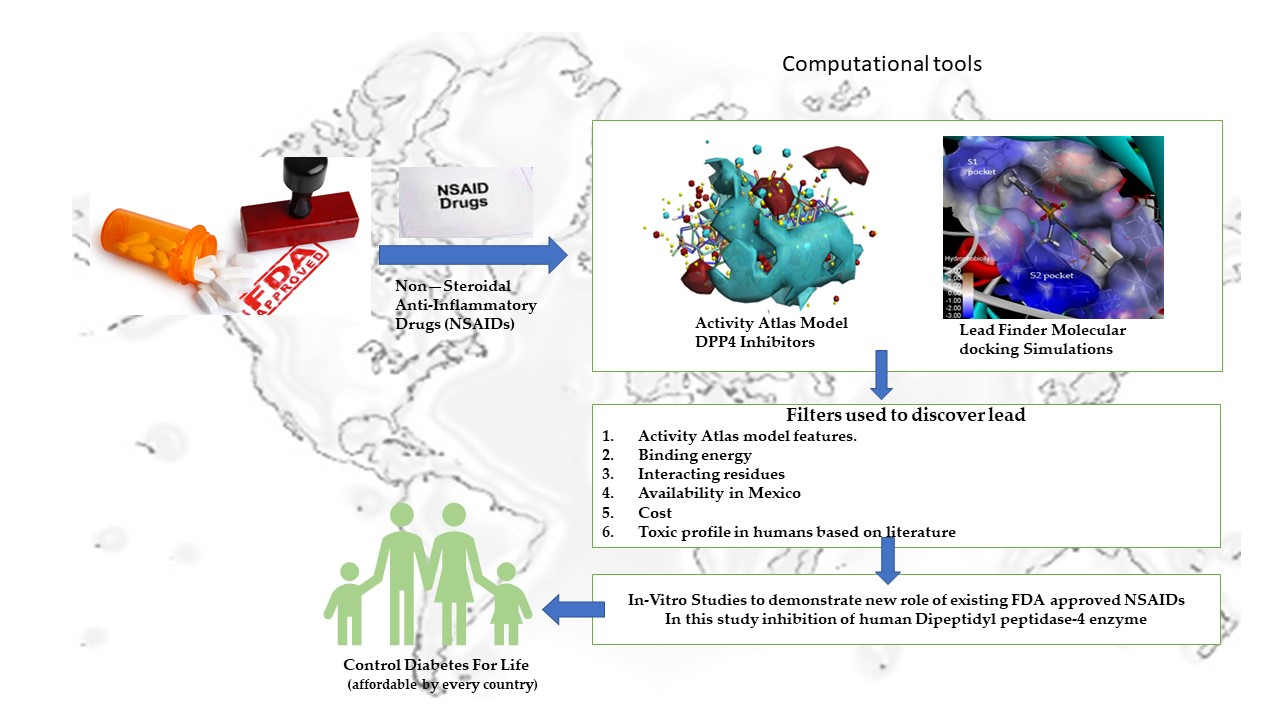
Repurposing of FDA-Approved NSAIDs for DPP-4 Inhibition as an Alternative for Diabetes Mellitus Treatment: Computational and in Vitro Study

 , , and
, , and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Compound Alignment, SAR Development, and Visualization of the Activity Atlas Model
2.3. DPP-4 Protein–FDA-Approved NSAID Molecular Docking Simulations
2.4. 3D-RISM Analysis to Investigate NSAID and DPP-4 Interactions
2.5. In Vitro DPP-4 Inhibition Assay
3. Results and Discussion
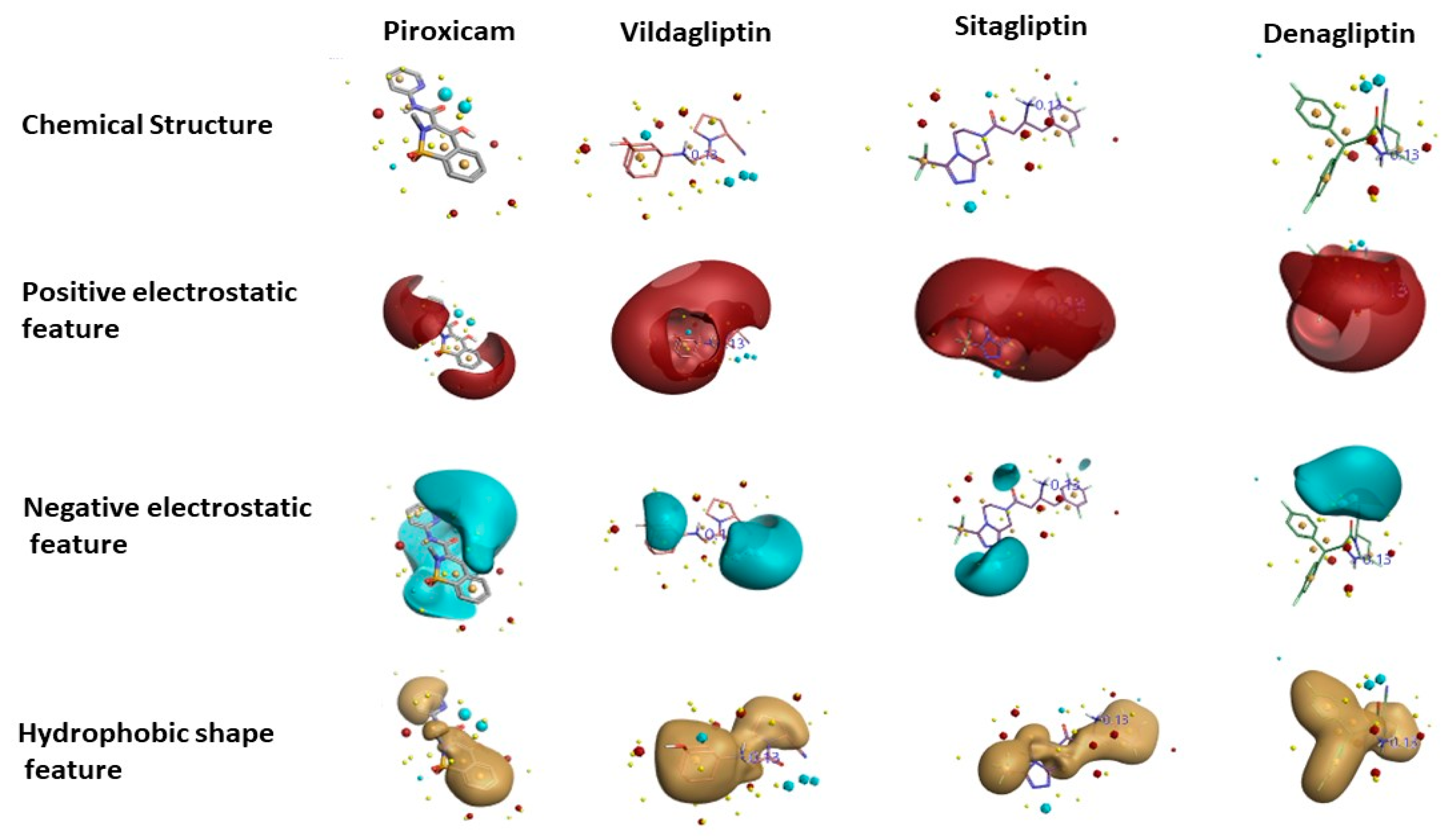
3.1. Activity Atlas Model Reveals the Structure–Activity Relationship (SAR) Mechanism
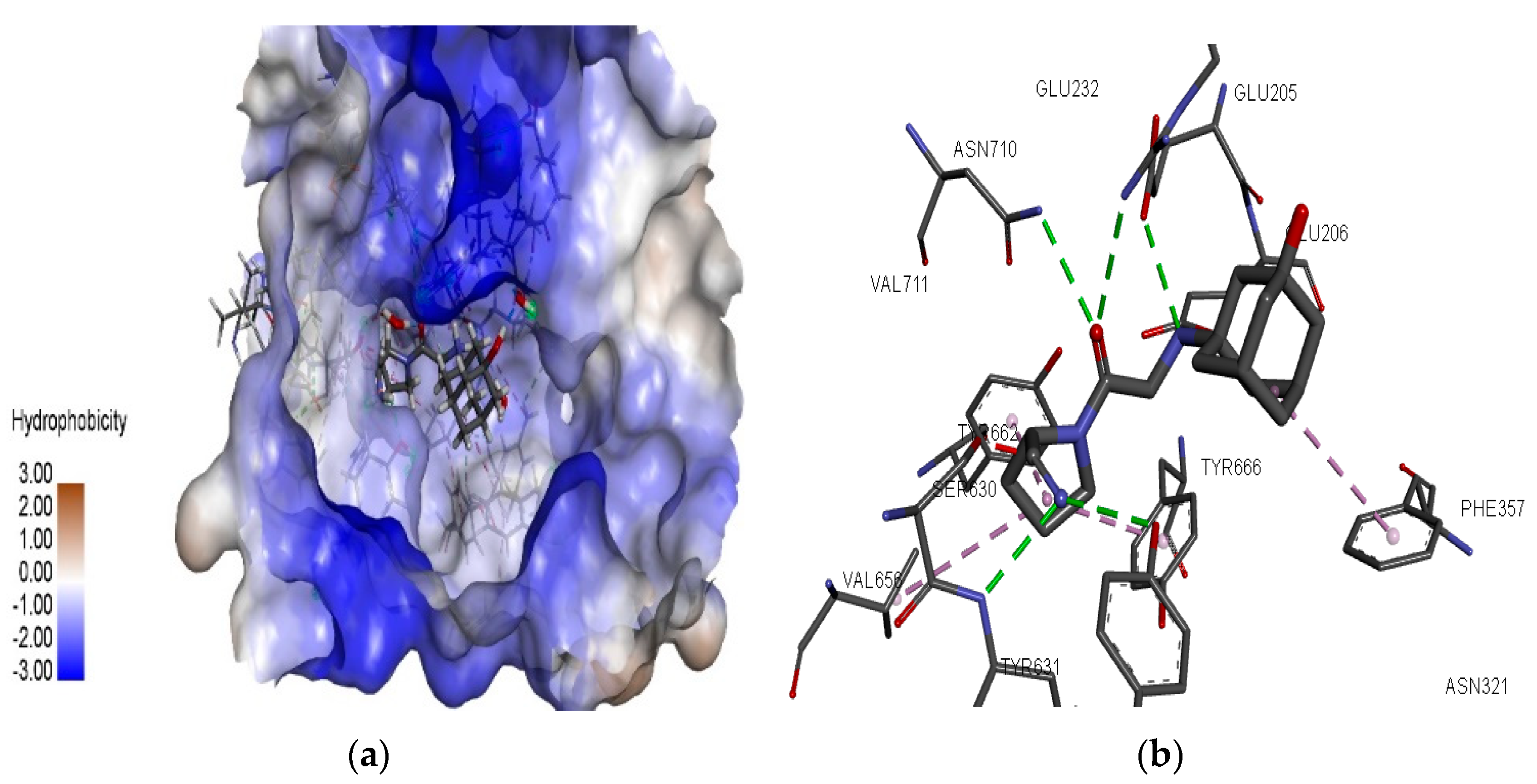
3.2. Molecular Docking Simulations
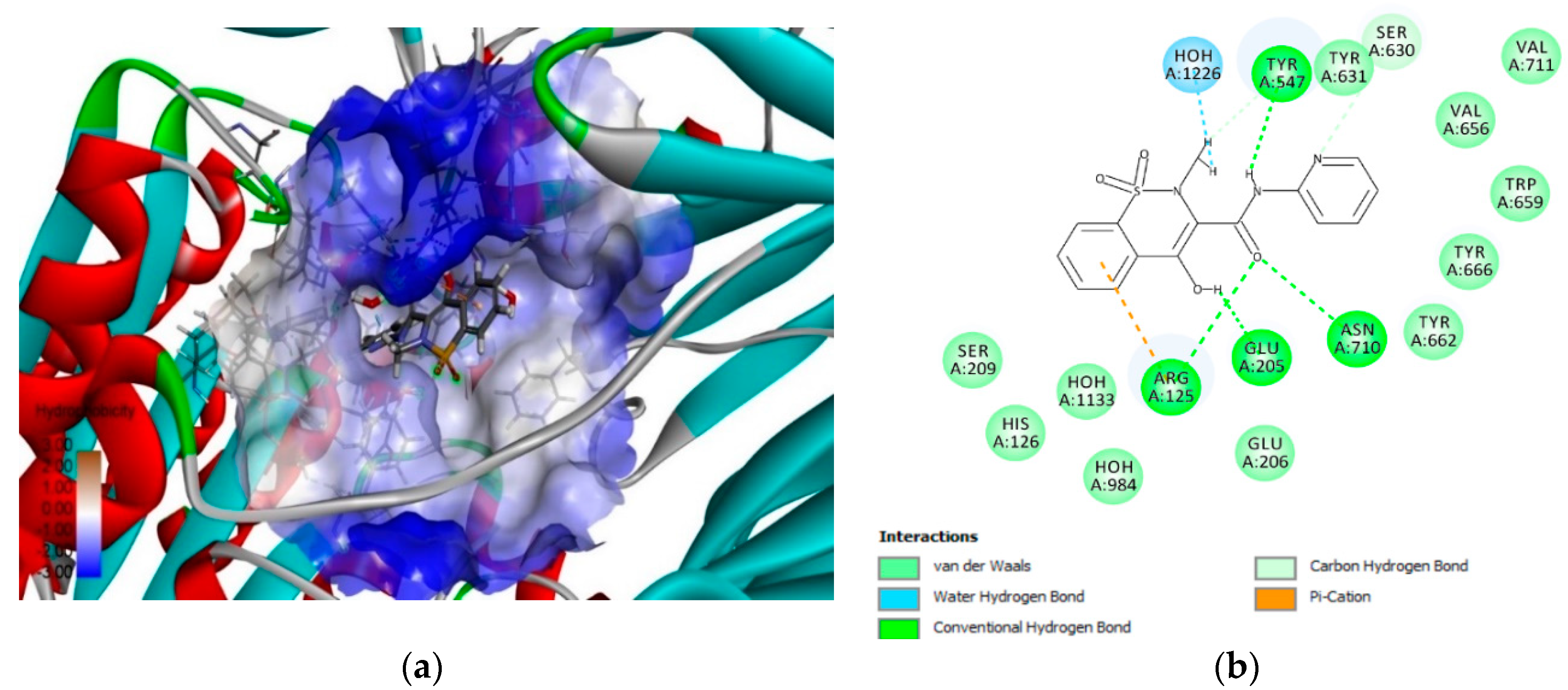
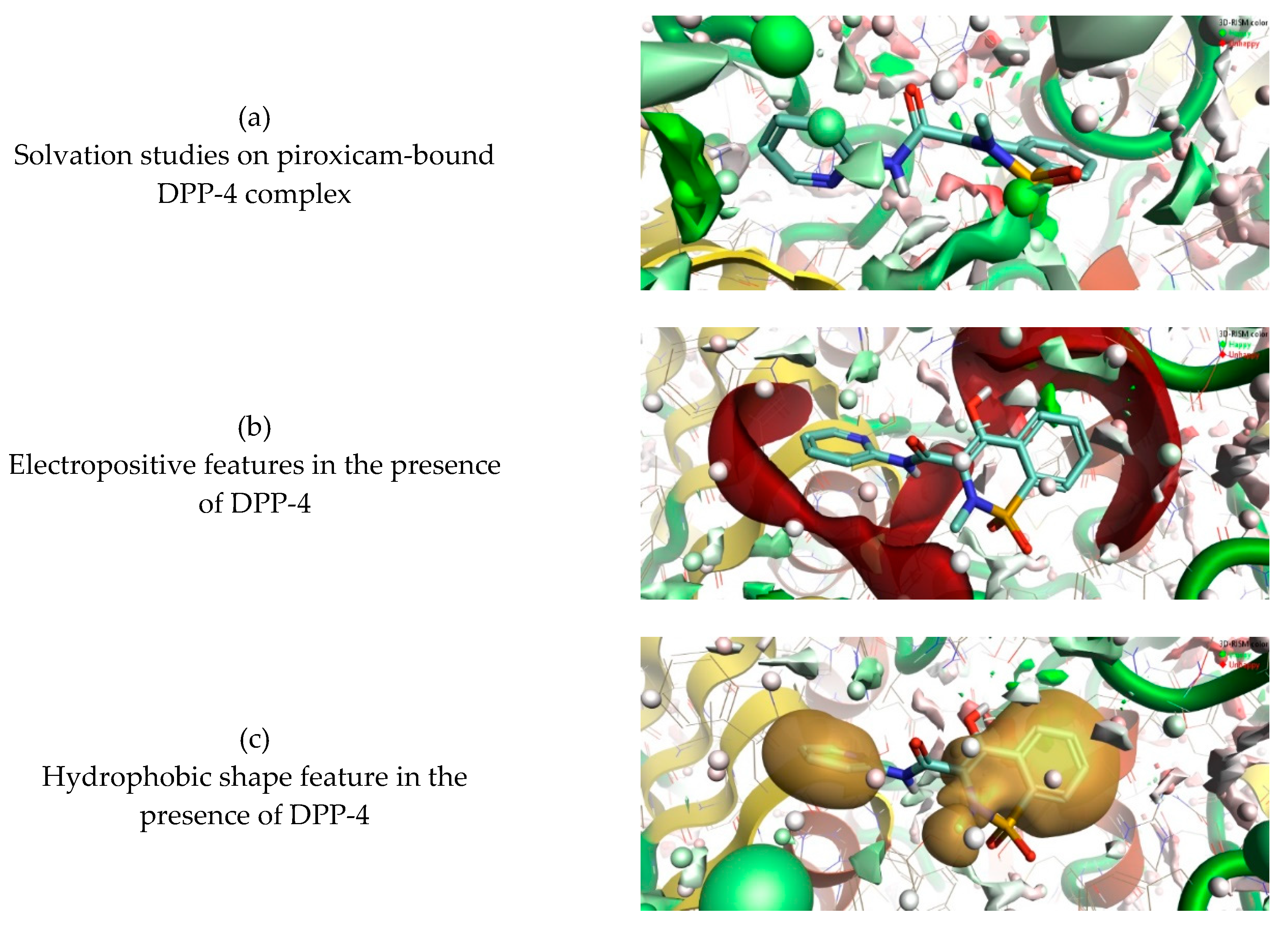
3.3. 3D-RISM Analysis of DPP-4–Piroxicam
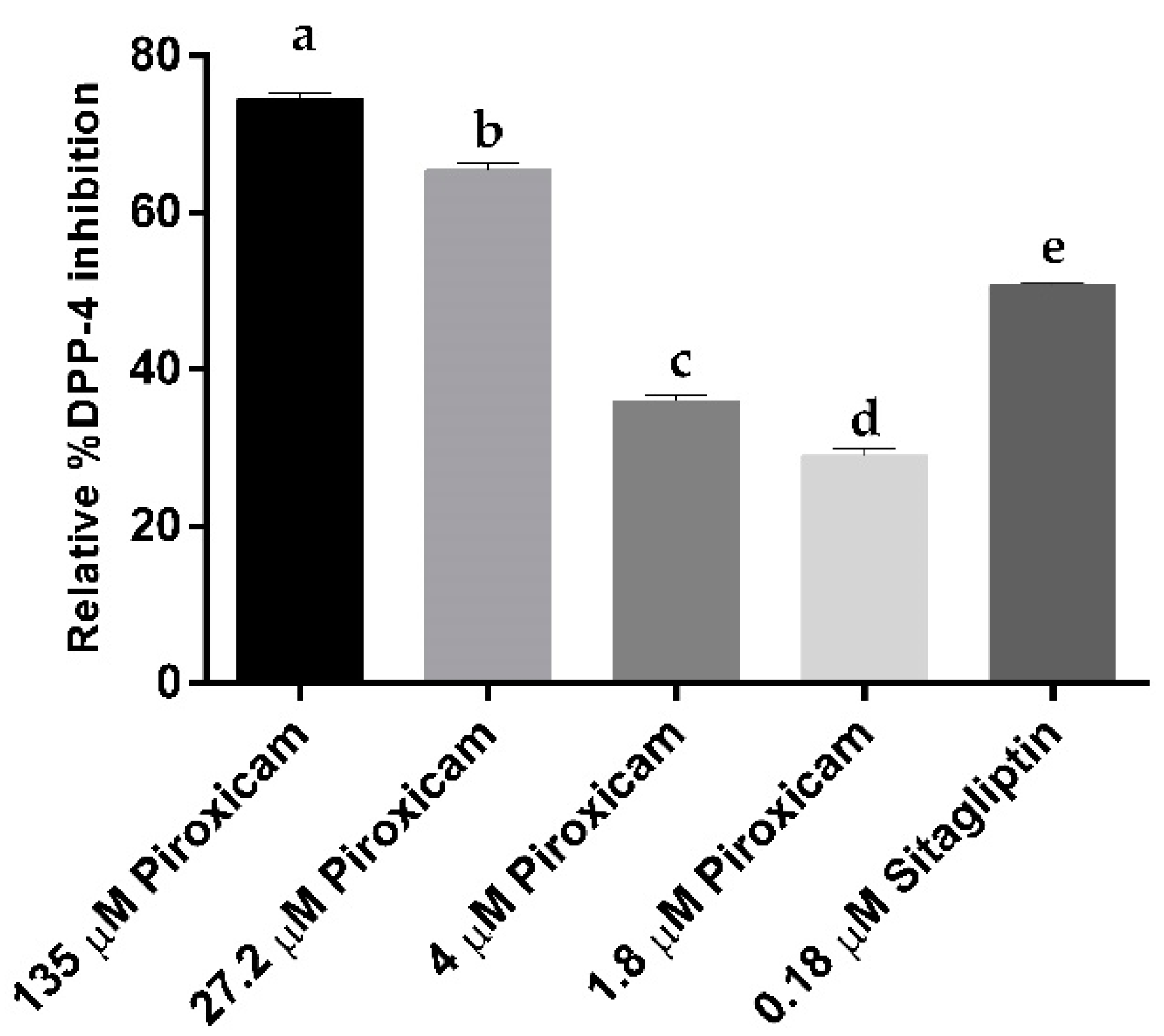
3.4. In Vitro Assay of Piroxicam Inhibition of Human Dipeptidyl Peptidase-4
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ozougwu, O. The pathogenesis and pathophysiology of type 1 and type 2 diabetes mellitus. J. Physiol. Pathophysiol. 2014. [Google Scholar] [CrossRef]
- Paschou, S.A.; Papadopoulou-Marketou, N.; Chrousos, G.P.; Kanaka-Gantenbein, C. On type 1 diabetes mellitus pathogenesis. Endocr. Connect. 2017. [Google Scholar] [CrossRef]
- DeFronzo, R.A. Pathogenesis of type 2 diabetes mellitus. Med. Clin. N. Am. 2004, 88, 787–835. [Google Scholar] [CrossRef] [PubMed]
- Bello-Chavolla, O.Y.; Rojas-Martinez, R.; Aguilar-Salinas, C.A.; Hernández-Avila, M. Epidemiology of diabetes mellitus in Mexico. Nutr. Rev. 2017. [Google Scholar] [CrossRef] [PubMed]
- Forouhi, N.G.; Wareham, N.J. Epidemiology of diabetes. Medicine (UK) 2014. [Google Scholar] [CrossRef]
- Ogurtsova, K.; da Rocha Fernandes, J.D.; Huang, Y.; Linnenkamp, U.; Guariguata, L.; Cho, N.H.; Cavan, D.; Shaw, J.E.; Makaroff, L.E. IDF Diabetes Atlas: Global estimates for the prevalence of diabetes for 2015 and 2040. Diabetes Res. Clin. Pract. 2017. [Google Scholar] [CrossRef]
- Papatheodorou, K.; Papanas, N.; Banach, M.; Papazoglou, D.; Edmonds, M. Complications of Diabetes 2016. J. Diabetes Res. 2016. [Google Scholar] [CrossRef]
- Yang, W.; Dall, T.M.; Beronjia, K.; Lin, J.; Semilla, A.P.; Chakrabarti, R.; Hogan, P.F.; Petersen, M.P. Economic costs of diabetes in the U.S. in 2017. Diabetes Care 2018. [Google Scholar] [CrossRef]
- Meza, R.; Barrientos-Gutierrez, T.; Rojas-Martinez, R.; Reynoso-Noverón, N.; Palacio-Mejia, L.S.; Lazcano-Ponce, E.; Hernández-Ávila, M. Burden of type 2 diabetes in Mexico: Past, current and future prevalence and incidence rates. Prev. Med. (Baltim) 2015. [Google Scholar] [CrossRef] [PubMed]
- Marín-Peñalver, J.J.; Martín-Timón, I.; Sevillano-Collantes, C.; Cañizo-Gómez, F.J. del Update on the treatment of type 2 diabetes mellitus. World J. Diabetes 2016. [Google Scholar] [CrossRef]
- Kim, W.; Egan, J.M. The Role of Incretins in Glucose Homeostasis and Diabetes Treatment. Pharmacol. Rev. 2008. [Google Scholar] [CrossRef] [PubMed]
- Mulvihill, E.E.; Drucker, D.J. Pharmacology, physiology, and mechanisms of action of dipeptidyl peptidase-4 inhibitors. Endocr. Rev. 2014, 35, 992–1019. [Google Scholar] [CrossRef]
- Zhong, J.; Rao, X.; Rajagopalan, S. An emerging role of dipeptidyl peptidase 4 (DPP4) beyond glucose control: Potential implications in cardiovascular disease. Atherosclerosis 2013, 226, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Drucker, D.J. The biology of incretin hormones. Cell Metab. 2006, 3, 153–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, B.R.; Kim, H.Y.; Choi, I.; Kim, J.B.; Jin, C.H.; Han, A.R. DPP-IV inhibitory potentials of flavonol glycosides isolated from the seeds of lens culinaris: In vitro and molecular docking analyses. Molecules 2018. [Google Scholar] [CrossRef]
- Schreiber, A.K. Diabetic neuropathic pain: Physiopathology and treatment. World J. Diabetes 2015. [Google Scholar] [CrossRef]
- Snyder, M.J.; Gibbs, L.M.; Lindsay, T.J. Treating painful diabetic peripheral neuropathy: An update. Am. Fam. Physician 2016. [Google Scholar] [CrossRef]
- Roelofs, P.D.D.M.; Deyo, R.A.; Koes, B.W.; Scholten, R.J.P.M.; Van Tulder, M.W. Non-steroidal anti-inflammatory drugs for low back pain. Cochrane Database Syst. Rev. 2008, 33, 1766–1774. [Google Scholar] [CrossRef] [PubMed]
- Tsai, H.-J.; Hsu, Y.-H.; Huang, Y.-W.; Chang, Y.-K.; Liu, J.-S.; Hsu, C.-C. Use of non-steroidal anti-inflammatory drugs and risk of chronic kidney disease in people with Type 2 diabetes mellitus, a nationwide longitudinal cohort study. Diabet. Med. 2015, 32, 382–390. [Google Scholar] [CrossRef]
- Goldfine, A.B.; Fonseca, V.; Jablonski, K.A.; Chen, Y.I.; Tipton, L.; Staten, M.A.; Shoelson, S.E. Salicylate (Salsalate) in Patients With Type 2 Diabetes. Ann. Intern. Med. 2014, 159, 1–12. [Google Scholar] [CrossRef]
- Mork, N.L.; Robertson, R.P. Effects of nonsteroidal antiinflammatory drugs in conventional dosage on glucose homeostasis in patients with diabetes. West. J. Med. 1983, 139, 46–49. [Google Scholar]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2018, 18, 41–58. [Google Scholar] [CrossRef]
- Novac, N. Challenges and opportunities of drug repositioning. Trends Pharmacol. Sci. 2013, 34, 267–272. [Google Scholar] [CrossRef]
- Jin, G.; Wong, S.T.C. Toward better drug repositioning: Prioritizing and integrating existing methods into efficient pipelines. Drug Discov. Today 2014, 19, 637–644. [Google Scholar] [CrossRef]
- Sleire, L.; Førde-Tislevoll, H.E.; Netland, I.A.; Leiss, L.; Skeie, B.S.; Enger, P.Ø. Drug repurposing in cancer. Pharmacol. Res. 2017, 124, 74–91. [Google Scholar] [CrossRef]
- Shim, J.S.; Liu, J.O. Recent advances in drug repositioning for the discovery of new anticancer drugs. Int. J. Biol. Sci. 2014, 10, 654. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.; Murray, J.L.; Rubin, D.H. Drug Repurposing: New Treatments for Zika Virus Infection? Trends Mol. Med. 2016, 22, 919–921. [Google Scholar] [CrossRef] [PubMed]
- Madrid, P.B.; Panchal, R.G.; Warren, T.K.; Shurtleff, A.C.; Endsley, A.N.; Green, C.E.; Kolokoltsov, A.; Davey, R.; Manger, I.D.; Gilfillan, L.; et al. Evaluation of Ebola Virus Inhibitors for Drug Repurposing. ACS Infect. Dis. 2016. [Google Scholar] [CrossRef]
- Goldstein, J.A.; Bastarache, L.A.; Denny, J.C.; Roden, D.M.; Pulley, J.M.; Aronoff, D.M. Calcium channel blockers as drug repurposing candidates for gestational diabetes: Mining large scale genomic and electronic health records data to repurpose medications. Pharmacol. Res. 2018. [Google Scholar] [CrossRef]
- Oral, E.A.; Reilly, S.M.; Gomez, A.V.; Meral, R.; Butz, L.; Ajluni, N.; Chenevert, T.L.; Korytnaya, E.; Neidert, A.H.; Hench, R.; et al. Inhibition of IKKɛ and TBK1 Improves Glucose Control in a Subset of Patients with Type 2 Diabetes. Cell Metab. 2017. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Lin, Y.; Wen, X.; Jorissen, R.N.; Gilson, M.K. BindingDB: A web-accessible database of experimentally determined protein-ligand binding affinities. Nucleic Acids Res. 2007. [Google Scholar] [CrossRef]
- Kramer, B.; Rarey, M.; Lengauer, T. Evaluation of the FlexX incremental construction algorithm for protein-ligand docking. Proteins Struct. Funct. Genet. 1999. [Google Scholar] [CrossRef]
- Skyner, R.E.; McDonagh, J.L.; Groom, C.R.; Van Mourik, T.; Mitchell, J.B.O. A review of methods for the calculation of solution free energies and the modelling of systems in solution. Phys. Chem. Chem. Phys. 2015, 17, 6174–6191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, N.; Hirata, F. A new method to determine electrostatic potential around a macromolecule in solution from molecular wave functions. J. Comput. Chem. 2006, 27, 453–462. [Google Scholar] [CrossRef]
- Williams, B.S. Nonopioid Analgesics: Nonsteroidal Antiinflammatory Drugs, Cyclooxygenase-2 Inhibitors, and Acetaminophen. In Essentials of Pain Medicine; Elsevier Inc.: Amsterdam, The Netherlands, 2018; ISBN 9780323401968. [Google Scholar]
- Jamali, F.; Brocks, D.R. Clinical Pharmacokinetics of Ketoprofen and Its Enantiomers. Clin. Pharmacokinet. 1990, 19, 197–217. [Google Scholar] [CrossRef] [PubMed]
- Turck, D.; Roth, W.; Busch, U. A Review of the Clinical Pharmacokinetics of Meloxicam. Rheumatology 2012. [Google Scholar] [CrossRef]
- Wagner, J.G. Pharmacokinetic Studies in Man. In Pharmacokinetics; European Medical Agency: Amsterdam, The Netherlands, 2012. [Google Scholar]
- Dhake, A.S.; Patwardhan, P.D.; Ramaswamy, V.; Tipnis, H.P. Pharmacokinetics of piroxicam in man. Indian Drugs 1990, 6, 46–55. [Google Scholar]
- Herman, G.A.; Stevens, C.; Van Dyck, K.; Bergman, A.; Yi, B.; De Smet, M.; Snyder, K.; Hilliard, D.; Tanen, M.; Tanaka, W.; et al. Pharmacokinetics and pharmacodynamics of sitagliptin, an inhibitor of dipeptidyl peptidase IV, in healthy subjects: Results from two randomized, double-blind, placebo-controlled studies with single oral doses. Clin. Pharmacol. Ther. 2005, 78, 675–688. [Google Scholar] [CrossRef]
- Smith, D.A.; Beaumont, K.; Maurer, T.S.; Di, L. Relevance of Half-Life in Drug Design. J. Med. Chem. 2018, 61, 4272–4282. [Google Scholar] [CrossRef]
- Diwan, P.V.; Sastry, M.S.P.; Satyanarayana, N.V. Potentiation of hypoglycemic response of glibenclamide by piroxicam in rats and humans. Indian J. Exp. Biol. 1992, 30, 317–319. [Google Scholar]
- Parry, G.J.; Hitoshi, K.O. Piroxicam may reduce the rate of progression of experimental diabetic neuropathy. Neurology 2012. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Drug | New Indication | Year Approved (for New Indication) |
|---|---|---|---|
| 1 | Methotrexate | Rheumatoid arthritis | 1999 |
| 2 | Topiramate | Migraine | 2004 |
| 3 | Cymbalta | Diabetic peripheral neuropathy | 2004 |
| 4 | Mifepristone | Cushing’s syndrome | 2012 |
| No. | Chemical Name | FlexX Score (kJ/mol) | Novelty Score |
|---|---|---|---|
| 1 | Celecoxib | −8.7 | Moderate |
| 2 | Valdecoxib | −17.2 | Very High |
| 3 | Rofecoxib | −10.6 | High |
| 4 | Diclofenac | −16.06 | High |
| 5 | Diflunisal | −15.7 | High |
| 6 | Etodolac | −10.19 | High |
| 7 | Fenoprofen | −13.4 | Moderate |
| 8 | Flurbiprofen | −15.4 | High |
| 9 | Ibuprofen | −16.06 | Moderate |
| 10 | Indomethacin | −13.47 | Very High |
| 11 | Ketoprofen | −20.9 | Very High |
| 12 | Ketorolac | −17.7 | Very High |
| 13 | Mefenamic Acid | −26.680 | Low |
| 14 | Meloxicam | −18.9 | Low |
| 15 | Nabumetone | −13.0 | Low |
| 16 | Naproxen | −15.4 | Moderate |
| 17 | Oxaprozin | −8.5 | Very High |
| 18 | Piroxicam | −19.9 | Low |
| 19 | Sulindac | −8.4 | Very High |
| 20 | Tolmetin | −18.0 | Very High |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chittepu, V.C.S.R.; Kalhotra, P.; Osorio-Gallardo, T.; Gallardo-Velázquez, T.; Osorio-Revilla, G. Repurposing of FDA-Approved NSAIDs for DPP-4 Inhibition as an Alternative for Diabetes Mellitus Treatment: Computational and in Vitro Study. Pharmaceutics 2019, 11, 238. https://doi.org/10.3390/pharmaceutics11050238
Chittepu VCSR, Kalhotra P, Osorio-Gallardo T, Gallardo-Velázquez T, Osorio-Revilla G. Repurposing of FDA-Approved NSAIDs for DPP-4 Inhibition as an Alternative for Diabetes Mellitus Treatment: Computational and in Vitro Study. Pharmaceutics. 2019; 11(5):238. https://doi.org/10.3390/pharmaceutics11050238
Chicago/Turabian StyleChittepu, Veera C. S. R., Poonam Kalhotra, Tzayhri Osorio-Gallardo, Tzayhri Gallardo-Velázquez, and Guillermo Osorio-Revilla. 2019. "Repurposing of FDA-Approved NSAIDs for DPP-4 Inhibition as an Alternative for Diabetes Mellitus Treatment: Computational and in Vitro Study" Pharmaceutics 11, no. 5: 238. https://doi.org/10.3390/pharmaceutics11050238