Systemic Design and Evaluation of Ticagrelor-Loaded Nanostructured Lipid Carriers for Enhancing Bioavailability and Antiplatelet Activity

 , and
, and 
Abstract
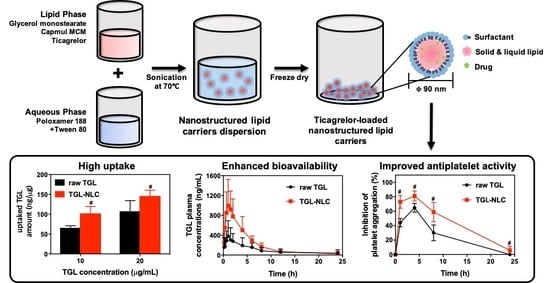
:
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. HPLC Analysis
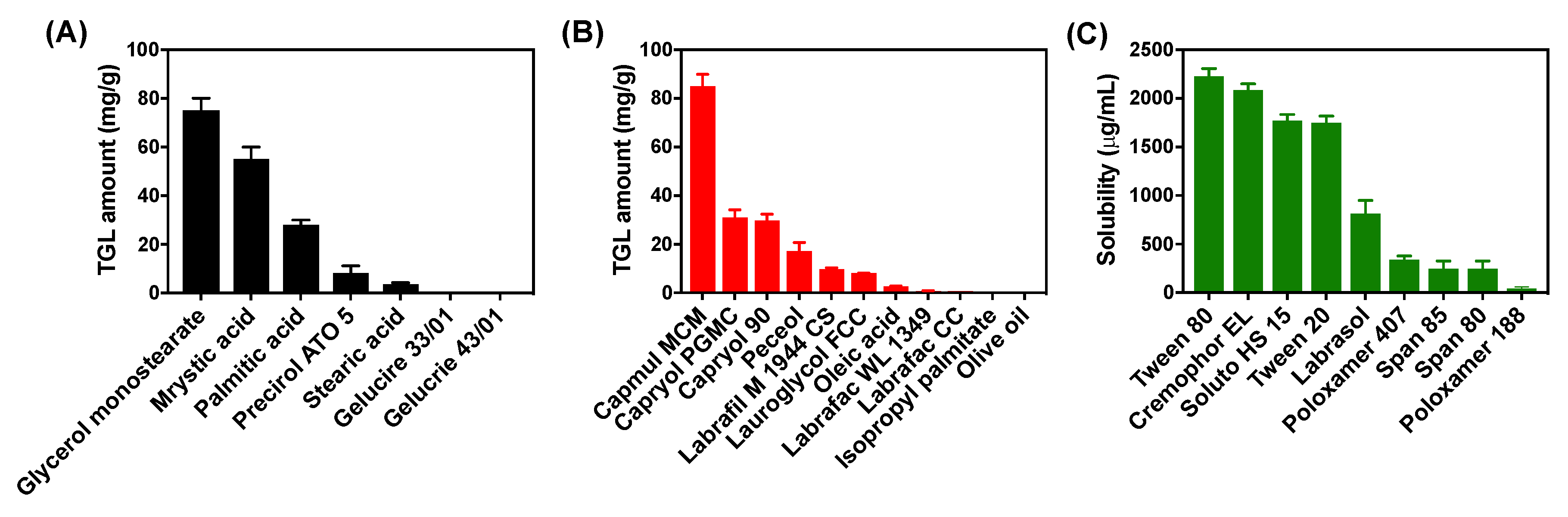
2.3. Solubility Study
2.4. Preparation of TGL-NLC
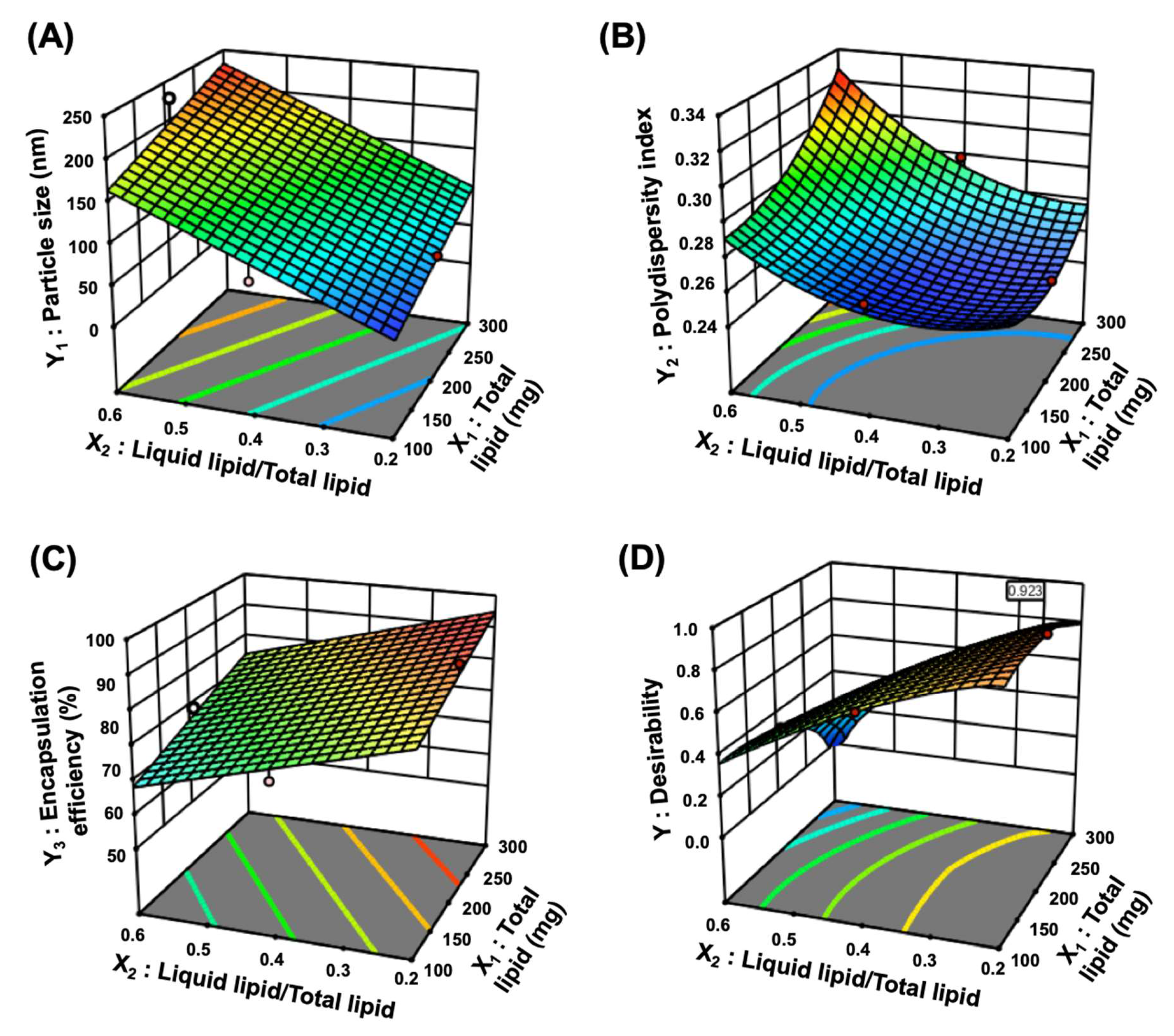
2.5. Optimization of TGL-NLC
2.5.1. Particle size (Y1) and Polydispersity Index (Y2)
2.5.2. Encapsulation Efficiency (Y3)
2.6. Characterization of Optimized TGL-NLC
2.7. Cell Studies
2.7.1. Cell Culture
2.7.2. Cytotoxicity Study
2.7.3. Cellular Uptake Study
2.8. Pharmacokinetic Study
2.8.1. Animal Study
2.8.2. LC-MS/MS analysis of TGL
2.8.3. Pharmacokinetic Data Analysis
2.9. Pharmacodynamic Study
2.10. Statistical Analysis
3. Results
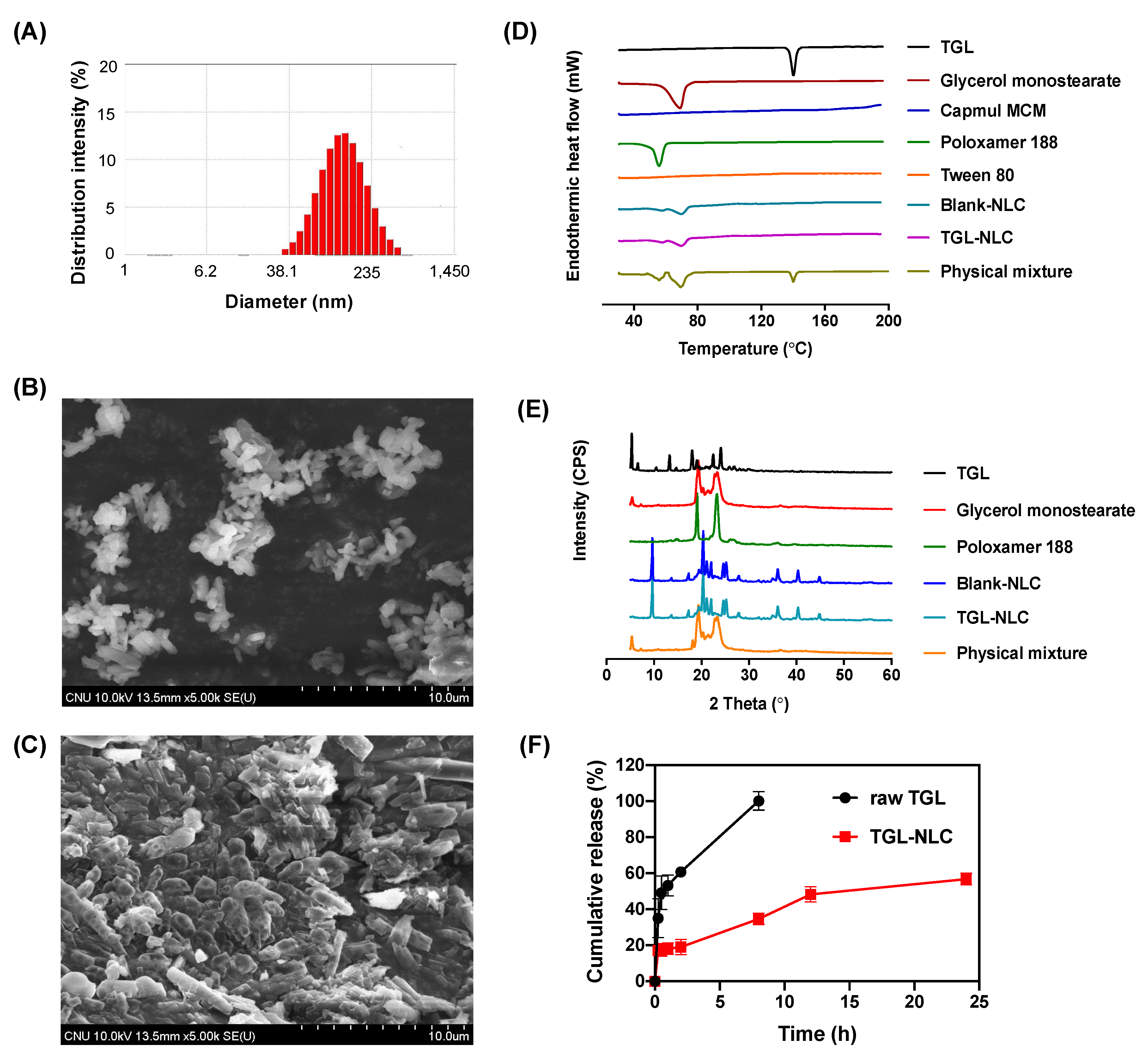
3.1. Optimization and Characterization of TGL-NLC
3.1.1. Solubility Study for TGL-NLC
3.1.2. Optimization of TGL-NLC
3.1.3. Characterization of Optimized TGL-NLC
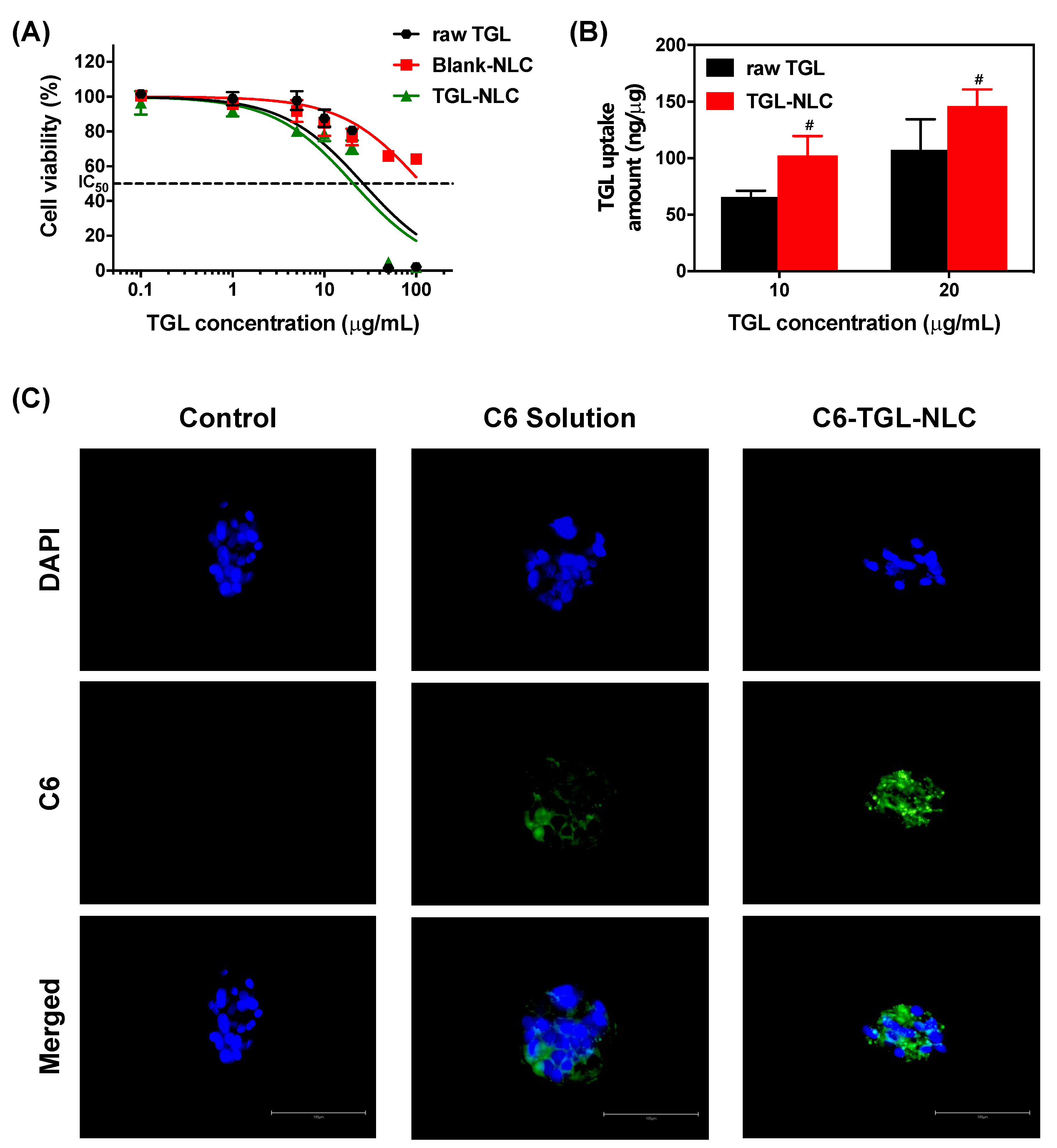
3.2. Cell Studies
3.2.1. Cytotoxicity Study
3.2.2. Cellular Uptake Study
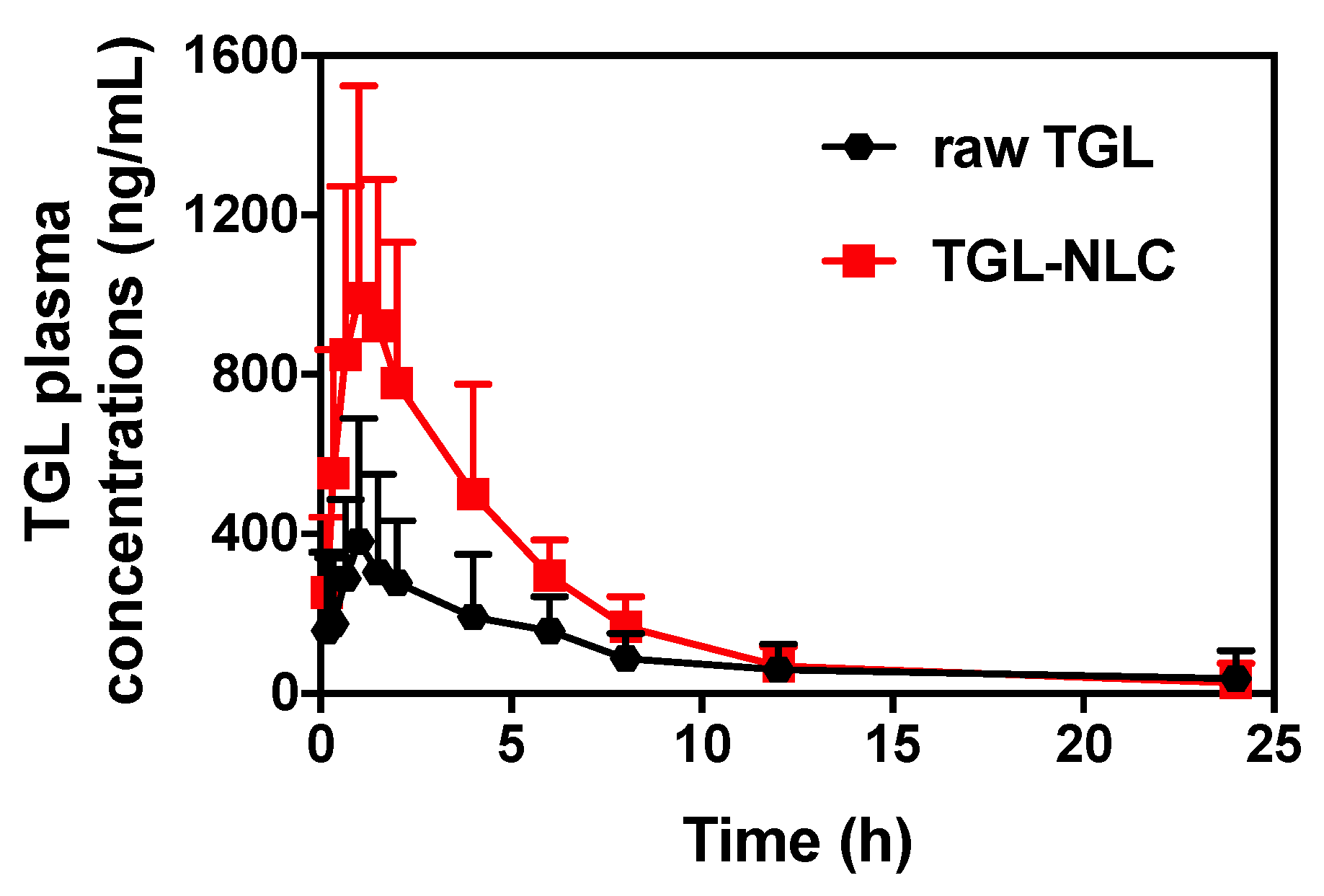
3.3. Pharmacokinetic Study
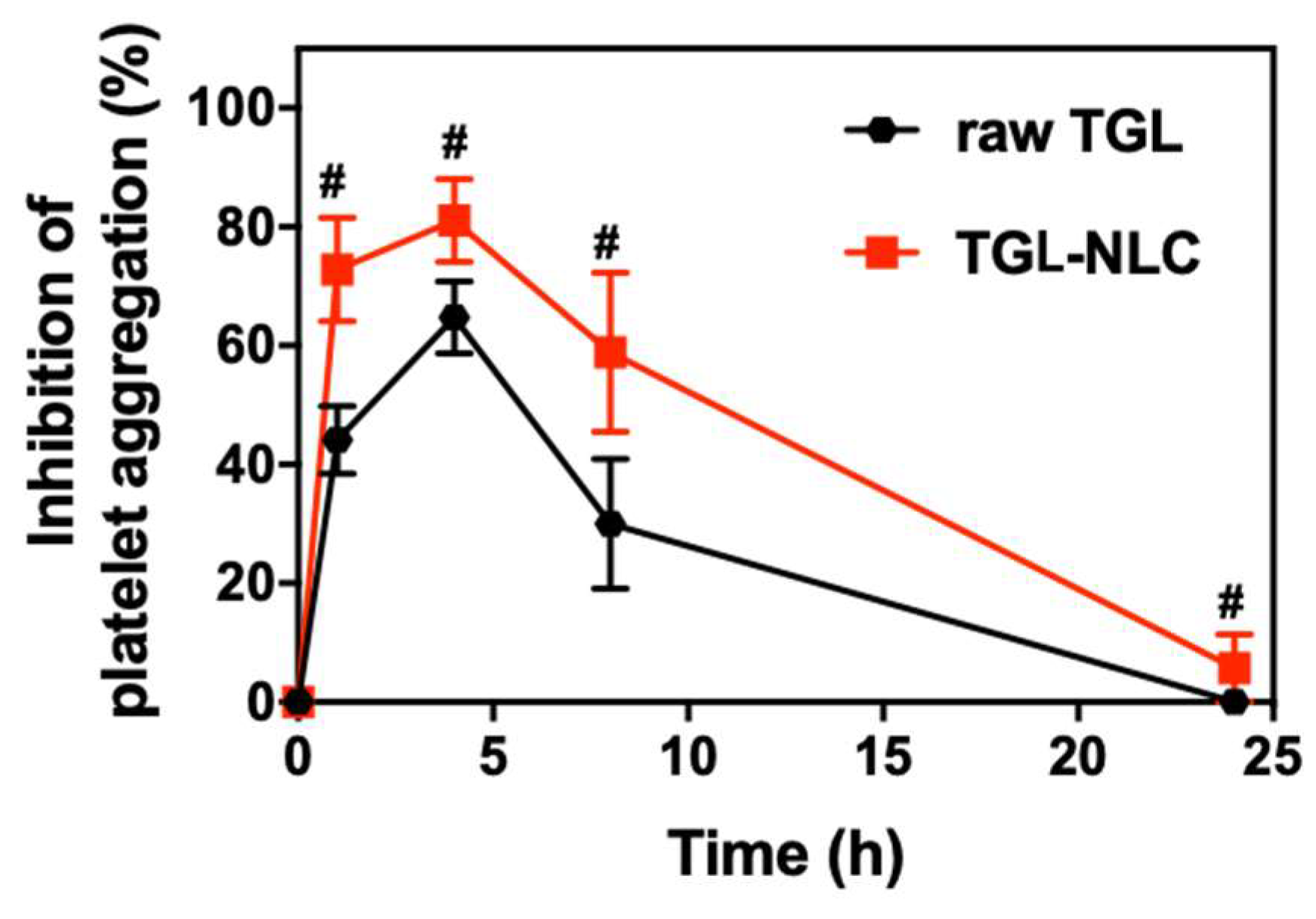
3.4. Pharmacodynamic Study
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Husted, S.; van Giezen, J.J. Ticagrelor: The first reversibly binding oral P2Y12 receptor antagonist. Cardiovasc. Ther. 2009, 27, 259–274. [Google Scholar] [CrossRef] [PubMed]
- Kubisa, M.J.; Jezewski, M.P.; Gasecka, A.; Siller-Matula, J.M.; Postula, M. Ticagrelor-toward more efficient platelet inhibition and beyond. Ther. Clin. Risk Manag. 2018, 14, 129–140. [Google Scholar] [CrossRef]
- Norgard, N.B.; Abu-Fadel, M. Comparison of prasugrel and clopidogrel in patients with acute coronary syndrome undergoing percutaneous coronary intervention. Vasc. Health Risk Manag. 2009, 5, 873–882. [Google Scholar] [CrossRef]
- Ghadi, R.; Dand, N. BCS class IV drugs: Highly notorious candidates for formulation development. J. Control. Release 2017, 248, 71–95. [Google Scholar] [CrossRef]
- Teng, R.; Maya, J. Absolute bioavailability and regional absorption of ticagrelor in healthy volunteers. J. Drug Assess. 2014, 3, 43–50. [Google Scholar] [PubMed] [Green Version]
- Mohammadi, M.R.; Nojoomi, A.; Mozafari, M.; Dubnika, A.; Inayathullah, M.; Rajadas, J. Nanomaterials engineering for drug delivery: A hybridization approach. J. Mater. Chem. B 2017, 5, 3995–4018. [Google Scholar] [CrossRef]
- Beloqui, A.; Solinis, M.A.; Rodriguez-Gascon, A.; Almeida, A.J.; Preat, V. Nanostructured lipid carriers: Promising drug delivery systems for future clinics. Nanomedicine 2016, 12, 143–161. [Google Scholar] [CrossRef] [PubMed]
- Na, Y.G.; Byeon, J.J.; Wang, M.; Huh, H.W.; Son, G.H.; Jeon, S.H.; Bang, K.H.; Kim, S.J.; Lee, H.J.; Lee, H.K.; et al. Strategic approach to developing a self-microemulsifying drug delivery system to enhance antiplatelet activity and bioavailability of ticagrelor. Int. J. Nanomed. 2019, 14, 1193–1212. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Lee, H.K.; Na, Y.G.; Bang, K.H.; Lee, H.J.; Wang, M.; Huh, H.W.; Cho, C.W. A novel composition of ticagrelor by solid dispersion technique for increasing solubility and intestinal permeability. Int. J. Pharm. 2019, 555, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Herneisey, M.; Liu, L.; Lambert, E.; Schmitz, N.; Loftus, S.; Janjic, J.M. Development of theranostic perfluorocarbon nanoemulsions as a model non-opioid pain nanomedicine using a quality by design (QbD) approach. AAPS PharmSciTech 2019, 20, 65. [Google Scholar] [CrossRef] [PubMed]
- Beg, S.; Kaur, R.; Khurana, R.K.; Rana, V.; Sharma, T.; Singh, B. QbD-based development of cationic self-nanoemulsifying drug delivery systems of paclitaxel with Improved biopharmaceutical attributes. AAPS PharmSciTech 2019, 20, 118. [Google Scholar] [CrossRef] [PubMed]
- Dahmash, E.Z.; Al-Khattawi, A.; Iyire, A.; Al-Yami, H.; Dennison, T.J.; Mohammed, A.R. Quality by design (QbD) based process optimisation to develop functionalised particles with modified release properties using novel dry particle coating technique. PLoS ONE 2018, 13, e0206651. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Jhawat, V. Quality by design (QbD) approach of pharmacogenomics in drug designing and formulation development for optimization of drug delivery systems. J. Control. Release 2017, 245, 15–26. [Google Scholar] [CrossRef]
- Kim, B.S.; Na, Y.G.; Choi, J.H.; Kim, I.; Lee, E.; Kim, S.Y.; Lee, J.Y.; Cho, C.W. The improvement of skin whitening of phenylethyl resorcinol by nanostructured lipid carriers. Nanomaterials 2017, 7, 241. [Google Scholar] [CrossRef]
- Baek, J.S.; Na, Y.G.; Cho, C.W. Sustained cytotoxicity of wogonin on breast cancer cells by encapsulation in solid lipid nanoparticles. Nanomaterials 2018, 8, 159. [Google Scholar] [CrossRef]
- Yu, S.H.; Tan, G.X.; Liu, D.D.; Yang, X.G.; Pan, W.S. Nanostructured lipid carrier (NLC)-based novel hydrogels as potential carriers for nepafenac applied after cataract surgery for the treatment of inflammation: Design, characterization and in vitro cellular inhibition and uptake studies. RSC Adv. 2017, 7, 16668–16677. [Google Scholar] [CrossRef]
- Guo, X.; Zhao, Z.; Chen, D.; Qiao, M.; Wan, F.; Cun, D.; Sun, Y.; Yang, M. Co-delivery of resveratrol and docetaxel via polymeric micelles to improve the treatment of drug-resistant tumors. Asian J. Pharm. Sci. 2019, 14, 78–85. [Google Scholar] [CrossRef]
- Hvas, A.M.; Favaloro, E.J. Platelet function analyzed by light transmission aggregometry. Methods Mol. Biol. 2017, 1646, 321–331. [Google Scholar]
- Uner, M. Preparation, characterization and physico-chemical properties of solid lipid nanoparticles (SLN) and nanostructured lipid carriers (NLC): Their benefits as colloidal drug carrier systems. Pharmazie 2006, 61, 375–386. [Google Scholar] [PubMed]
- Poovi, G.; Damodharan, N. Lipid nanoparticles: A challenging approach for oral delivery of BCS Class-II drugs. Future J. Pharm. Sci. 2018, 4, 191–205. [Google Scholar] [CrossRef]
- Sharma, N.; Madan, P.; Lin, S.S. Effect of process and formulation variables on the preparation of parenteral paclitaxel-loaded biodegradable polymeric nanoparticles: A co-surfactant study. Asian J. Pharm. Sci. 2016, 11, 404–416. [Google Scholar] [CrossRef] [Green Version]
- Souto, E.B.; Mehnert, W.; Muller, R.H. Polymorphic behaviour of Compritol (R) 888 ATO as bulk lipid and as SLN and NLC. J. Microencapsul. 2006, 23, 417–433. [Google Scholar] [CrossRef] [PubMed]
- Schmidts, T.; Dobler, D.; Nissing, C.; Runkel, F. Influence of hydrophilic surfactants on the properties of multiple W/O/W emulsions. J. Colloid Interface Sci. 2009, 338, 184–192. [Google Scholar] [CrossRef]
- Patel, K.; Padhye, S.; Nagarsenker, M. Duloxetine HCl lipid nanoparticles: Preparation, characterization, and dosage form design. AAPS PharmSciTech 2012, 13, 125–133. [Google Scholar] [CrossRef]
- Liu, S.S.; Ho, P.C. Formulation optimization of scutellarin-loaded HP-beta-CD/chitosan nanoparticles using response surface methodology with Box–Behnken design. Asian J. Pharm. Sci. 2017, 12, 378–385. [Google Scholar] [CrossRef]
- Shahbazi, M.A.; Santos, H.A. Improving oral absorption via drug-loaded nanocarriers: Absorption mechanisms, intestinal models and rational fabrication. Curr. Drug Metab. 2013, 14, 28–56. [Google Scholar] [CrossRef]
- Danaei, M.; Dehghankhold, M.; Ataei, S.; Hasanzadeh Davarani, F.; Javanmard, R.; Dokhani, A.; Khorasani, S.; Mozafari, M. Impact of particle size and polydispersity index on the clinical applications of lipidic nanocarrier systems. Pharmaceutics 2018, 10, 57. [Google Scholar] [CrossRef] [PubMed]
- Baek, J.S.; Cho, C.W. Surface modification of solid lipid nanoparticles for oral delivery of curcumin: Improvement of bioavailability through enhanced cellular uptake, and lymphatic uptake. Eur. J. Pharm. Biopharm. 2017, 117, 132–140. [Google Scholar] [CrossRef] [PubMed]
- Son, G.H.; Lee, H.J.; Na, Y.G.; Lee, H.K.; Kim, S.J.; Huh, H.W.; Kim, K.T.; Kang, J.S.; Kim, Y.H.; Myung, C.S.; et al. Formulation and statistical analysis of an herbal medicine tablet containing Morus alba leaf extracts. J. Pharm. Investig. 2018, 1–10. [Google Scholar] [CrossRef]
- Festing, M.F.; Altman, D.G. Guidelines for the design and statistical analysis of experiments using laboratory animals. ILAR J. 2002, 43, 244–258. [Google Scholar] [CrossRef]
- Mourabet, M.; El Rhilassi, A.; El Boujaady, H.; Bennani-Ziatni, M.; Taitai, A. Use of response surface methodology for optimization of fluoride adsorption in an aqueous solution by Brushite. Arab. J. Chem. 2017, 10, S3292–S3302. [Google Scholar] [CrossRef] [Green Version]
- Schneider, A.; Hommel, G.; Blettner, M. Linear regression analysis: Part 14 of a series on evaluation of scientific publications. Dtsch. Arztebl. Int. 2010, 107, 776–782. [Google Scholar]
- Bewick, V.; Cheek, L.; Ball, J. Statistics review 7: Correlation and regression. Crit. Care 2003, 7, 451–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Figueiredo Filho, D.B.; Júnior, J.A.S.; Rocha, E.C. What is R2 all about? Leviathan (São Paulo) 2011, 3, 60–68. [Google Scholar] [CrossRef]
- Mahmood, S.; Taher, M.; Mandal, U.K. Experimental design and optimization of raloxifene hydrochloride loaded nanotransfersomes for transdermal application. Int. J. Nanomed. 2014, 9, 4331–4346. [Google Scholar] [Green Version]
- Yeom, D.W.; Song, Y.S.; Kim, S.R.; Lee, S.G.; Kang, M.H.; Lee, S.; Choi, Y.W. Development and optimization of a self-microemulsifying drug delivery system for atorvastatin calcium by using D-optimal mixture design. Int. J. Nanomed. 2015, 10, 3865–3878. [Google Scholar]
- Gan, L.; Zhang, C.; Wu, F.J.; Li, H.; Zhang, W.P.; Zhang, Q.J. Microencapsulated nanostructured lipid carriers as delivery system for rutin. Mater. Technol. 2018, 33, 357–363. [Google Scholar] [CrossRef]
- Bhaskar, K.; Anbu, J.; Ravichandiran, V.; Venkateswarlu, V.; Rao, Y.M. Lipid nanoparticles for transdermal delivery of flurbiprofen: Formulation, in vitro, ex vivo and in vivo studies. Lipids Health Dis. 2009, 8, 6. [Google Scholar] [CrossRef] [PubMed]
- Seta, Y.; Higuchi, F.; Kawahara, Y.; Nishimura, K.; Okada, R. Design and preparation of captopril sustained-release dosage forms and their biopharmaceutical properties. Int. J. Pharm. 1988, 41, 245–254. [Google Scholar] [CrossRef]
- Khan, S.; Baboota, S.; Ali, J.; Khan, S.; Narang, R.S.; Narang, J.K. Nanostructured lipid carriers: An emerging platform for improving oral bioavailability of lipophilic drugs. Int. J. Pharm. Investig. 2015, 5, 182–191. [Google Scholar] [Green Version]
- Sambuy, Y.; Angelis, I.; Ranaldi, G.; Scarino, M.L.; Stammati, A.; Zucco, F. The Caco-2 cell line as a model of the intestinal barrier: Influence of cell and culture-related factors on Caco-2 cell functional characteristics. Cell Biol. Toxicol. 2005, 21, 1–26. [Google Scholar] [CrossRef]
- Doktorovova, S.; Souto, E.B.; Silva, A.M. Nanotoxicology applied to solid lipid nanoparticles and nanostructured lipid carriers - a systematic review of in vitro data. Eur. J. Pharm. Biopharm. 2014, 87, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Tabata, Y.; Ikada, Y. Phagocytosis of Polymer Microspheres by Macrophages. Adv. Polym. Sci. 1990, 94, 107–141. [Google Scholar]
- Ye, H.L.; Shen, Z.Q.; Yu, L.; Wei, M.; Li, Y. Manipulating nanoparticle transport within blood flow through external forces: An exemplar of mechanics in nanomedicine. Proc. Math. Phys. Eng. Sci. 2018, 474, 20170845. [Google Scholar] [CrossRef]
- Hathout, R.M.; Mansour, S.; Mortada, N.D.; Geneidi, A.S.; Guy, R.H. Uptake of microemulsion components into the stratum corneum and their molecular effects on skin barrier function. Mol. Pharm. 2010, 7, 1266–1273. [Google Scholar] [CrossRef]
- Tian, C.; Asghar, S.; Wu, Y.; Chen, Z.; Jin, X.; Yin, L.; Huang, L.; Ping, Q.; Xiao, Y. Improving intestinal absorption and oral bioavailability of curcumin via taurocholic acid-modified nanostructured lipid carriers. Int. J. Nanomed. 2017, 12, 7897–7911. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Factors | Range | |
| Low Limit (mg) | High Limit (mg) | |
| X1: Total lipid amount | 100 | 300 |
| X2: Ratio of liquid lipid/total lipid | 0.2 | 0.6 |
| X3: Percentage of surfactant | 1 | 3 |
| Responses | Goal | |
| Y1: Particle size (nm) | Minimize | |
| Y2: Polydispersity index | Minimize | |
| Y3: Encapsulation efficiency (%) | Maximize | |
| Responses | Suggested Model | Model p-Value | Lack of Fit p-Value | R2 | Adjusted R2 | Adequate Precision |
|---|---|---|---|---|---|---|
| Y1: Particle size (nm) | Linear | <0.0001 | 0.7090 | 0.8570 | 0.8241 | 17.2218 |
| Y2: Polydispersity index | Quadratic | 0.0139 | 0.4403 | 0.8740 | 0.7119 | 7.8183 |
| Y3: Encapsulation efficiency (%) | Linear | <0.0001 | 0.7622 | 0.8430 | 0.8068 | 15.0182 |
| Run | Factors | Responses | ||||
|---|---|---|---|---|---|---|
| X1 | X2 | X3 | Y1 | Y2 | Y3 | |
| Total Lipid Amount (mg) | Ratio of Liquid Lipid/Total Lipid | Percentage of Surfactant (%) | Particle Size (nm) | Polydispersity Index | Encapsulation Efficiency (%) | |
| 1 | 200 | 0.6 | 1 | 151.2 ± 6.5 | 0.311 ± 0.031 | 84.25 ± 3.15 |
| 2 | 200 | 0.4 | 2 | 104.2 ± 3.5 | 0.303 ± 0.012 | 86.14 ± 1.42 |
| 3 | 200 | 0.4 | 2 | 114.2 ± 3.4 | 0.312 ± 0.031 | 85.32 ± 2.41 |
| 4 | 200 | 0.4 | 2 | 102.3 ± 8.1 | 0.317 ± 0.021 | 83.12 ± 3.36 |
| 5 | 100 | 0.4 | 3 | 84.2 ± 3.6 | 0.331 ± 0.024 | 78.26 ± 1.48 |
| 6 | 200 | 0.6 | 3 | 124.8 ± 5.7 | 0.277 ± 0.019 | 78.24 ± 2.85 |
| 7 | 300 | 0.2 | 2 | 104.8 ± 8.1 | 0.331 ± 0.027 | 95.12 ± 3.14 |
| 8 | 200 | 0.4 | 2 | 121.1 ± 7.1 | 0.342 ± 0.018 | 90.42 ± 1.45 |
| 9 | 100 | 0.6 | 2 | 132.1 ± 8.3 | 0.347 ± 0.014 | 74.26 ± 2.85 |
| 10 | 200 | 0.2 | 3 | 80.3 ± 3.4 | 0.36 ± 0.025 | 87.36 ± 1.64 |
| 11 | 300 | 0.4 | 1 | 115.2 ± 3.2 | 0.319 ± 0.037 | 91.34 ± 1.75 |
| 12 | 200 | 0.4 | 2 | 93.5 ± 2.7 | 0.308 ± 0.021 | 83.25 ± 2.34 |
| 13 | 100 | 0.4 | 1 | 91.5 ± 1.9 | 0.285 ± 0.033 | 82.64 ± 3.15 |
| 14 | 100 | 0.2 | 2 | 76.1 ± 3.1 | 0.361 ± 0.022 | 83.14 ± 1.48 |
| 15 | 300 | 0.4 | 3 | 124.3 ± 1.4 | 0.277 ± 0.027 | 81.26 ± 2.95 |
| 16 | 300 | 0.6 | 2 | 149.2 ± 2.5 | 0.36 ± 0.024 | 82.15 ± 3.48 |
| 17 | 200 | 0.2 | 1 | 88.1 ± 3.5 | 0.279 ± 0.018 | 95.14 ± 4.52 |
| Optimized Factors | Responses | 95 % CI * Low Predicted Value | Predicted Value | 95% CI * High Predicted Value | Actual Value | Error Percentage (%) |
|---|---|---|---|---|---|---|
| X1: 189.3 mg | Y1: Particle size (nm) | 74.4 | 85.8 | 97.3 | 87.6 ± 6.6 | 2.1 |
| X2: 0.2 | Y2: Polydispersity index | 0.244 | 0.276 | 0.308 | 0.259 ± 0.013 | 6.2 |
| X3: 1.0% | Y3: Encapsulation efficiency (%) | 90.1 | 93.1 | 96.2 | 92.1 ± 3.1 | 1.1 |
| Pharmacokinetic Parameters | Samples | |
|---|---|---|
| Raw TGL | TGL-NLC | |
| Tmax (h) | 2.65 ± 0.82 | 1.20 ± 0.12 |
| Cmax (ng/mL) | 461.75 ± 88.77 | 1050.44 ± 170.14 # |
| AUC0–∞ (ng·h/mL) | 2103.01 ± 283.36 | 5362.43 ± 808.51 # |
| T1/2 (h) | 3.46 ± 0.56 | 4.80 ± 1.21 |
| RBA (%) vs. raw TGL | 254.99 | |
| Parameter | Samples | |
|---|---|---|
| Raw TGL | TGL-NLC | |
| AUIC0–24 (%⋅h) | 615.0 ± 91.9 | 1064.2 ± 121.5 # |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Son, G.-H.; Na, Y.-G.; Huh, H.W.; Wang, M.; Kim, M.-K.; Han, M.-G.; Byeon, J.-J.; Lee, H.-K.; Cho, C.-W. Systemic Design and Evaluation of Ticagrelor-Loaded Nanostructured Lipid Carriers for Enhancing Bioavailability and Antiplatelet Activity. Pharmaceutics 2019, 11, 222. https://doi.org/10.3390/pharmaceutics11050222
Son G-H, Na Y-G, Huh HW, Wang M, Kim M-K, Han M-G, Byeon J-J, Lee H-K, Cho C-W. Systemic Design and Evaluation of Ticagrelor-Loaded Nanostructured Lipid Carriers for Enhancing Bioavailability and Antiplatelet Activity. Pharmaceutics. 2019; 11(5):222. https://doi.org/10.3390/pharmaceutics11050222
Chicago/Turabian StyleSon, Gi-Ho, Young-Guk Na, Hyun Wook Huh, Miao Wang, Min-Ki Kim, Min-Gu Han, Jin-Ju Byeon, Hong-Ki Lee, and Cheong-Weon Cho. 2019. "Systemic Design and Evaluation of Ticagrelor-Loaded Nanostructured Lipid Carriers for Enhancing Bioavailability and Antiplatelet Activity" Pharmaceutics 11, no. 5: 222. https://doi.org/10.3390/pharmaceutics11050222