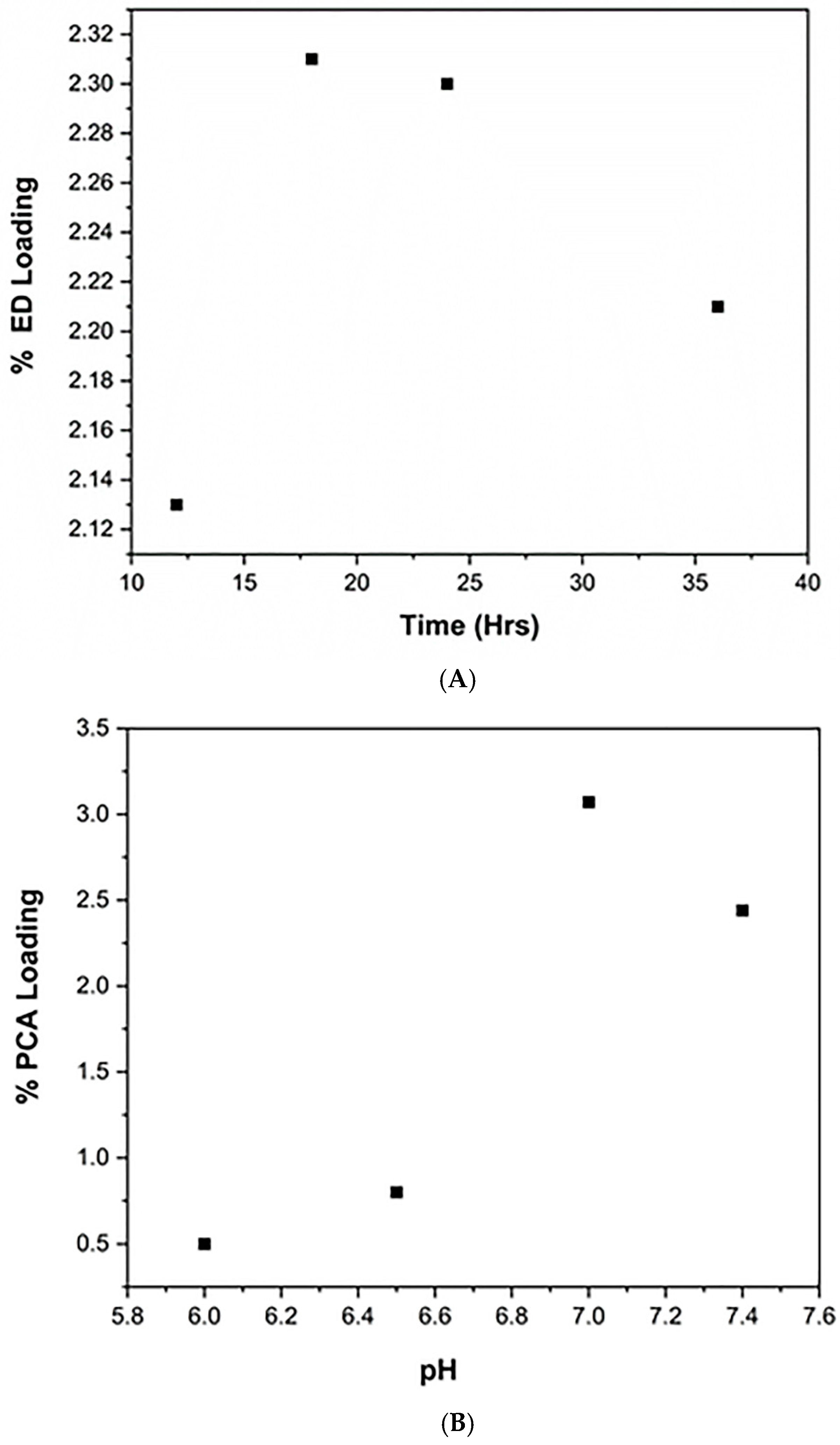
3.2. Optimization of Diamine MPI and PCA Coupling
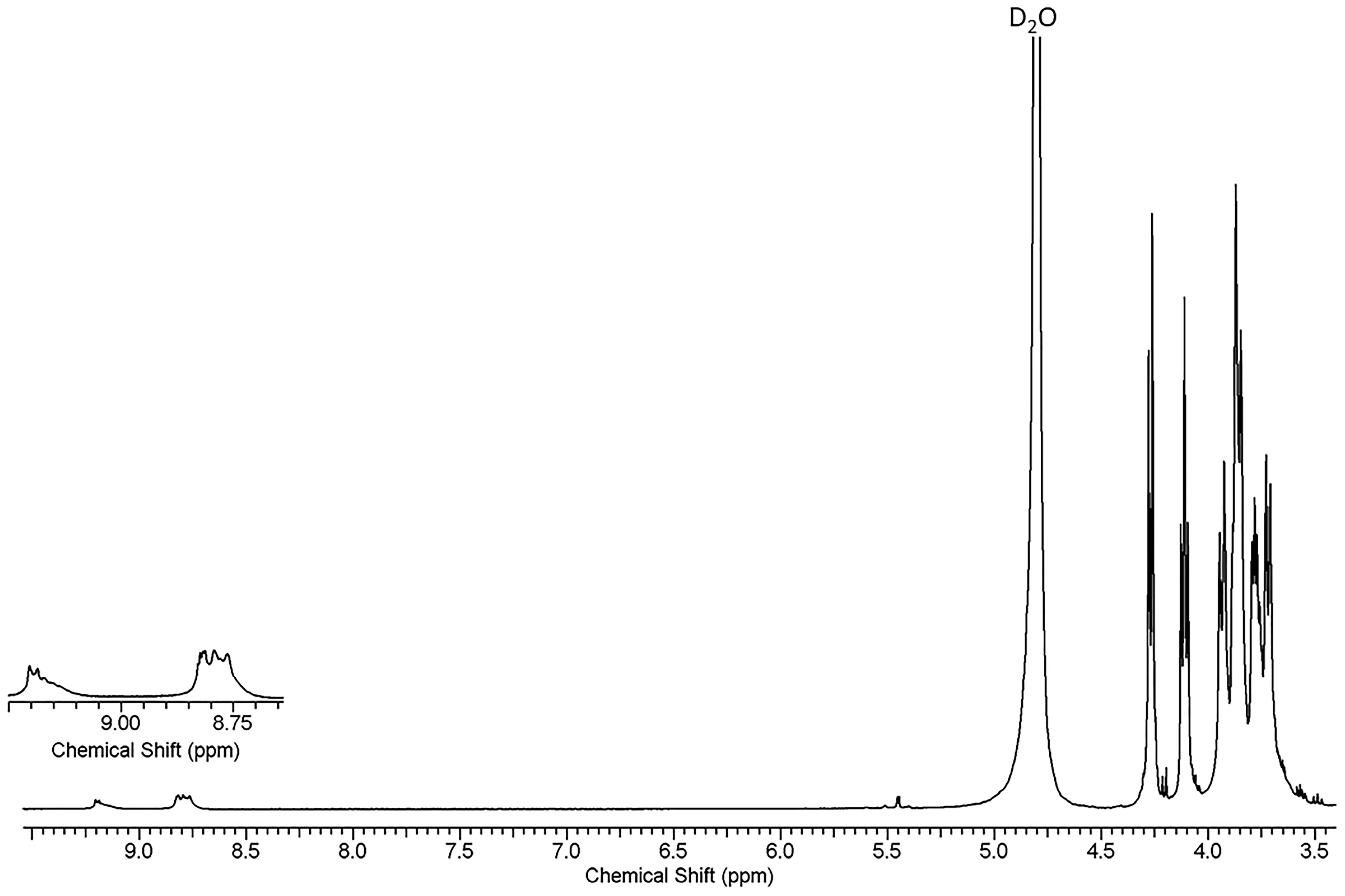
3.3. Characterization Using 1h NMR Spectroscopy
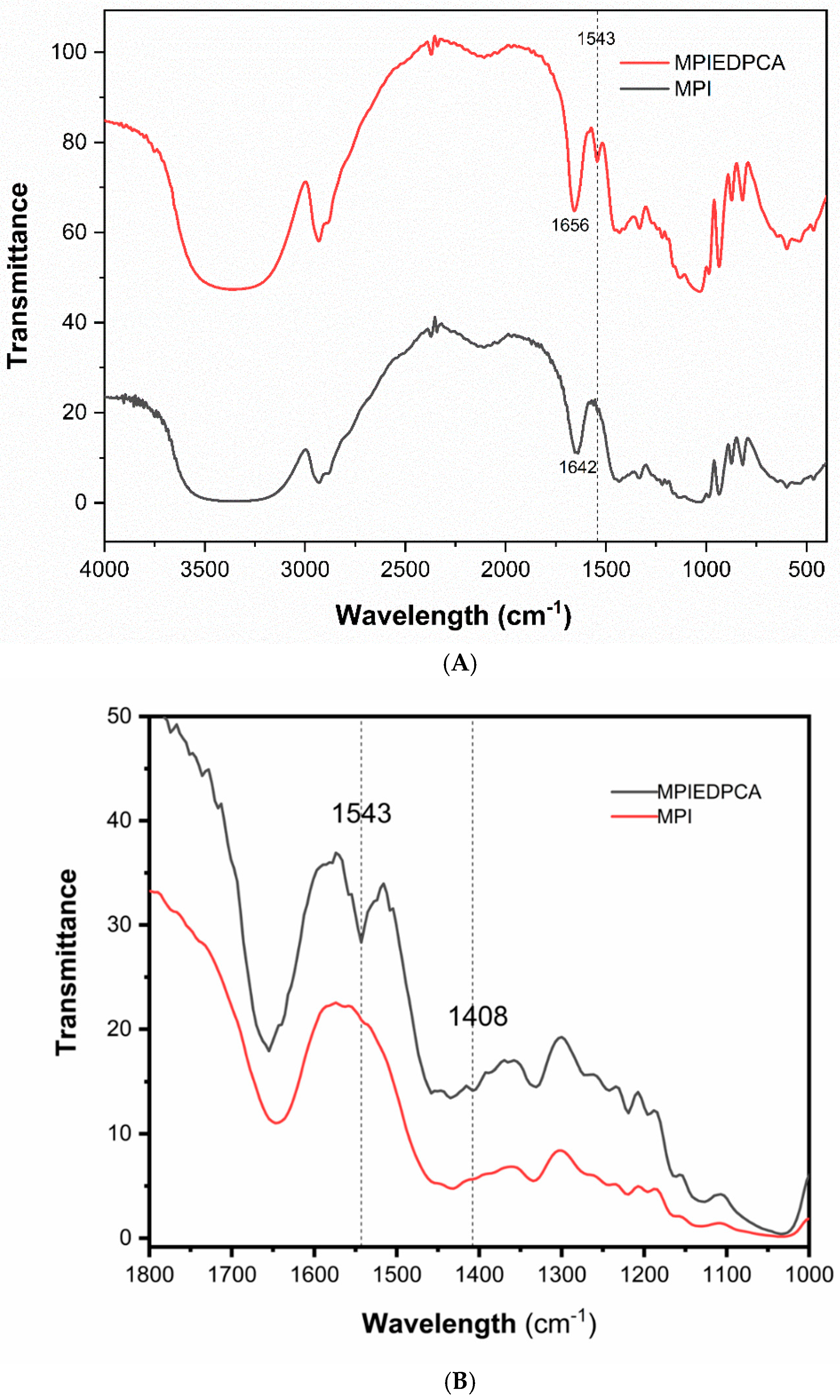
3.4. FTIR
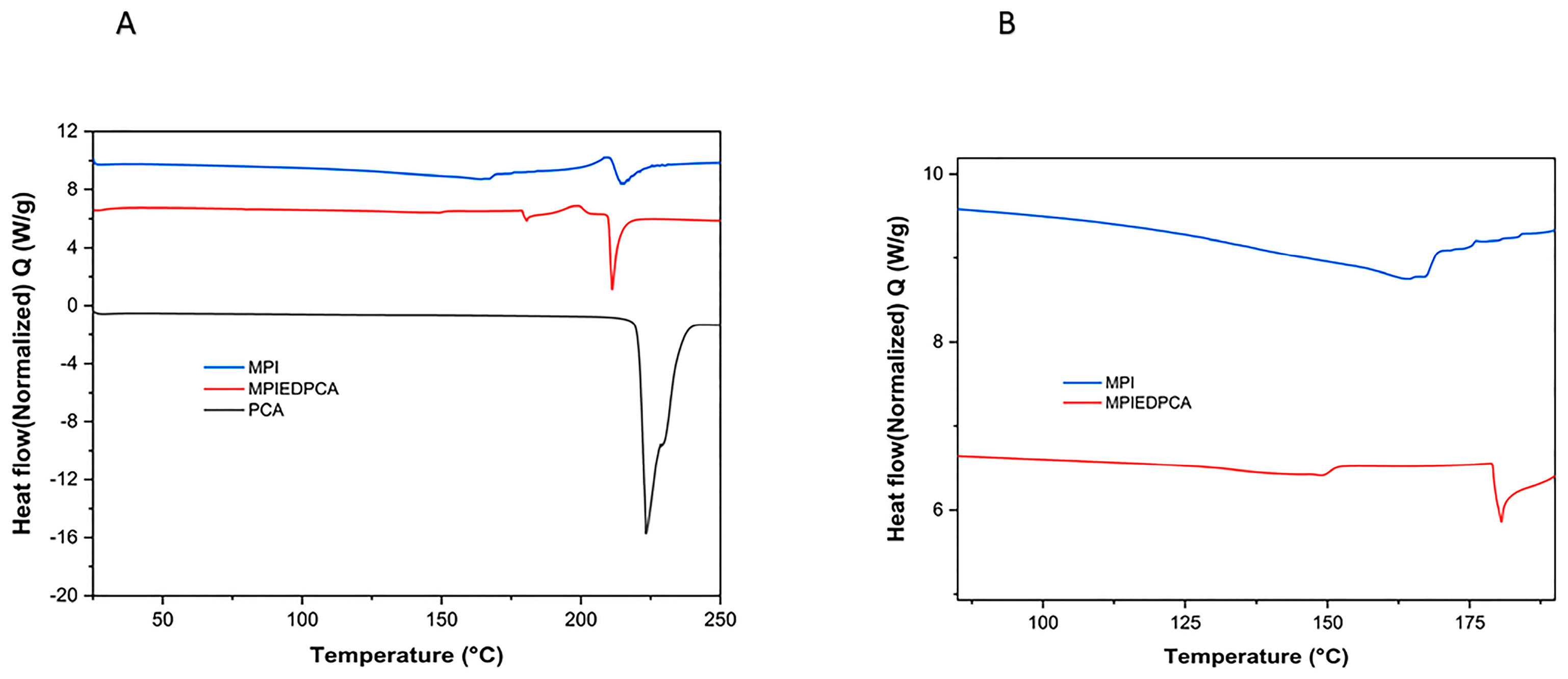
3.5. DSC
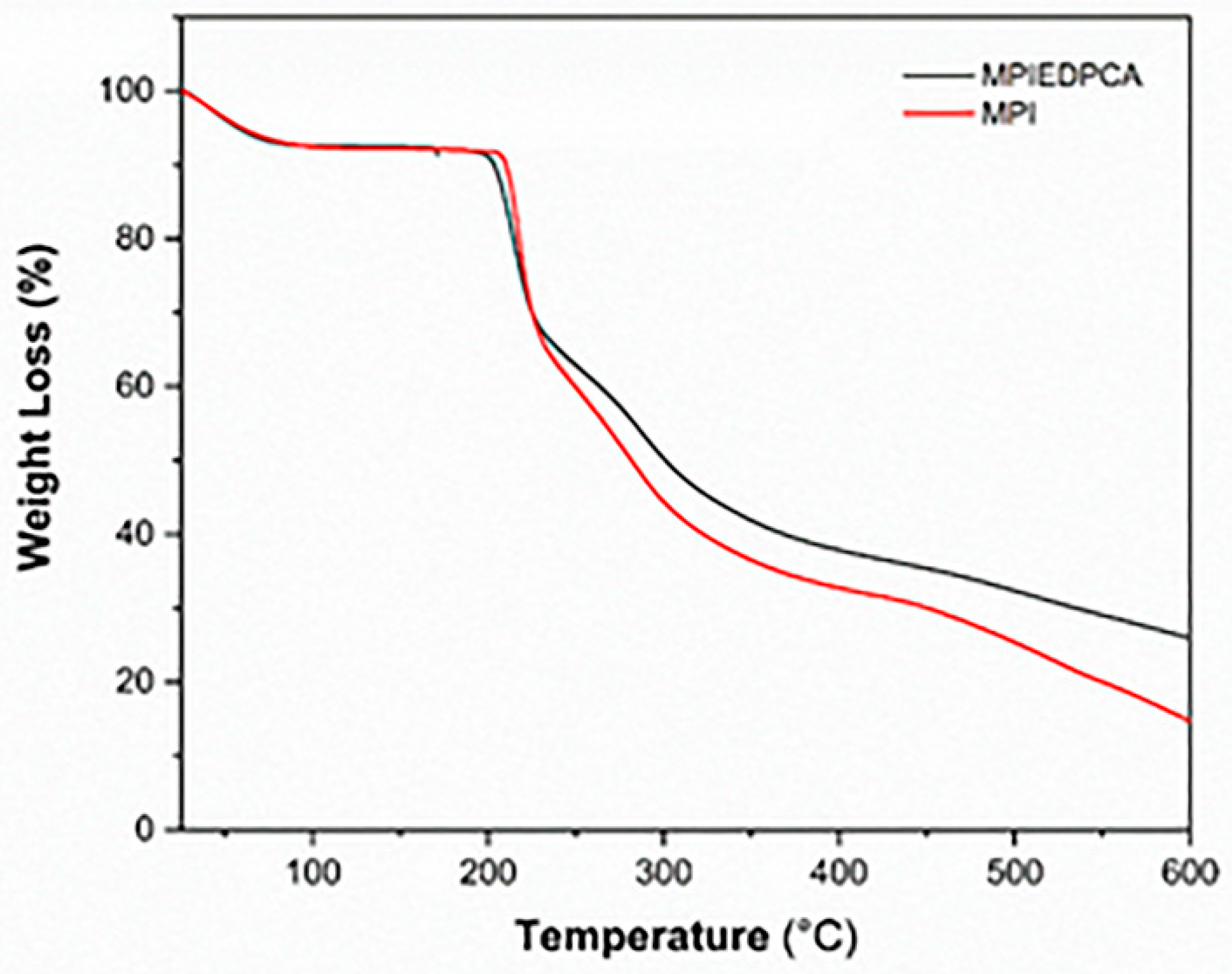
3.6. TGA
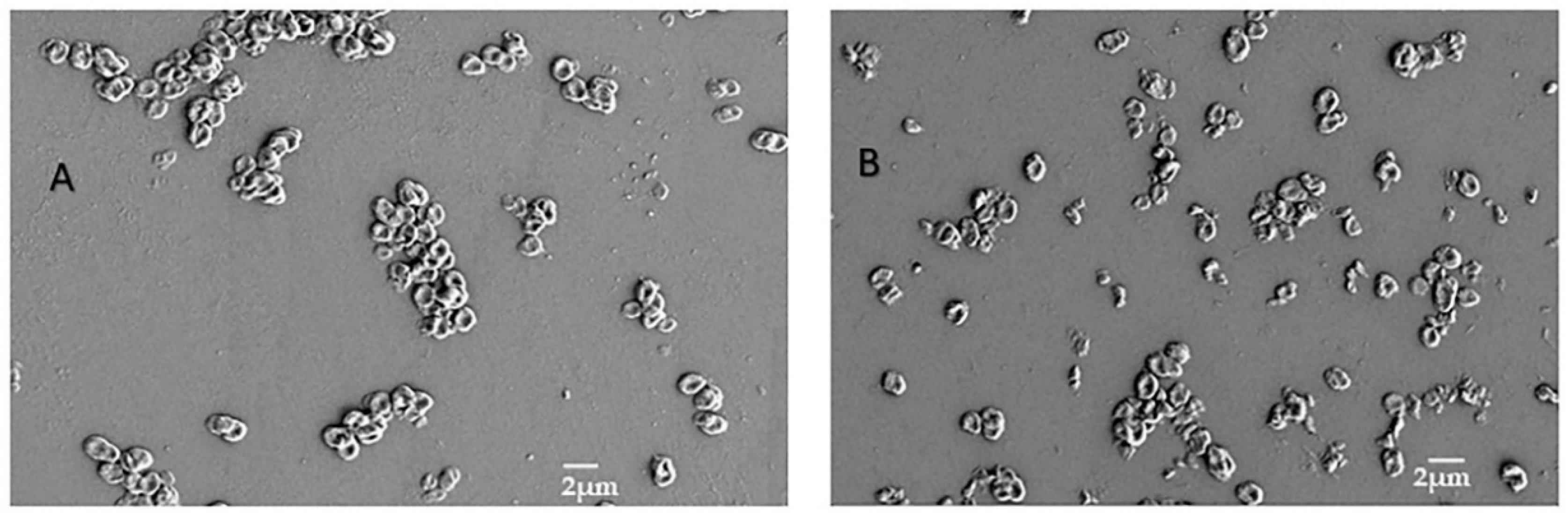
3.7. SEM
3.8. Zeta Potential
3.9. Drug Loading and pH-Triggered PCA Release
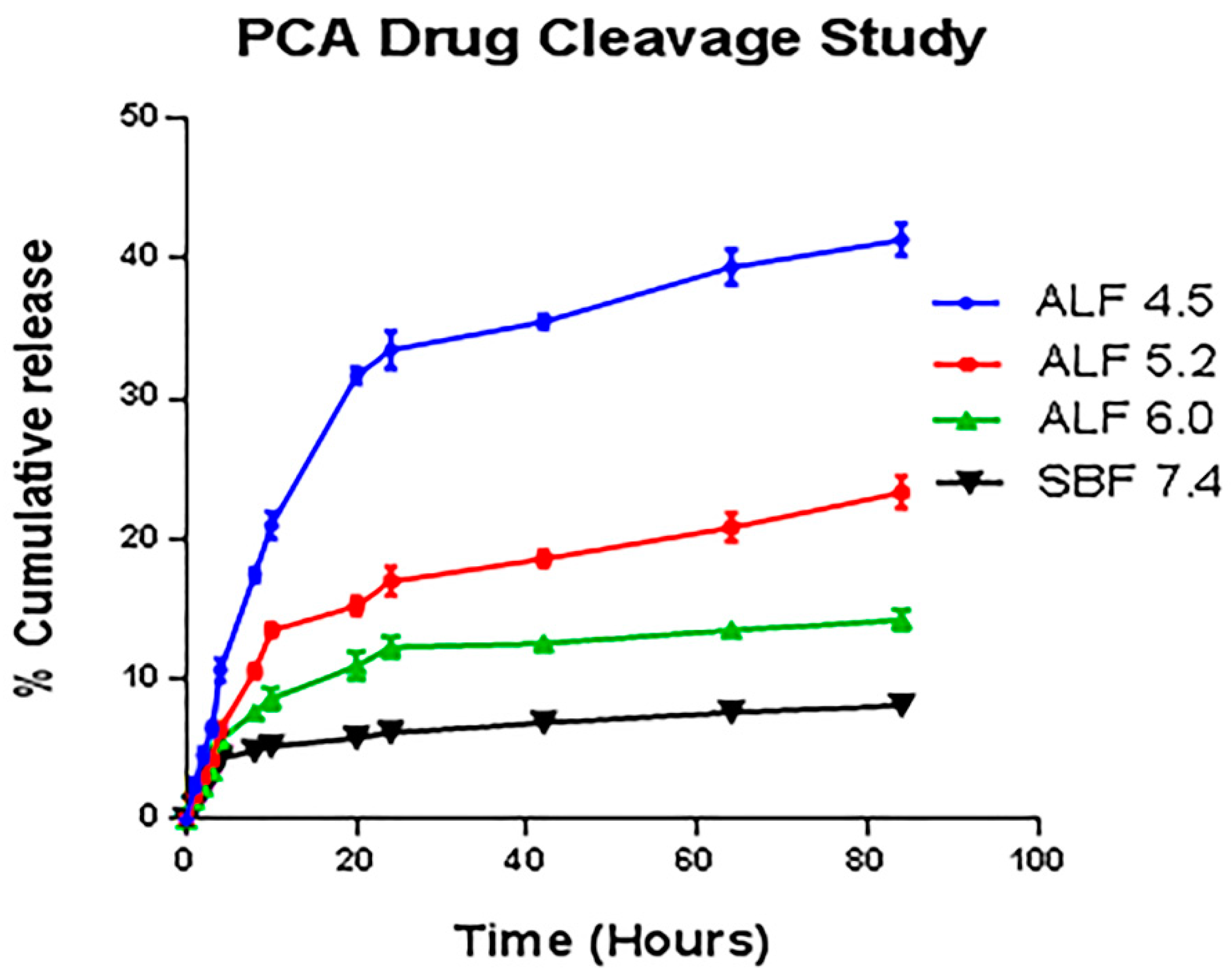
3.10. Mechanism of PCA Drug Release
3.11. Kinetic Release Study of PCA
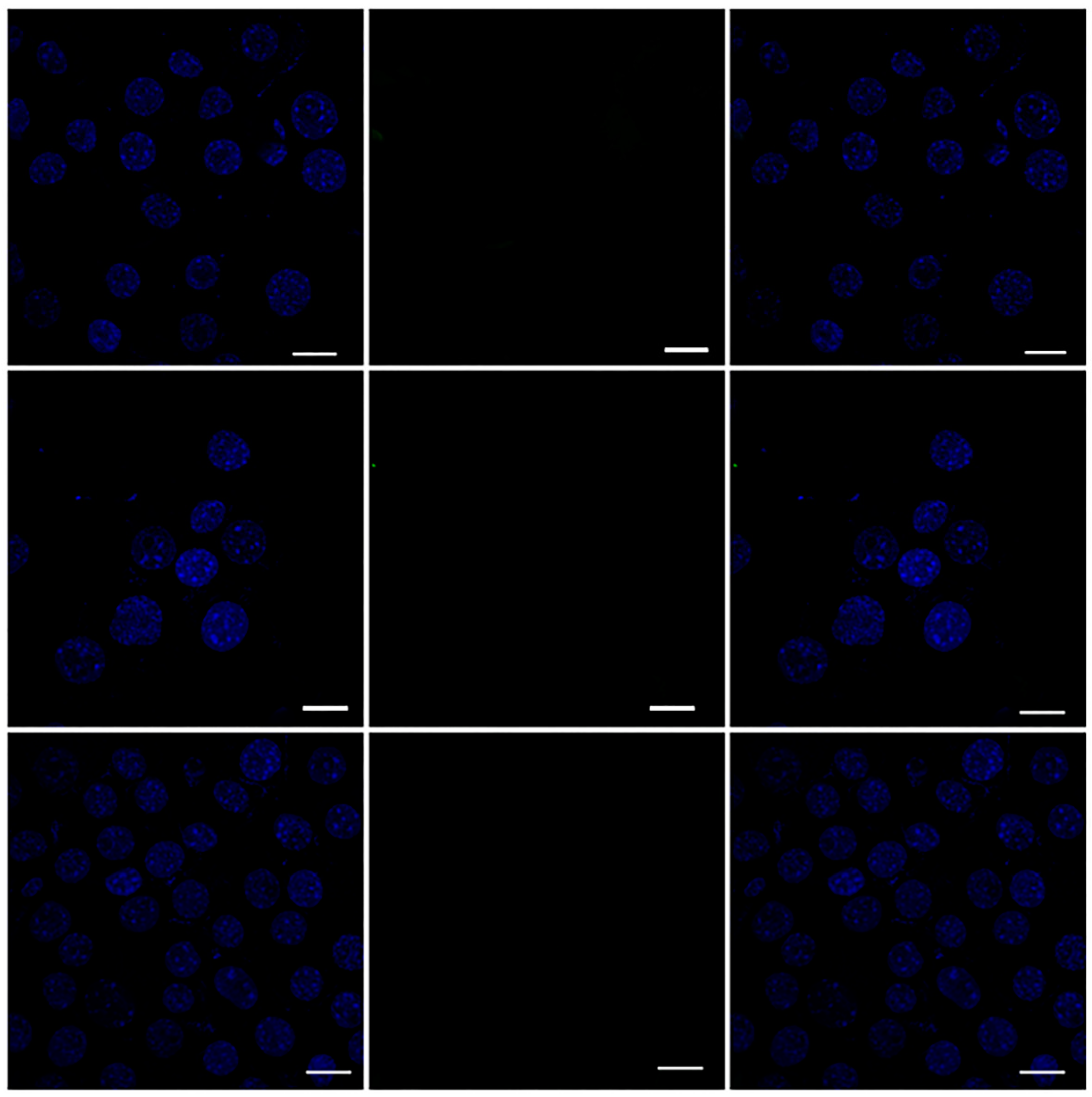
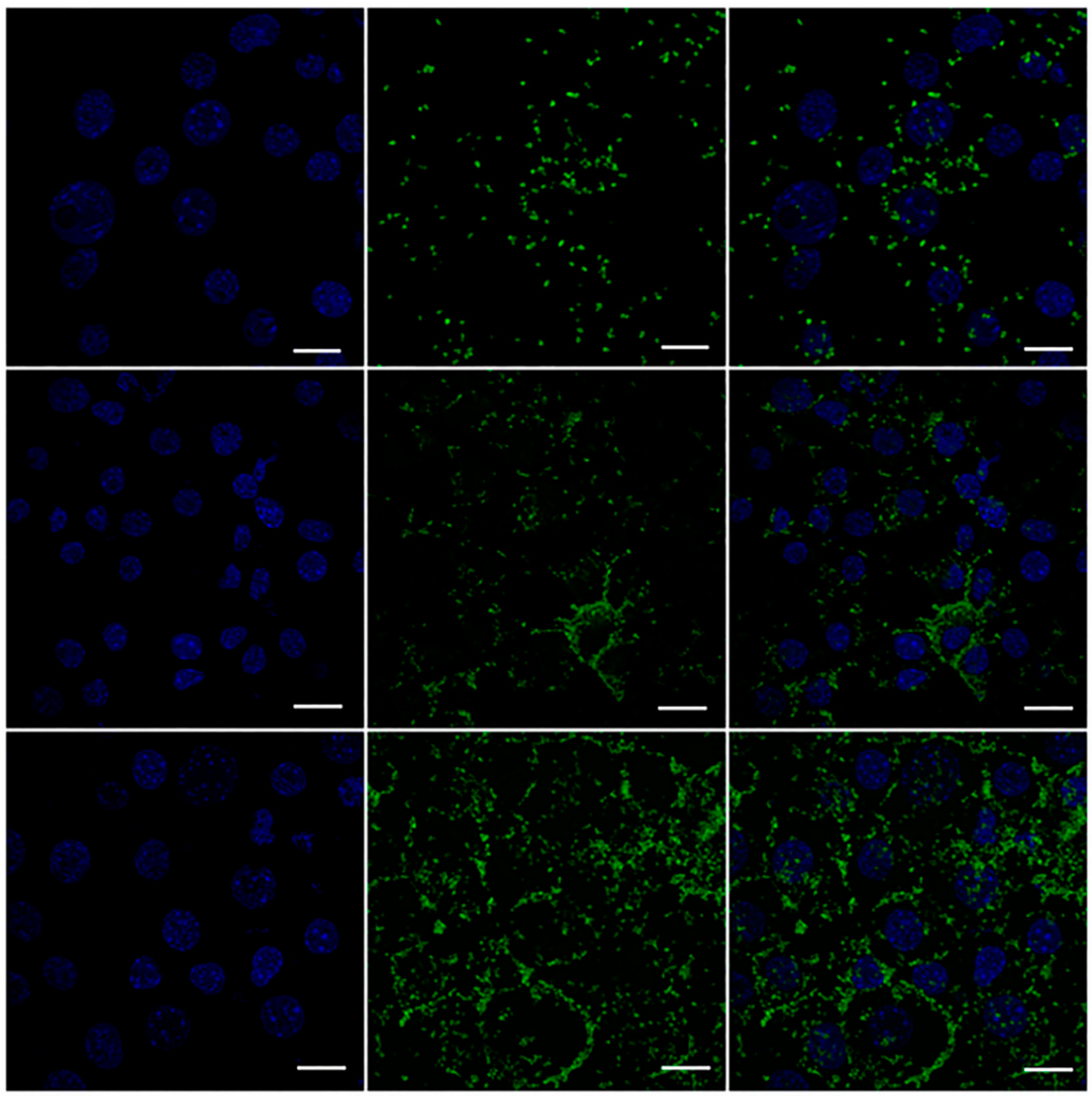
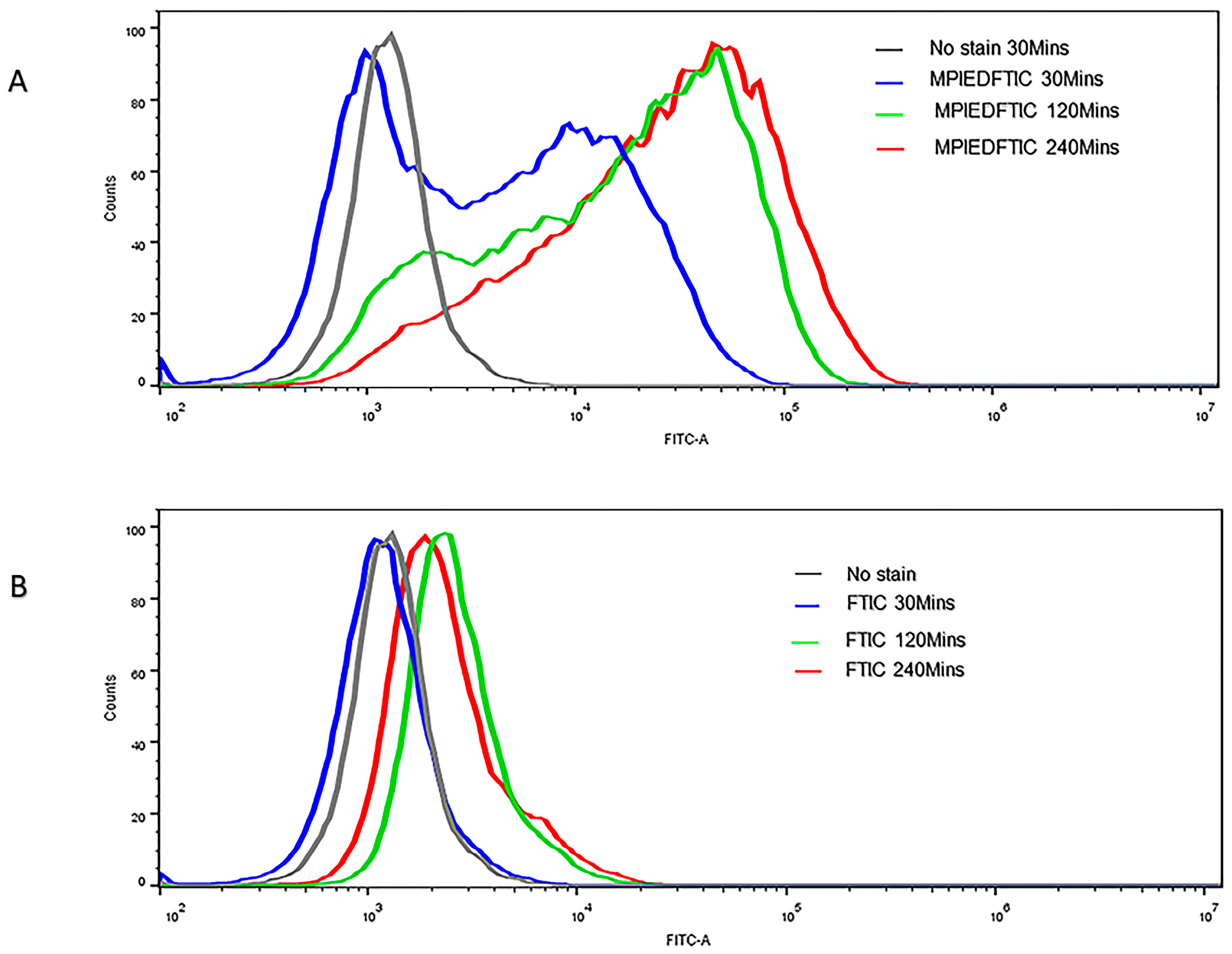
3.12. Efficient Cellular Uptake of MPIEDFITC by RAW 264.7 Macrophage Cells
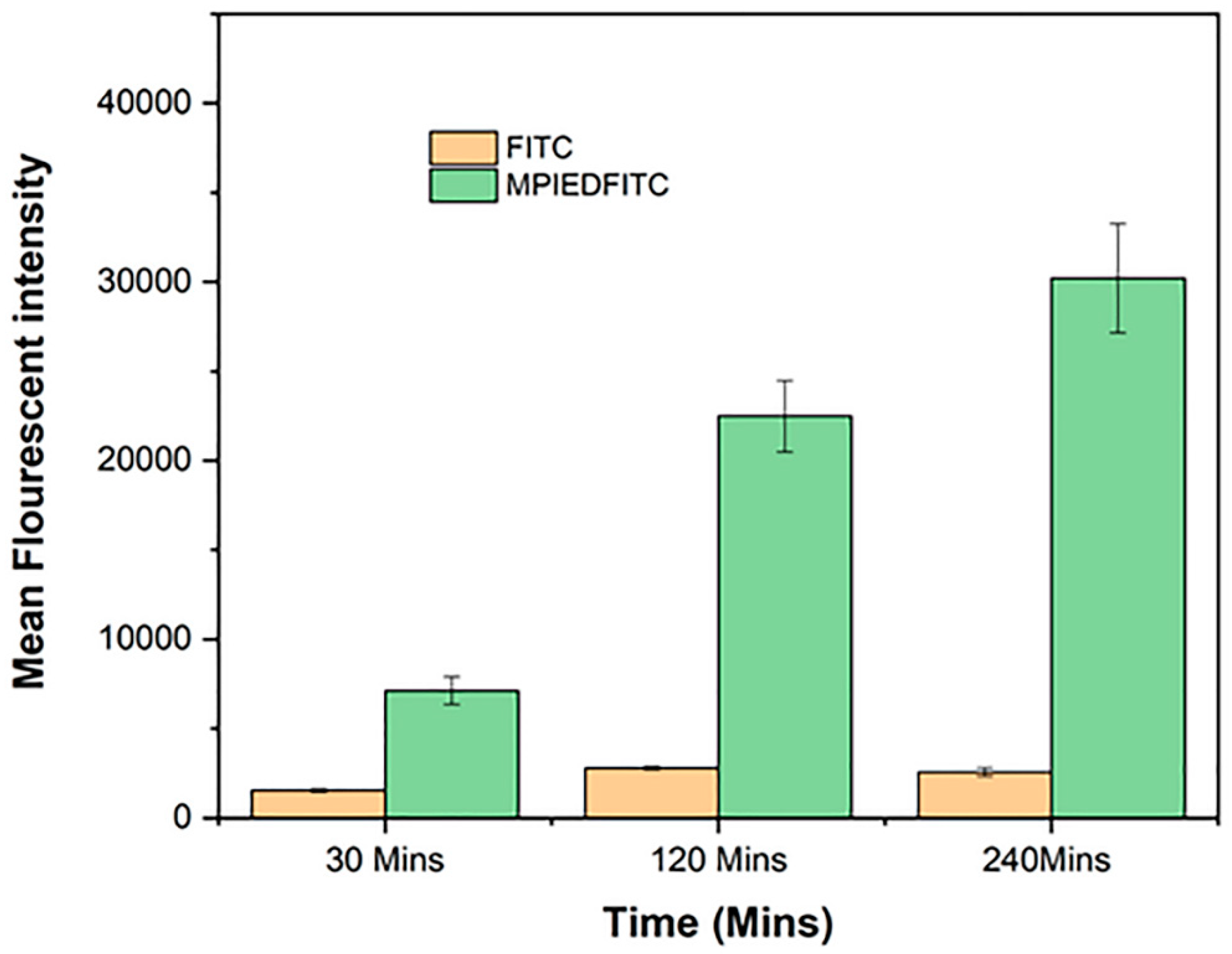
3.13. Quantification of RAW 264.7 Macrophage Cellular Uptake
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- World Health Organisation. Global Tuberculosis Report 2017; World Health Organisation: Geneva, Switzerland, 2017. [Google Scholar]
- World Health Organization. Global Tuberculosis Report 2015; World Health Organization: Geneva, Switzerland, 2015. [Google Scholar]
- Kwan, C.K.; Ernst, J.D. Hiv and tuberculosis: A deadly human syndemic. Clin. Microbiol. Rev. 2011, 24, 351–376. [Google Scholar] [CrossRef]
- Etard, J.F.; Ndiaye, I.; Thierry-Mieg, M.; Gueye, N.F.; Gueye, P.M.; Laniece, I.; Dieng, A.B.; Diouf, A.; Laurent, C.; Mboup, S.; et al. Mortality and causes of death in adults receiving highly active antiretroviral therapy in senegal: A 7-year cohort study. Aids 2006, 20, 1181–1189. [Google Scholar] [CrossRef]
- Martinson, N.A.; Hoffmann, C.J.; Chaisson, R.E. Epidemiology of tuberculosis and hiv: Recent advances in understanding and responses. Proc. Am. Thorac. Soc. 2011, 8, 288–293. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, N.R.; Moll, A.; Sturm, A.W.; Pawinski, R.; Govender, T.; Lalloo, U.; Zeller, K.; Andrews, J.; Friedland, G. Extensively drug-resistant tuberculosis as a cause of death in patients co-infected with tuberculosis and hiv in a rural area of south africa. Lancet 2006, 368, 1575–1580. [Google Scholar] [CrossRef]
- Shah, N.S.; Wright, A.; Bai, G.-H.; Barrera, L.; Boulahbal, F.; Martín-Casabona, N.; Drobniewski, F.; Gilpin, C.; Havelková, M.; Lepe, R.; et al. Worldwide emergence of extensively drug-resistant tuberculosis. Emerg. Infect. Dis. 2007, 13, 380–387. [Google Scholar] [CrossRef] [PubMed]
- Torraca, V.; Masud, S.; Spaink, H.P.; Meijer, A.H. Macrophage-pathogen interactions in infectious diseases: New therapeutic insights from the zebrafish host model. Dis. Models Mech. 2014, 7, 785–797. [Google Scholar] [CrossRef]
- Baena, A.; Porcelli, S.A. Evasion and subversion of antigen presentation by mycobacterium tuberculosis. Tissue Antigens 2009, 74, 189–204. [Google Scholar] [CrossRef]
- Behar, S.M.; Divangahi, M.; Remold, H.G. Evasion of innate immunity by mycobacterium tuberculosis: Is death an exit strategy? Nat. Rev. Microbiol. 2010, 8, 668–674. [Google Scholar] [CrossRef]
- Peyron, P.; Vaubourgeix, J.; Poquet, Y.; Levillain, F.; Botanch, C.; Bardou, F.; Daffe, M.; Emile, J.F.; Marchou, B.; Cardona, P.J.; et al. Foamy macrophages from tuberculous patients’ granulomas constitute a nutrient-rich reservoir for m. Tuberculosis persistence. PLoS Pathog. 2008, 4, e1000204. [Google Scholar] [CrossRef]
- Ryndak, M.B.; Chandra, D.; Laal, S. Understanding dissemination of mycobacterium tuberculosis from the lungs during primary infection. J. Med. Microbiol. 2016. [Google Scholar] [CrossRef]
- Moliva, J.I.; Turner, J.; Torrelles, J.B. Prospects in mycobacterium bovis bacille calmette et guérin (bcg) vaccine diversity and delivery: Why does bcg fail to protect against tuberculosis? Vaccine 2015, 33, 5035–5041. [Google Scholar] [CrossRef]
- Zhang, Y.; Shi, W.; Zhang, W.; Mitchison, D. Mechanisms of pyrazinamide action and resistance. Microbiol. Spectr. 2014, 2. [Google Scholar] [CrossRef] [PubMed]
- Cynamon, M.H.; Klemens, S.P.; Chou, T.S.; Gimi, R.H.; Welch, J.T. Antimycobacterial activity of a series of pyrazinoic acid esters. J. Med. Chem. 1992, 35, 1212–1215. [Google Scholar] [CrossRef]
- Cynamon, M.H.; Gimi, R.; Gyenes, F.; Sharpe, C.A.; Bergmann, K.E.; Han, H.J.; Gregor, L.B.; Rapolu, R.; Luciano, G.; Welch, J.T. Pyrazinoic acid esters with broad spectrum in vitro antimycobacterial activity. J. Med. Chem. 1995, 38, 3902–3907. [Google Scholar] [CrossRef]
- Korbelik, M.; Cooper, P.D. Potentiation of photodynamic therapy of cancer by complement: The effect of γ-inulin. Br. J. Cancer 2007, 96, 67–72. [Google Scholar] [CrossRef]
- Batrakova, E.V.; Gendelman, H.E.; Kabanov, A.V. Cell-mediated drug delivery. Expert Opin. Drug Deliv. 2011, 8, 415–433. [Google Scholar] [CrossRef] [Green Version]
- Murdoch, C.; Giannoudis, A.; Lewis, C.E. Mechanisms regulating the recruitment of macrophages into hypoxic areas of tumors and other ischemic tissues. Blood 2004, 104, 2224–2234. [Google Scholar] [CrossRef]
- Gordon, S.; Taylor, P.R. Monocyte and macrophage heterogeneity. Nat. Rev. Immunol. 2005, 5, 953–964. [Google Scholar] [CrossRef]
- Tiwari, S.; Chaturvedi, A.P.; Tripathi, Y.B.; Mishra, B. Macrophage-specific targeting of isoniazid through mannosylated gelatin microspheres. AAPS Pharmscitech. 2011, 12, 900–908. [Google Scholar] [CrossRef]
- Barrow, E.L.W.; Winchester, G.A.; Staas, J.K.; Quenelle, D.C.; Barrow, W.W. Use of microsphere technology for targeted delivery of rifampin to mycobacterium tuberculosis-infected macrophages. Antimicrob. Agents Chemother. 1998, 42, 2682–2689. [Google Scholar] [CrossRef]
- Nowacek, A.S.; Miller, R.L.; McMillan, J.; Kanmogne, G.; Kanmogne, M.; Mosley, R.L.; Ma, Z.; Graham, S.; Chaubal, M.; Werling, J.; et al. Nanoart synthesis, characterization, uptake, release and toxicology for human monocyte-macrophage drug delivery. Nanomedicine 2009, 4, 903–917. [Google Scholar] [CrossRef]
- Dou, H.; Morehead, J.; Destache, C.J.; Kingsley, J.D.; Shlyakhtenko, L.; Zhou, Y.; Chaubal, M.; Werling, J.; Kipp, J.; Rabinow, B.E.; et al. Laboratory investigations for the morphologic, pharmacokinetic, and anti-retroviral properties of indinavir nanoparticles in human monocyte-derived macrophages. Virology 2007, 358, 148–158. [Google Scholar] [CrossRef]
- Killion, J.J.; Fidler, I.J. Therapy of cancer metastasis by tumoricidal activation of tissue macrophages using liposome-encapsulated immunomodulators. Pharmacol. Ther. 1998, 78, 141–154. [Google Scholar] [CrossRef]
- Johansen, P.T.; Zucker, D.; Parhamifar, L.; Pourhassan, H.; Madsen, D.V.; Henriksen, J.R.; Gad, M.; Barberis, A.; Maj, R.; Andresen, T.L.; et al. Monocyte targeting and activation by cationic liposomes formulated with a tlr7 agonist. Expert Opin. Drug Deliv. 2015, 12, 1045–1058. [Google Scholar] [CrossRef]
- Schultze, J.L.; Schmieder, A.; Goerdt, S. Macrophage activation in human diseases. Semin. Immunol. 2015, 27, 249–256. [Google Scholar] [CrossRef]
- Su, Y.; Xie, Z.; Kim, G.B.; Dong, C.; Yang, J. Design strategies and applications of circulating cell-mediated drug delivery systems. ACS Biomater. Sci. Eng. 2015, 1, 201–217. [Google Scholar] [CrossRef]
- Garapaty, A.; Champion, J.A. Tunable particles alter macrophage uptake based on combinatorial effects of physical properties. Bioeng. Transl. Med. 2017, 2, 92–101. [Google Scholar] [CrossRef] [Green Version]
- Kaur, N.; Gupta, A.K. Applications of inulin and oligofructose in health and nutrition. J. Biosci. 2002, 27, 703–714. [Google Scholar] [CrossRef]
- Barclay, T.; Ginic-Markovic, M.; Cooper, P.; Petrovsky, N. Inulin—A versatile polysaccharide with multiple pharmaceutical and food chemical uses. J. Excip. Food Chem. 2010, 1, 27–50. [Google Scholar]
- Cooper, P.D.; Petrovsky, N. Delta inulin: A novel, immunologically active, stable packing structure comprising beta-d-[2→1] poly(fructo-furanosyl) alpha-d-glucose polymers. Glycobiology 2011, 21, 595–606. [Google Scholar] [CrossRef]
- Cooper, P.D.; Carter, M. The anti-melanoma activity of inulin in mice. Mol. Immunol. 1986, 23, 903–908. [Google Scholar] [CrossRef]
- Gordon, D.L.; Sajkov, D.; Woodman, R.J.; Honda-Okubo, Y.; Cox, M.M.; Heinzel, S.; Petrovsky, N. Randomized clinical trial of immunogenicity and safety of a recombinant h1n1/2009 pandemic influenza vaccine containing advax polysaccharide adjuvant. Vaccine 2012, 30, 5407–5416. [Google Scholar] [CrossRef]
- Heddle, R.; Russo, P.; Petrovsky, N.; Hanna, R.; Smith, A. Immunotherapy—2076. A controlled study of delta inulin-adjuvanted honey bee venom immunotherapy. World Allergy Organ. 2013, 6, 441. [Google Scholar] [CrossRef]
- Gordon, D.; Kelley, P.; Heinzel, S.; Cooper, P.; Petrovsky, N. Immunogenicity and safety of advax, a novel polysaccharide adjuvant based on delta inulin, when formulated with hepatitis b surface antigen: A randomized controlled phase 1 study. Vaccine 2014, 32, 6469–6477. [Google Scholar] [CrossRef]
- Lobigs, M.; Pavy, M.; Hall, R.A.; Lobigs, P.; Cooper, P.; Komiya, T.; Toriniwa, H.; Petrovsky, N. An inactivated vero cell-grown japanese encephalitis vaccine formulated with advax, a novel inulin-based adjuvant, induces protective neutralizing antibody against homologous and heterologous flaviviruses. J. Gen. Virol. 2010, 91, 1407–1417. [Google Scholar] [CrossRef]
- Honda-Okubo, Y.; Saade, F.; Petrovsky, N. Advax™, a polysaccharide adjuvant derived from delta inulin, provides improved influenza vaccine protection through broad-based enhancement of adaptive immune responses. Vaccine 2012, 30, 5373–5381. [Google Scholar] [CrossRef]
- Cooper, P.D.; Barclay, T.G.; Ginic-Markovic, M.; Petrovsky, N. The polysaccharide inulin is characterized by an extensive series of periodic isoforms with varying biological actions. Glycobiology 2013, 23, 1164–1174. [Google Scholar] [CrossRef]
- Marques, M.; Löbenberg, R.; Almukainzi, M. Simulated biological fluids with possible application in dissolution testing. Dissolution Technol. 2011, 18, 15–28. [Google Scholar] [CrossRef]
- Schacht, E.; Ruys, L.; Vermeersch, J.; Remon, J.P.; Duncan, R. Use of polysaccharides as drug carriers. Dextran and inulin derivatives of procainamide. Ann. N. Y. Acad. Sci. 1985, 446, 199–212. [Google Scholar] [CrossRef]
- Dash, R.; Elder, T.; Ragauskas, A.J. Grafting of model primary amine compounds to cellulose nanowhiskers through periodate oxidation. Cellulose 2012, 19, 2069–2079. [Google Scholar] [CrossRef]
- Ec, A.; Simon, N.; Akpan, I. Synthesis, ftir and electronic spectra studies of metal (ii) complexes of pyrazine-2-carboxylic acid derivative. Med. Chem. 2017, 7, 321. [Google Scholar]
- Fares, M.M.; Salem, M.S.; Khanfar, M. Inulin and poly(acrylic acid) grafted inulin for dissolution enhancement and preliminary controlled release of poorly water-soluble irbesartan drug. Int. J. Pharm. 2011, 410, 206–211. [Google Scholar] [CrossRef] [PubMed]
- Barclay, T.G.; Rajapaksha, H.; Thilagam, A.; Qian, G.; Ginic-Markovic, M.; Cooper, P.D.; Gerson, A.; Petrovsky, N. Physical characterization and in silico modeling of inulin polymer conformation during vaccine adjuvant particle formation. Carbohydr. Polym. 2016, 143, 108–115. [Google Scholar] [CrossRef] [Green Version]
- Wu, W.; Yao, W.; Wang, X.; Xie, C.; Zhang, J.; Jiang, X. Bioreducible heparin-based nanogel drug delivery system. Biomaterials 2015, 39, 260–268. [Google Scholar] [CrossRef] [PubMed]
- Simões, M.F.; Valente, E.; Gómez, M.J.R.; Anes, E.; Constantino, L. Lipophilic pyrazinoic acid amide and ester prodrugs: Stability, activation and activity against m. Tuberculosis. Eur. J. Pharm. Sci. 2009, 37, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Zitko, J.; Servusova, B.; Paterova, P.; Mandikova, J.; Kubicek, V.; Kucera, R.; Hrabcova, V.; Kunes, J.; Soukup, O.; Dolezal, M. Synthesis, antimycobacterial activity and in vitro cytotoxicity of 5-chloro-n-phenylpyrazine-2-carboxamides. Molecules 2013, 18, 14807–14825. [Google Scholar] [CrossRef] [PubMed]
- Durham, P.G.; Zhang, Y.; German, N.; Mortensen, N.; Dhillon, J.; Mitchison, D.A.; Fourie, P.B.; Hickey, A.J. Spray dried aerosol particles of pyrazinoic acid salts for tuberculosis therapy. [corrected]. Mol. Pharm. 2015, 12, 2574–2581. [Google Scholar] [CrossRef] [PubMed]
- Leone, R.d.S.; Colman, T.A.D.; Schnitzler, E.; Ellendersen, L.N.; Masson, M.L. Evolved gas analysis (coupled tg–dsc–ftir) applied to the thermal behaviour of inulin. J. Anal. Appl. Pyrolysis 2014, 108, 323–326. [Google Scholar] [CrossRef]
- Santillan-Urquiza, E.; Arteaga-Cardona, F.; Hernandez-Herman, E.; Pacheco-Garcia, P.F.; Gonzalez-Rodriguez, R.; Coffer, J.L.; Mendoza-Alvarez, M.E.; Velez-Ruiz, J.F.; Mendez-Rojas, M.A. Inulin as a novel biocompatible coating: Evaluation of surface affinities toward cahpo4, alpha-Fe2O3, zno, CahPo4@zno and alpha-Fe2O3@ZNO nanoparticles. J. Colloid Interface Sci. 2015, 460, 339–348. [Google Scholar] [CrossRef]
- Dan, A.; Ghosh, S.; Moulik, S.P. Physicochemical studies on the biopolymer inulin: A critical evaluation of its self-aggregation, aggregate-morphology, interaction with water, and thermal stability. Biopolymers 2009, 91, 687–699. [Google Scholar] [CrossRef]
- Maia, J.; Carvalho, R.A.; Coelho, J.F.J.; Simões, P.N.; Gil, M.H. Insight on the periodate oxidation of dextran and its structural vicissitudes. Polymer 2011, 52, 258–265. [Google Scholar] [CrossRef]
- Cooper, P.D.; McComb, C.; Steele, E.J. The adjuvanticity of algammulin, a new vaccine adjuvant. Vaccine 1991, 9, 408–415. [Google Scholar] [CrossRef]
- Motskin, M.; Wright, D.M.; Muller, K.; Kyle, N.; Gard, T.G.; Porter, A.E.; Skepper, J.N. Hydroxyapatite nano and microparticles: Correlation of particle properties with cytotoxicity and biostability. Biomaterials 2009, 30, 3307–3317. [Google Scholar] [CrossRef]
- Gratton, S.E.; Ropp, P.A.; Pohlhaus, P.D.; Luft, J.C.; Madden, V.J.; Napier, M.E.; DeSimone, J.M. The effect of particle design on cellular internalization pathways. Proc. Natl. Acad. Sci. USA 2008, 105, 11613–11618. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; McCrate, J.M.; Lee, J.C.M.; Li, H. The role of surface charge on the uptake and biocompatibility of hydroxyapatite nanoparticles with osteoblast cells. Nanotechnology 2011, 22, 105708. [Google Scholar] [CrossRef]
- Chung, T.H.; Wu, S.H.; Yao, M.; Lu, C.W.; Lin, Y.S.; Hung, Y.; Mou, C.Y.; Chen, Y.C.; Huang, D.M. The effect of surface charge on the uptake and biological function of mesoporous silica nanoparticles in 3t3-l1 cells and human mesenchymal stem cells. Biomaterials 2007, 28, 2959–2966. [Google Scholar] [CrossRef]
- Villanueva, A.; Canete, M.; Roca, A.G.; Calero, M.; Veintemillas-Verdaguer, S.; Serna, C.J.; Morales Mdel, P.; Miranda, R. The influence of surface functionalization on the enhanced internalization of magnetic nanoparticles in cancer cells. Nanotechnology 2009, 20, 115103. [Google Scholar] [CrossRef]
- Ge, Y.; Zhang, Y.; Xia, J.; Ma, M.; He, S.; Nie, F.; Gu, N. Effect of surface charge and agglomerate degree of magnetic iron oxide nanoparticles on kb cellular uptake in vitro. Colloids Surf. B Biointerfaces 2009, 73, 294–301. [Google Scholar] [CrossRef]
- Smith, R.M.; Hansen, D.E. The ph-rate profile for the hydrolysis of a peptide bond. J. Am. Chem. 1998, 120, 8910–8913. [Google Scholar] [CrossRef]
- Radzicka, A.; Wolfenden, R. Rates of uncatalyzed peptide bond hydrolysis in neutral solution and the transition state affinities of proteases. J. Am. Chem. 1996, 118, 6105–6109. [Google Scholar] [CrossRef]
- Yoshioka, S.; Stella, V.J. Chemical stability of drug substances. In Stability of Drugs and Dosage Forms; Kluwer Academic Publishers: New York, NY, USA, 2002. [Google Scholar]
- Fife, T.H.; Squillacote, V.L. Intramolecular nucleophilic attack of carboxyl on amides. the metal ion inhibited hydrolysis of n-(2-pyridyl)phthalamic acid and n-(2-phenanthrolyl)phthalamic acid. J. Am. Chem. Soc. 1977, 99, 3762–3769. [Google Scholar] [CrossRef]
- Mo, R.; Gu, Z. Tumor microenvironment and intracellular signal-activated nanomaterials for anticancer drug delivery. Biochem. Pharm. 2016, 19, 274–283. [Google Scholar] [CrossRef]
- Wong, P.T.; Choi, S.K. Mechanisms of drug release in nanotherapeutic delivery systems. Chem. Rev. 2015, 115, 3388–3432. [Google Scholar] [CrossRef]
- Brandsen, B.M.; Hesser, A.R.; Castner, M.A.; Chandra, M.; Silverman, S.K. DNA-catalyzed hydrolysis of esters and aromatic amides. J. Am. Chem. Soc. 2013, 135, 16014–16017. [Google Scholar] [CrossRef]
- Dang, W.; Colvin, O.M.; Brem, H.; Saltzman, W.M. Covalent coupling of methotrexate to dextran enhances the penetration of cytotoxicity into a tissue-like matrix. Cancer Res. 1994, 54, 1729–1735. [Google Scholar]
- Suh, J.; Yoon, S.S.; Oh, E.; Kang, C.H.; Lee, E. Intramolecular nucleophilicity of amide groups—Implications on catalytic roles of amide groups in enzymatic-reactions. Bioorg. Chem. 1988, 16, 245–257. [Google Scholar] [CrossRef]
- Cherukuvada, S.; Thakuria, R.; Nangia, A. Pyrazinamide polymorphs: Relative stability and vibrational spectroscopy. Cryst. Growth Des. 2010, 10, 3931–3941. [Google Scholar] [CrossRef]
- Kabanda, M.M.; Van Tan, T.; Tran, Q.T.; Ebenso, E.E. A computational study of pyrazinamide: Tautomerism, acid-base properties, micro-solvation effects and acid hydrolysis mechanism. Comput. Theor. Chem. 2014, 1046, 30–41. [Google Scholar] [CrossRef]
- Wang, L.; Barclay, T.; Song, Y.; Joyce, P.; Sakala, I.G.; Petrovsky, N.; Garg, S. Investigation of the biodistribution, breakdown and excretion of delta inulin adjuvant. Vaccine 2017, 35, 4382–4388. [Google Scholar] [CrossRef]
- Counoupas, C.; Pinto, R.; Nagalingam, G.; Britton, W.J.; Petrovsky, N.; Triccas, J.A. Delta inulin-based adjuvants promote the generation of polyfunctional cd4(+) t cell responses and protection against mycobacterium tuberculosis infection. Sci. Rep. 2017, 7, 8582. [Google Scholar] [CrossRef]













© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Afinjuomo, F.; Barclay, T.G.; Parikh, A.; Song, Y.; Chung, R.; Wang, L.; Liu, L.; Hayball, J.D.; Petrovsky, N.; Garg, S. Design and Characterization of Inulin Conjugate for Improved Intracellular and Targeted Delivery of Pyrazinoic Acid to Monocytes. Pharmaceutics 2019, 11, 243. https://doi.org/10.3390/pharmaceutics11050243
Afinjuomo F, Barclay TG, Parikh A, Song Y, Chung R, Wang L, Liu L, Hayball JD, Petrovsky N, Garg S. Design and Characterization of Inulin Conjugate for Improved Intracellular and Targeted Delivery of Pyrazinoic Acid to Monocytes. Pharmaceutics. 2019; 11(5):243. https://doi.org/10.3390/pharmaceutics11050243
Chicago/Turabian StyleAfinjuomo, Franklin, Thomas G. Barclay, Ankit Parikh, Yunmei Song, Rosa Chung, Lixin Wang, Liang Liu, John D. Hayball, Nikolai Petrovsky, and Sanjay Garg. 2019. "Design and Characterization of Inulin Conjugate for Improved Intracellular and Targeted Delivery of Pyrazinoic Acid to Monocytes" Pharmaceutics 11, no. 5: 243. https://doi.org/10.3390/pharmaceutics11050243