Bacteria-Targeted Clindamycin Loaded Polymeric Nanoparticles: Effect of Surface Charge on Nanoparticle Adhesion to MRSA, Antibacterial Activity, and Wound Healing

 ,
,
Abstract
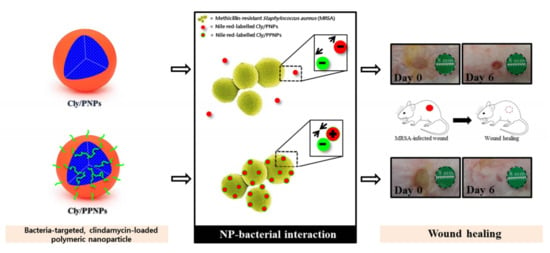
:
1. Introduction
2. Materials and Methods
2.1. Materials
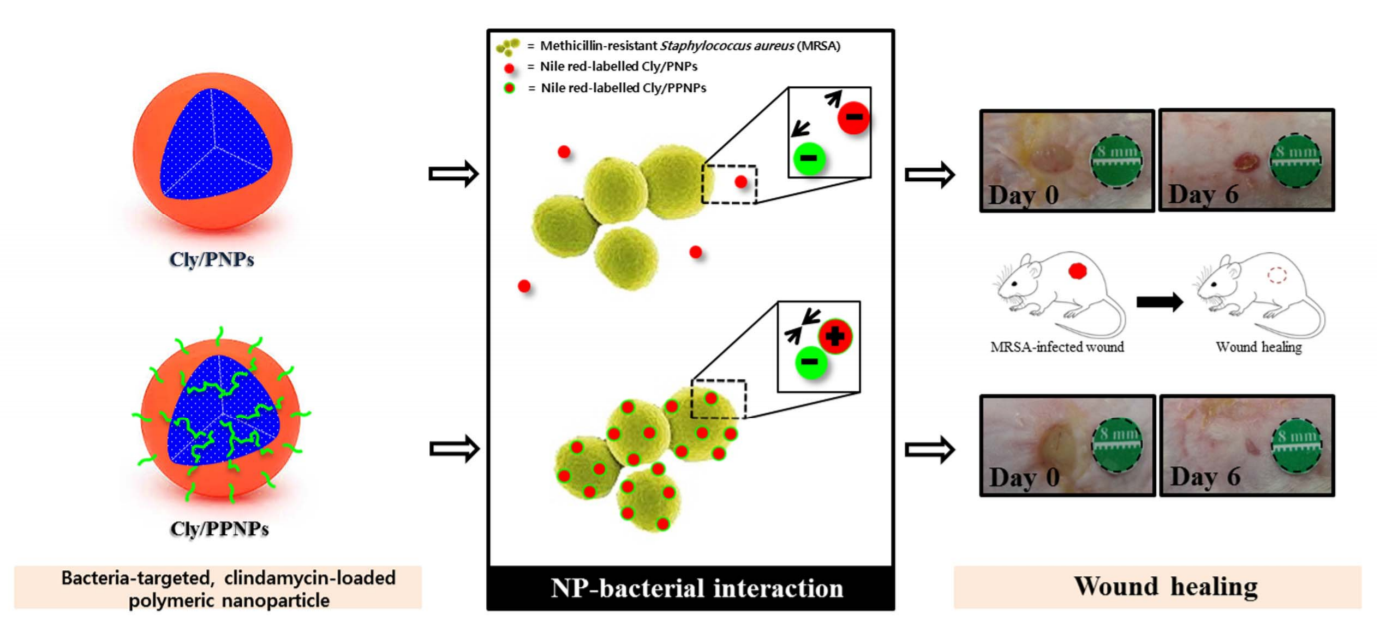
2.2. NP Preparation
2.3. Characterization of NP
2.3.1. Scanning Electron Microscopy (SEM)
2.3.2. Particle Size and Surface Charge Analysis
2.3.3. Drug Loading
2.4. In Vitro Drug Release
2.5. NP Adhesion to the Bacteria
2.6. In Vitro Antibacterial Study
2.7. In Vitro Cytotoxicity Study
2.8. In Vivo Wound Healing Assay
2.9. Histological Processing
2.10. Reduction of Wound Bacterial Burden
2.11. Statistical Analysis
3. Results
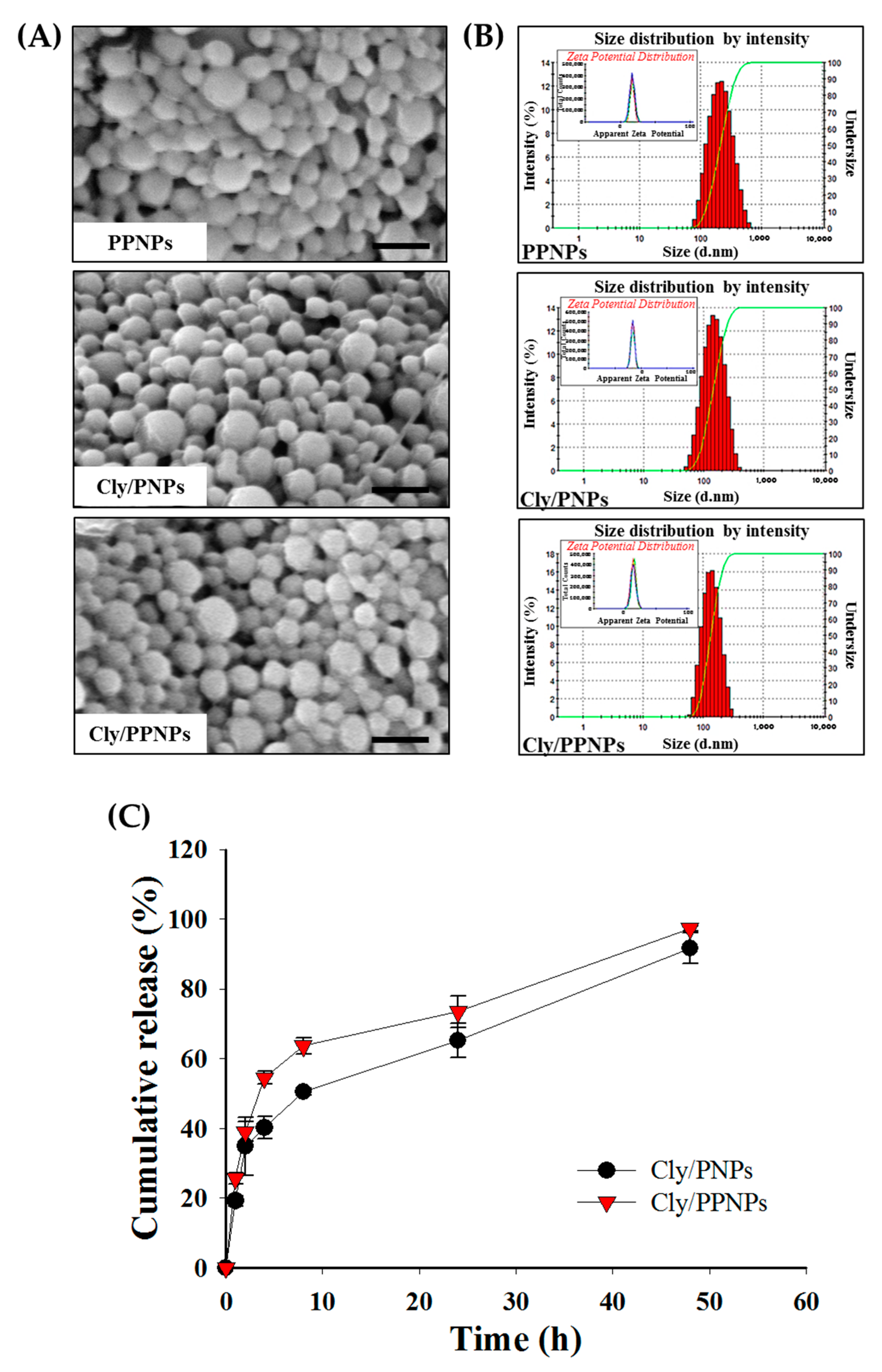
3.1. Characterization of NPs
3.2. In Vitro Drug Release
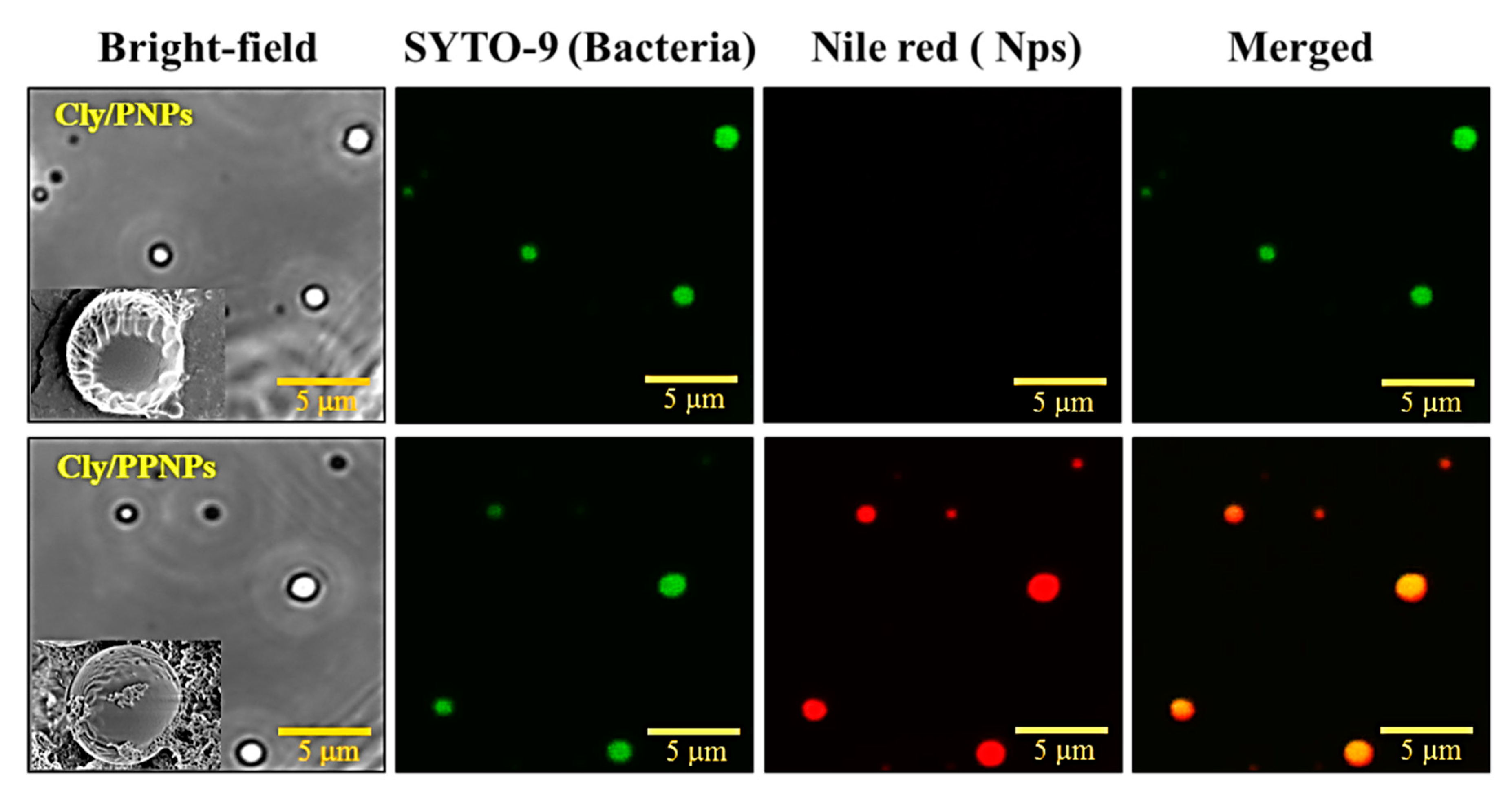
3.3. Adhesion of NPs to MRSA
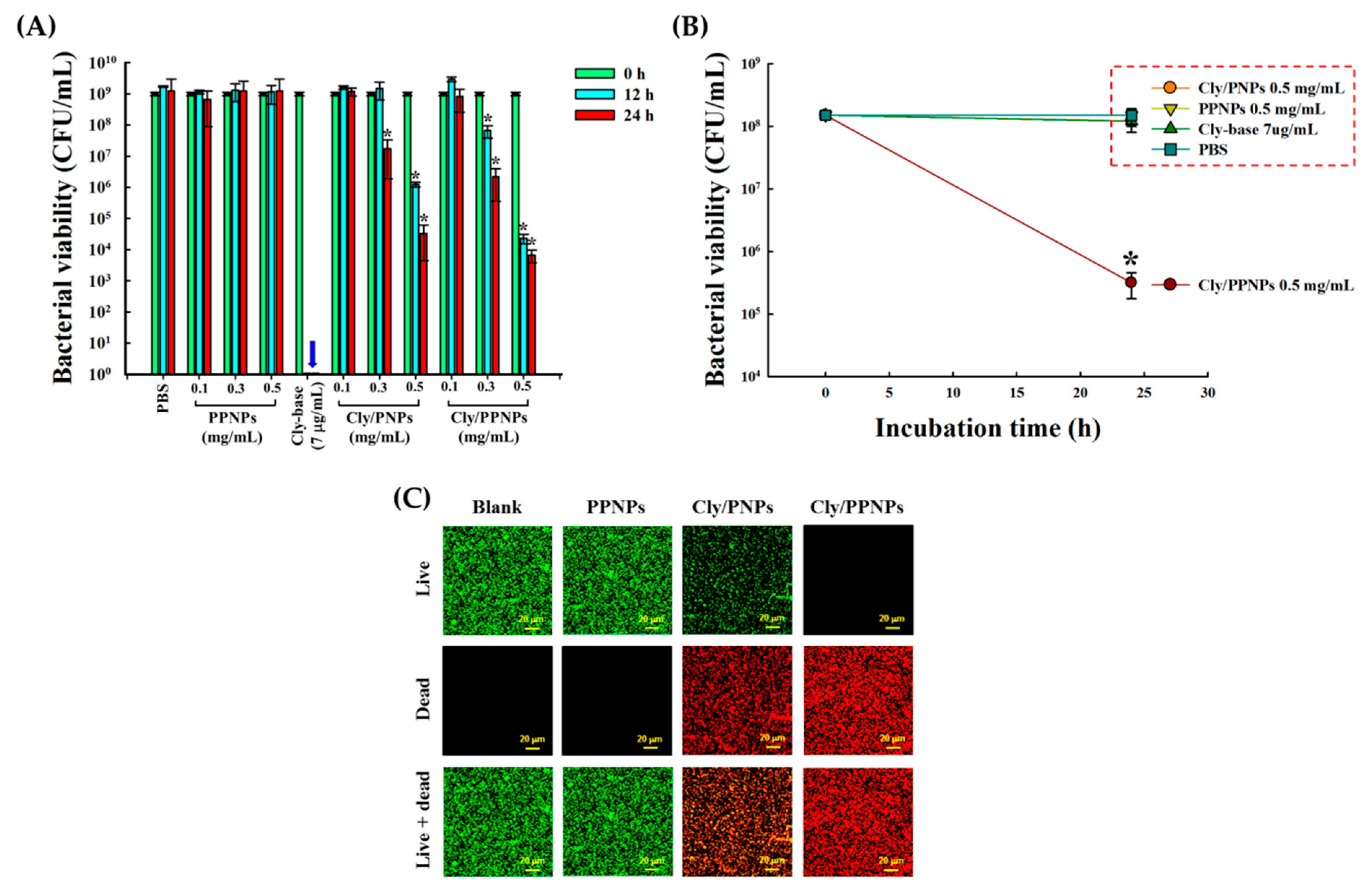
3.4. In Vitro Antibacterial Activity of NP
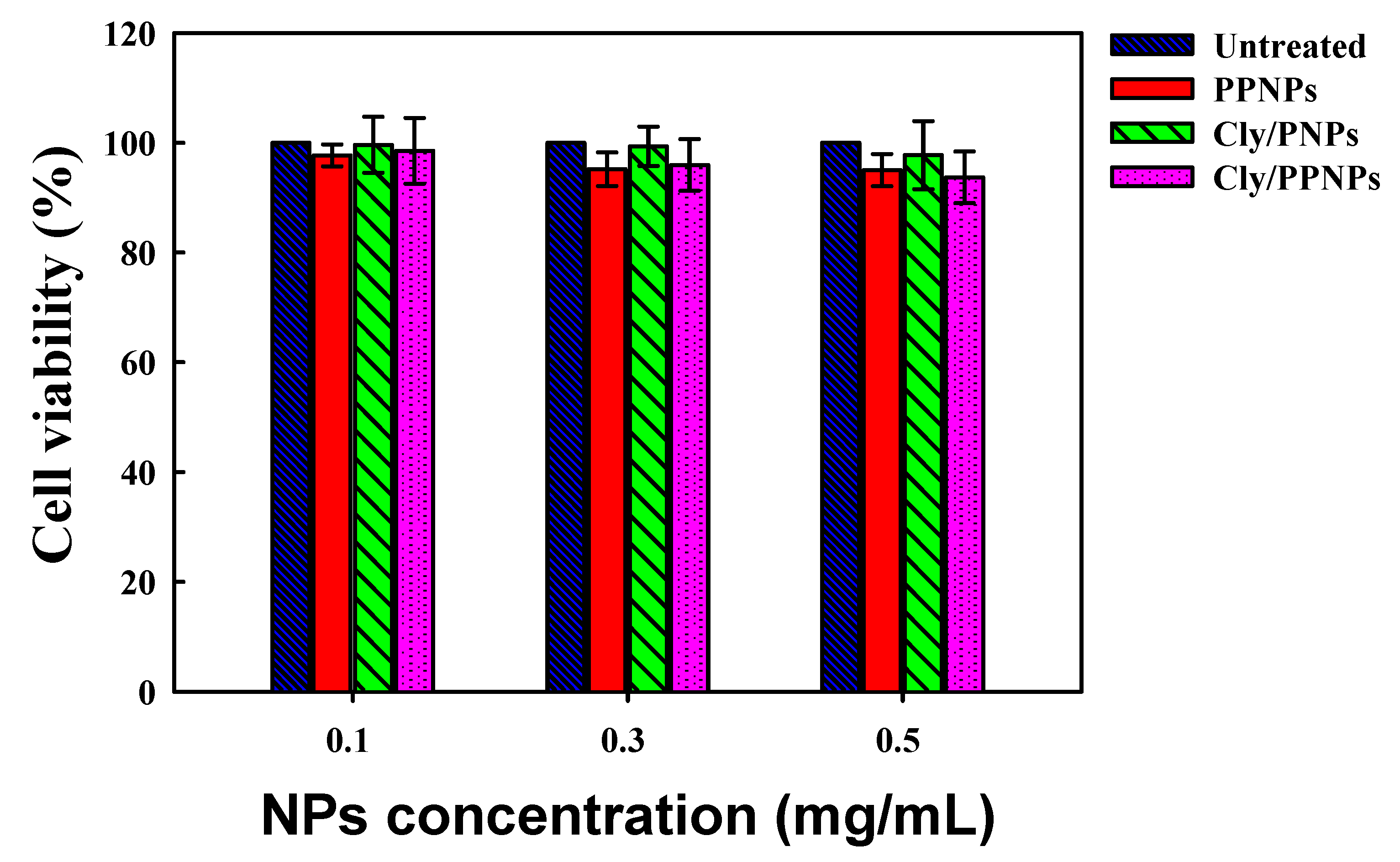
3.5. In Vitro Cytotoxicity Study
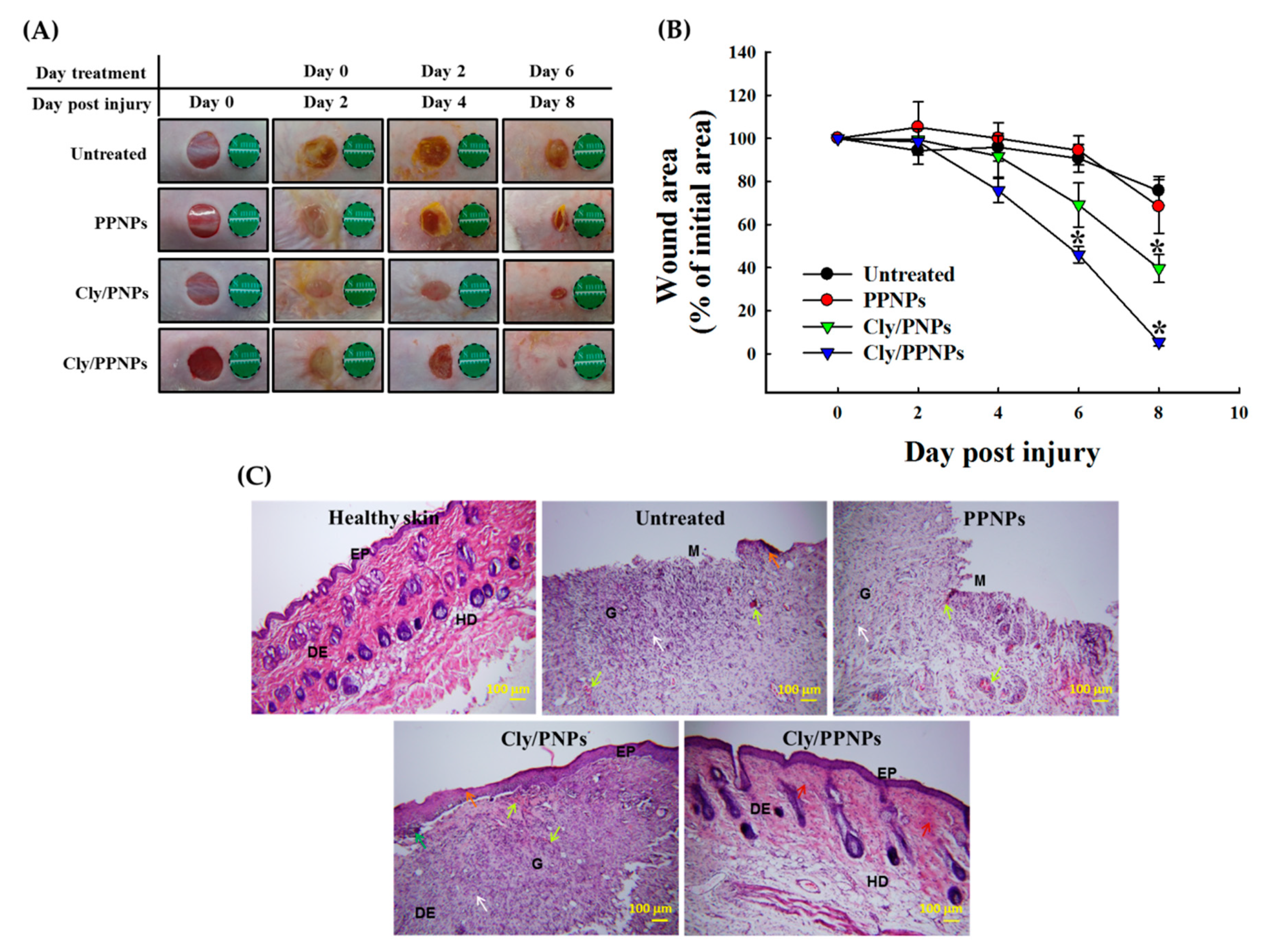
3.6. In Vivo Wound Healing Assay
3.7. Histological Analysis
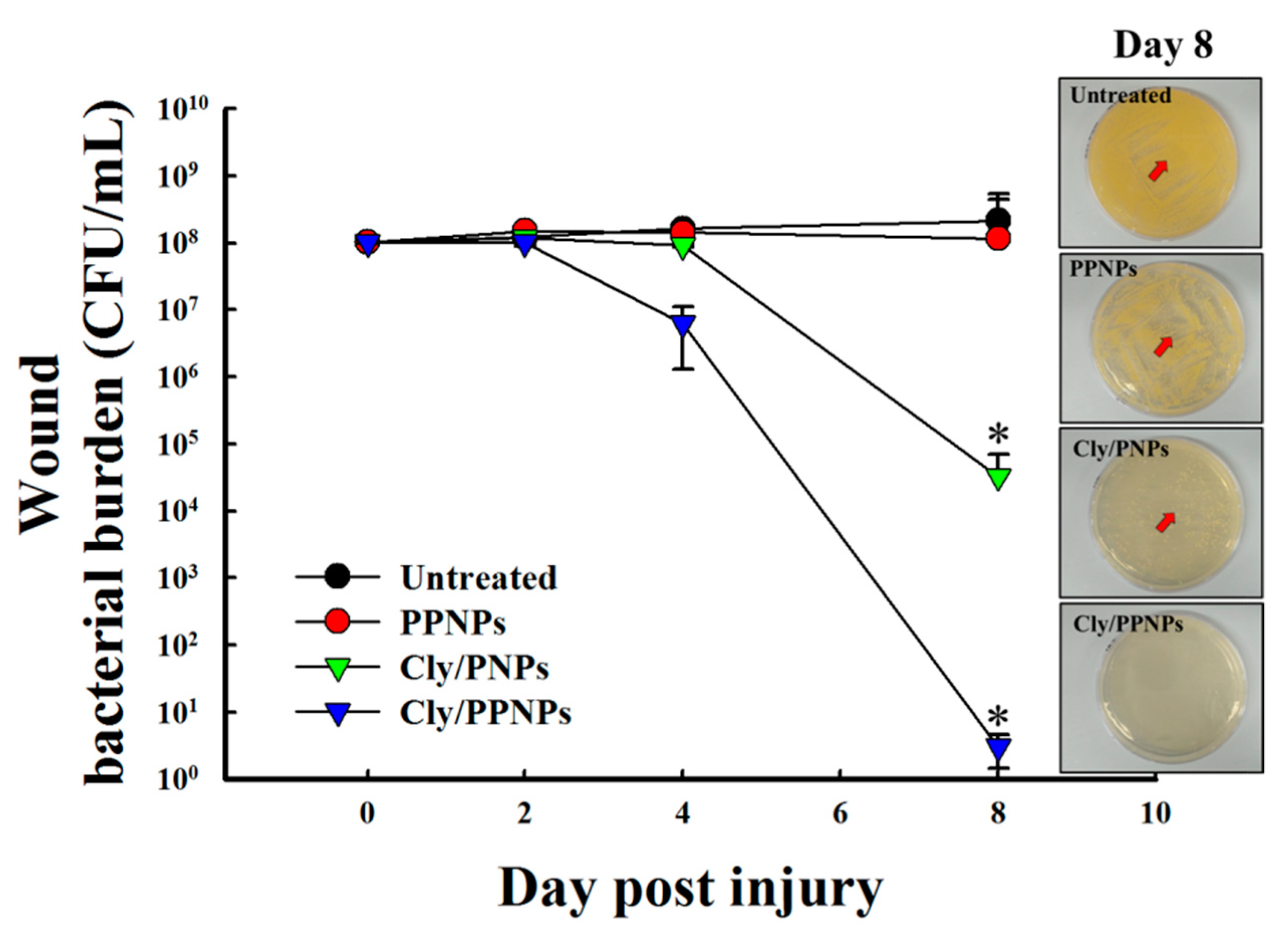
3.8. Reduction of Wound Bacterial Burden
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Hlaing, S.P.; Kim, J.; Lee, J.; Hasan, N.; Cao, J.; Naeem, M.; Lee, E.H.; Shin, J.H.; Jung, Y.; Lee, B.-L.; et al. S-Nitrosoglutathione loaded poly(lactic-co-glycolic acid) microparticles for prolonged nitric oxide release and enhanced healing of methicillin-resistant Staphylococcus aureus-infected wounds. Eur. J. Pharm. Biopharm. 2018, 132, 94–102. [Google Scholar] [CrossRef]
- Edwards, R.; Harding, K.G. Bacteria and wound healing. Curr. Opin. Infect. Dis. 2004, 17, 91–96. [Google Scholar] [CrossRef]
- Robson, M.C. Wound infection: A failure of wound healing caused by an imbalance of bacteria. Surg. Clin. N. Am. 1997, 77, 637–650. [Google Scholar] [CrossRef]
- Zervos, M.J.; Freeman, K.; Vo, L.; Haque, N.; Pokharna, H.; Raut, M.; Kim, M. Epidemiology and outcomes of complicated skin and soft tissue infections in hospitalized patients. J. Clin. Microbiol. 2012, 50, 238–245. [Google Scholar] [CrossRef] [PubMed]
- Rajan, S. Skin and soft-tissue infections: Classifying and treating a spectrum. Clevel. Clin. Q. 2012, 79, 57–66. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration. Guidance for Industry. Acute Bacterial Skin and Skin Structure Infections: Developing Drugs for Treatment; Food and Drug Administration: Silver Spring, MD, USA, 2013. Available online: http://www.fda.gov/downloads/Drugs/./Guidances/ucm071185.pdf (accessed on 16 January 2019).
- Klevens, R.M.; Morrison, M.A.; Nadle, J.; Petit, S.; Gershman, K.; Ray, S.; Harrison, L.H.; Lynfield, R.; Dumyati, G.; Townes, J.M.; et al. Invasive methicillin-resistant Staphylococcus aureus infections in the United States. JAMA 2007, 298, 1763–1771. [Google Scholar] [CrossRef]
- Andersson, H.; Lindholm, C.; Fossum, B. MRSA—Global threat and personal disaster: patients’ experiences. Int. Nurs. Rev. 2011, 58, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Hewagama, S.; Spelman, T.; Einsiedel, L. Staphylococcus aureus bacteraemia at Alice Springs Hospital, Central Australia, 2003–2006. Intern. Med. J. 2012, 42, 505–512. [Google Scholar] [CrossRef] [PubMed]
- Fridkin, S.K.; Hageman, J.C.; Morrison, M.; Sanza, L.T.; Como-Sabetti, K.; Jernigan, J.A.; Harriman, K.; Harrison, L.H.; Lynfield, R.; Farley, M.M. Methicillin-resistant Staphylococcus aureus disease in three communities. N. Engl. J. Med. 2005, 352, 1436–1444. [Google Scholar] [CrossRef]
- Cosgrove, S.E.; Sakoulas, G.; Perencevich, E.N.; Schwaber, M.J.; Karchmer, A.W.; Carmeli, Y. Comparison of mortality associated with methicillin-resistant and methicillin-susceptible Staphylococcus aureus bacteremia: A meta-analysis. Clin. Infect. Dis. 2003, 36, 53–59. [Google Scholar] [CrossRef]
- Tom, S.; Galbraith, J.; Valiquette, L.; Jacobsson, G.; Collignon, P.; Schønheyder, H.; Søgaard, M.; Kennedy, K.; Knudsen, J.; Østergaard, C.; et al. Case fatality ratio and mortality rate trends of community-onset Staphylococcus aureus bacteraemia. Clin. Microbiol. Infect. 2014, 20, 0630–0632. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention. Outpatient Management of Skin and Soft Tissue Infections in the Era of Community-Associated MRSA. 2009. Available online: http://www.cdc.gov/ncidod/dhqp/pdf/ar/AMA_Flyer_Final.pdf (accessed on 16 January 2019).
- Martínez-Aguilar, G.; Hammerman, W.A.; Mason, E.O., Jr.; Kaplan, S.L. Clindamycin treatment of invasive infections caused by community-acquired, methicillin-resistant and methicillin-susceptible Staphylococcus aureus in children. Pediatr. Infect. Dis. J. 2003, 22, 593–599. [Google Scholar] [CrossRef]
- Spížek, J.; Řezanka, T. Lincosamides: Chemical structure, biosynthesis, mechanism of action, resistance, and applications. Biochem. Pharmacol. 2017, 133, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Iyer, S.; Jones, D.H. Community-acquired methicillin-resistant Staphylococcus aureus skin infection: A retrospective analysis of clinical presentation and treatment of a local outbreak. J. Am. Acad. Dermatol. 2004, 50, 854–858. [Google Scholar] [CrossRef] [PubMed]
- Frei, C.R.; Miller, M.L.; Lewis, J.S.; Lawson, K.A.; Hunter, J.M.; Oramasionwu, C.U.; Talbert, R.L. Trimethoprim-sulfamethoxazole or clindamycin for community-associated MRSA (CA-MRSA) skin infections. J. Am. Board Fam. Med. 2010, 23, 714–719. [Google Scholar] [CrossRef]
- Uskoković, V.; Desai, T.A. Simultaneous bactericidal and osteogenic effect of nanoparticulate calcium phosphate powders loaded with clindamycin on osteoblasts infected with Staphylococcus aureus. Mater. Sci. Eng. C 2014, 37, 210–222. [Google Scholar] [CrossRef]
- Hajipour, M.J.; Fromm, K.M.; Ashkarran, A.A.; de Aberasturi, D.J.; de Larramendi, I.R.; Rojo, T.; Serpooshan, V.; Parak, W.J.; Mahmoudi, M. Antibacterial properties of nanoparticles. Trends Biotechnol. 2012, 30, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Rajput, N.; Bankar, A. Bio-inspired gold nanoparticles synthesis and their anti-biofilm efficacy. J. Pharm. Investig. 2017, 47, 521–530. [Google Scholar] [CrossRef]
- Reis, C.P.; Neufeld, R.J.; Ribeiro, A.J.; Veiga, F. Nanoencapsulation I. Methods for preparation of drug-loaded polymeric nanoparticles. Nanomed. Nanotechnol. Biol. Med. 2006, 2, 8–21. [Google Scholar] [CrossRef]
- Radovic-Moreno, A.F.; Lu, T.K.; Puscasu, V.A.; Yoon, C.J.; Langer, R.; Farokhzad, O.C. Surface charge-switching polymeric nanoparticles for bacterial cell wall-targeted delivery of antibiotics. ACS Nano 2012, 6, 4279–4287. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.H.; Han, H.-K. Nanomedicines: Current status and future perspectives in aspect of drug delivery and pharmacokinetics. J. Pharm. Investig. 2018, 48, 43–60. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.H.; Lee, S.G.; Kang, M.J.; Lee, S.; Choi, Y.W. Surface modification of lipid-based nanocarriers for cancer cell-specific drug targeting. J. Pharm. Investig. 2017, 47, 203–227. [Google Scholar] [CrossRef]
- Zeb, A.; Arif, S.T.; Malik, M.; Shah, F.A.; Din, F.U.; Qureshi, O.S.; Lee, E.-S.; Lee, G.-Y.; Kim, J.-K. Potential of nanoparticulate carriers for improved drug delivery via skin. J. Pharm. Investig. 2018, 1–33. [Google Scholar] [CrossRef]
- Zhang, L.; Pornpattananangkul, D.; Hu, C.-M.; Huang, C.-M. Development of nanoparticles for antimicrobial drug delivery. Curr. Med. Chem. 2010, 17, 585–594. [Google Scholar] [CrossRef] [PubMed]
- Levy, S.B.; Marshall, B. Antibacterial resistance worldwide: Causes, challenges and responses. Nat. Med. 2004, 10, S122–S129. [Google Scholar] [CrossRef]
- Huh, A.J.; Kwon, Y.J. “Nanoantibiotics”: A new paradigm for treating infectious diseases using nanomaterials in the antibiotics resistant era. J. Control. Release 2011, 156, 128–145. [Google Scholar] [CrossRef] [PubMed]
- Abbaspour, M.; Makhmalzadeh, B.S.; Arastoo, Z.; Jahangiri, A.; Shiralipour, R. Effect of anionic polymers on drug loading and release from clindamycin phosphate solid lipid nanoparticles. Trop. J. Pharm. Res. 2013, 12, 477–482. [Google Scholar] [CrossRef]
- Vukomanović, M.; Zavašnik-Bergant, T.; Bračko, I.; Škapin, S.D.; Ignjatović, N.; Radmilović, V.; Uskoković, D. Poly (d, l-lactide-co-glycolide)/hydroxyapatite core–shell nanospheres. Part 3: Properties of hydroxyapatite nano-rods and investigation of a distribution of the drug within the composite. Colloids Surf. B 2011, 87, 226–235. [Google Scholar] [CrossRef]
- Durán, N.; Marcato, P.; De Conti, R.; Alves, O.; Brocchi, M. Silver nanoparticles: Control of pathogens, toxicity and cytotoxicity. Nanotoxicology 2008, 2, S32. [Google Scholar]
- Nurhasni, H.; Cao, J.; Choi, M.; Kim, I.; Lee, B.L.; Jung, Y.; Yoo, J.-W. Nitric oxide-releasing poly (lactic-co-glycolic acid)-polyethylenimine nanoparticles for prolonged nitric oxide release, antibacterial efficacy, and in vivo wound healing activity. Int. J. Nanomed. 2015, 10, 3065–3080. [Google Scholar]
- Choi, J.-S.; Cao, J.; Naeem, M.; Noh, J.; Hasan, N.; Choi, H.-K.; Yoo, J.-W. Size-controlled biodegradable nanoparticles: Preparation and size-dependent cellular uptake and tumor cell growth inhibition. Colloids Surf. B 2014, 122, 545–551. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.-S.; Seo, K.; Yoo, J.-W. Recent advances in PLGA particulate systems for drug delivery. J. Pharm. Investig. 2012, 42, 155–163. [Google Scholar] [CrossRef]
- Ma, X.; Williams, R.O. Polymeric nanomedicines for poorly soluble drugs in oral delivery systems: An update. J. Pharm. Investig. 2018, 48, 61–75. [Google Scholar] [CrossRef]
- Cao, J.; Choi, J.-S.; Oshi, M.A.; Lee, J.; Hasan, N.; Kim, J.; Yoo, J.-W. Development of PLGA micro-and nanorods with high capacity of surface ligand conjugation for enhanced targeted delivery. Asian J. Pharm. Sci. 2019, 14, 86–94. [Google Scholar] [CrossRef]
- Naeem, M.; Bae, J.; Oshi, M.A.; Kim, M.-S.; Moon, H.R.; Lee, B.L.; Im, E.; Jung, Y.; Yoo, J.-W. Colon-targeted delivery of cyclosporine A using dual-functional Eudragit® FS30D/PLGA nanoparticles ameliorates murine experimental colitis. Int. J. Nanomed. 2018, 13, 1225–1240. [Google Scholar] [CrossRef] [PubMed]
- McDougal, L.K.; Steward, C.D.; Killgore, G.E.; Chaitram, J.M.; McAllister, S.K.; Tenover, F.C. Pulsed-field gel electrophoresis typing of oxacillin-resistant Staphylococcus aureus isolates from the United States: Establishing a national database. J. Clin. Microbiol. 2003, 41, 5113–5120. [Google Scholar] [CrossRef]
- Fredenberg, S.; Wahlgren, M.; Reslow, M.; Axelsson, A. The mechanisms of drug release in poly (lactic-co-glycolic acid)-based drug delivery systems—A review. Int. J. Pharm. 2011, 415, 34–52. [Google Scholar] [CrossRef] [PubMed]
- Chereddy, K.K.; Vandermeulen, G.; Préat, V. PLGA based drug delivery systems: Promising carriers for wound healing activity. Wound Repair Regen. 2016, 24, 223–236. [Google Scholar] [CrossRef] [PubMed]
- Son, G.-H.; Lee, B.-J.; Cho, C.-W. Mechanisms of drug release from advanced drug formulations such as polymeric-based drug-delivery systems and lipid nanoparticles. J. Pharm. Investig. 2017, 47, 287–296. [Google Scholar] [CrossRef]
- Sosnik, A.; Carcaboso, Á.M.; Glisoni, R.J.; Moretton, M.A.; Chiappetta, D.A. New old challenges in tuberculosis: Potentially effective nanotechnologies in drug delivery. Adv. Drug Deliv. Rev. 2010, 62, 547–559. [Google Scholar] [CrossRef] [PubMed]
- Weidenmaier, C.; Peschel, A. Teichoic acids and related cell-wall glycopolymers in Gram-positive physiology and host interactions. Nat. Rev. Microbiol. 2008, 6, 276–287. [Google Scholar] [CrossRef]
- Rudramurthy, G.R.; Swamy, M.K.; Sinniah, U.R.; Ghasemzadeh, A. Nanoparticles: Alternatives against drug-resistant pathogenic microbes. Molecules 2016, 21, 836. [Google Scholar] [CrossRef] [PubMed]
- Khalil, H.; Chen, T.; Riffon, R.; Wang, R.; Wang, Z. Synergy between polyethylenimine and different families of antibiotics against a resistant clinical isolate of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2008, 52, 1635–1641. [Google Scholar] [CrossRef]
- Rezaee, M.; Gholami, L.; Gildeh, M.S.; Ramezani, M.; Oskuee, R.K. Charge reduction: An efficient strategy to reduce toxicity and increase the transfection efficiency of high molecular weight polyethylenimine. J. Pharm. Investig. 2019, 49, 105–114. [Google Scholar] [CrossRef]
- Chung, Y.-C.; Wang, H.-L.; Chen, Y.-M.; Li, S.-L. Effect of abiotic factors on the antibacterial activity of chitosan against waterborne pathogens. Bioresour. Technol. 2003, 88, 179–184. [Google Scholar] [CrossRef]
- Fang, B.; Gon, S.; Park, M.; Kumar, K.-N.; Rotello, V.M.; Nusslein, K.; Santore, M.M. Bacterial adhesion on hybrid cationic nanoparticle–polymer brush surfaces: Ionic strength tunes capture from monovalent to multivalent binding. Colloids Surf. B 2011, 87, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Dillen, K.; Bridts, C.; Van der Veken, P.; Cos, P.; Vandervoort, J.; Augustyns, K.; Stevens, W.; Ludwig, A. Adhesion of PLGA or Eudragit®/PLGA nanoparticles to Staphylococcus and Pseudomonas. Int. J. Pharm. 2008, 349, 234–240. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Xu, K.; Wang, H.; Tan, P.J.; Fan, W.; Venkatraman, S.S.; Li, L.; Yang, Y.-Y. Self-assembled cationic peptide nanoparticles as an efficient antimicrobial agent. Nat. Nanotechnol. 2009, 4, 457–463. [Google Scholar] [CrossRef]
- Wong, T.; McGrath, J.; Navsaria, H. The role of fibroblasts in tissue engineering and regeneration. Br. J. Dermatol. 2007, 156, 1149–1155. [Google Scholar] [CrossRef]
- Aarabi, S.; Bhatt, K.A.; Shi, Y.; Paterno, J.; Chang, E.I.; Loh, S.A.; Holmes, J.W.; Longaker, M.T.; Yee, H.; Gurtner, G.C. Mechanical load initiates hypertrophic scar formation through decreased cellular apoptosis. FASEB J. 2007, 21, 3250–3261. [Google Scholar] [CrossRef]
- Bae, S.H.; Bae, Y.C.; Nam, S.B.; Choi, S.J. A skin fixation method for decreasing the influence of wound contraction on wound healing in a rat model. Arch. Plast. Surg. 2012, 39, 457–462. [Google Scholar] [CrossRef] [PubMed]
- Porporato, P.E.; Payen, V.L.; De Saedeleer, C.J.; Préat, V.; Thissen, J.-P.; Feron, O.; Sonveaux, P. Lactate stimulates angiogenesis and accelerates the healing of superficial and ischemic wounds in mice. Angiogenesis 2012, 15, 581–592. [Google Scholar] [CrossRef] [PubMed]
- Hunt, T.K.; Conolly, W.B.; Aronson, S.B.; Goldstein, P. Anaerobic metabolism and wound healing: An hypothesis for the initiation and cessation of collagen synthesis in wounds. Am. J. Surg. 1978, 135, 328–332. [Google Scholar] [CrossRef]
- Chereddy, K.K.; Coco, R.; Memvanga, P.B.; Ucakar, B.; des Rieux, A.; Vandermeulen, G.; Préat, V. Combined effect of PLGA and curcumin on wound healing activity. J. Control. Release 2013, 171, 208–215. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NPs | Drug Loading (% w/w) | Size (nm) | PDI | Zeta Potential (mV) | |
|---|---|---|---|---|---|
| DLS | SEM | ||||
| PPNPs | Not determined | 193 ± 38 | 184 ± 36 | 0.15 | +17 ± 0.50 |
| Cly/PNPs | 1.43 ± 0.46 | 132 ± 41 | 141 ± 43 | 0.14 | −16 ± 0.20 |
| Cly/PPNPs | 1.31 ± 0.26 | 126 ± 33 | 147 ± 37 | 0.10 | +13 ± 0.60 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hasan, N.; Cao, J.; Lee, J.; Hlaing, S.P.; Oshi, M.A.; Naeem, M.; Ki, M.-H.; Lee, B.L.; Jung, Y.; Yoo, J.-W. Bacteria-Targeted Clindamycin Loaded Polymeric Nanoparticles: Effect of Surface Charge on Nanoparticle Adhesion to MRSA, Antibacterial Activity, and Wound Healing. Pharmaceutics 2019, 11, 236. https://doi.org/10.3390/pharmaceutics11050236
Hasan N, Cao J, Lee J, Hlaing SP, Oshi MA, Naeem M, Ki M-H, Lee BL, Jung Y, Yoo J-W. Bacteria-Targeted Clindamycin Loaded Polymeric Nanoparticles: Effect of Surface Charge on Nanoparticle Adhesion to MRSA, Antibacterial Activity, and Wound Healing. Pharmaceutics. 2019; 11(5):236. https://doi.org/10.3390/pharmaceutics11050236
Chicago/Turabian StyleHasan, Nurhasni, Jiafu Cao, Juho Lee, Shwe Phyu Hlaing, Murtada A. Oshi, Muhammad Naeem, Min-Hyo Ki, Bok Luel Lee, Yunjin Jung, and Jin-Wook Yoo. 2019. "Bacteria-Targeted Clindamycin Loaded Polymeric Nanoparticles: Effect of Surface Charge on Nanoparticle Adhesion to MRSA, Antibacterial Activity, and Wound Healing" Pharmaceutics 11, no. 5: 236. https://doi.org/10.3390/pharmaceutics11050236