Preparation and Physicochemical Stability of Liquid Oral Dosage Forms Free of Potentially Harmful Excipient Designed for Pediatric Patients

 , and
, and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Feasibility Study
- -
- First, the exact amount of API powder was weighed in order to obtain the targeted concentration.
- -
- API powder was added to 13 g of Syrspend® SF PH4 Dry and triturated in a mortar until homogeneity was achieved.
- -
- Subsequently, sterile water was gently added, while stirring continuously, until reaching a final volume of 200 mL.
- -
- Finally, the suspension was bottled in a 20 mL amber type I glass container.
2.3. Stability Study
2.4. Analytical Method Development and Validation
2.4.1. Calibration Curve
2.4.2. Linearity and Matrix Effect
2.4.3. Accuracy and Limit of Quantification
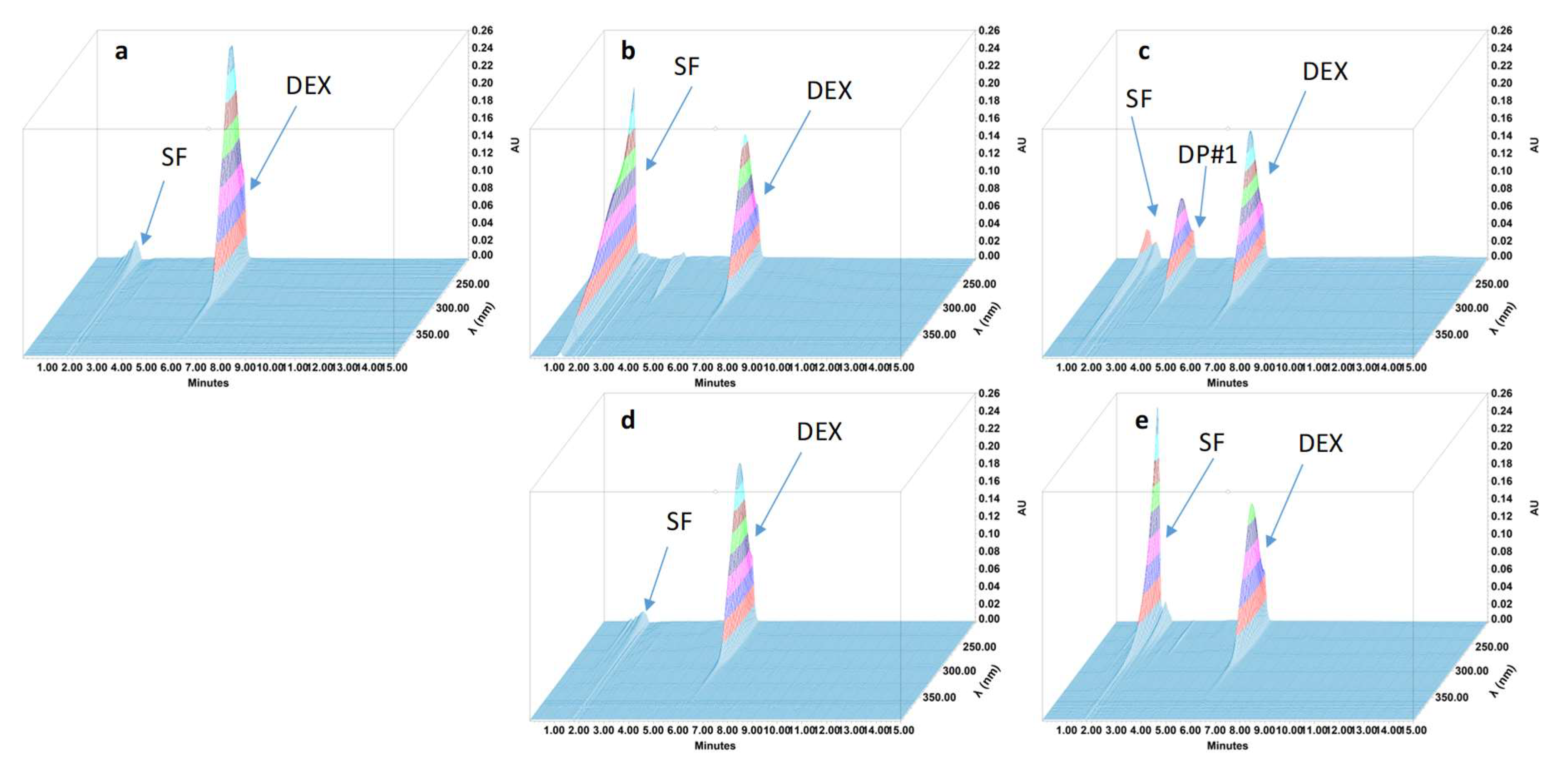
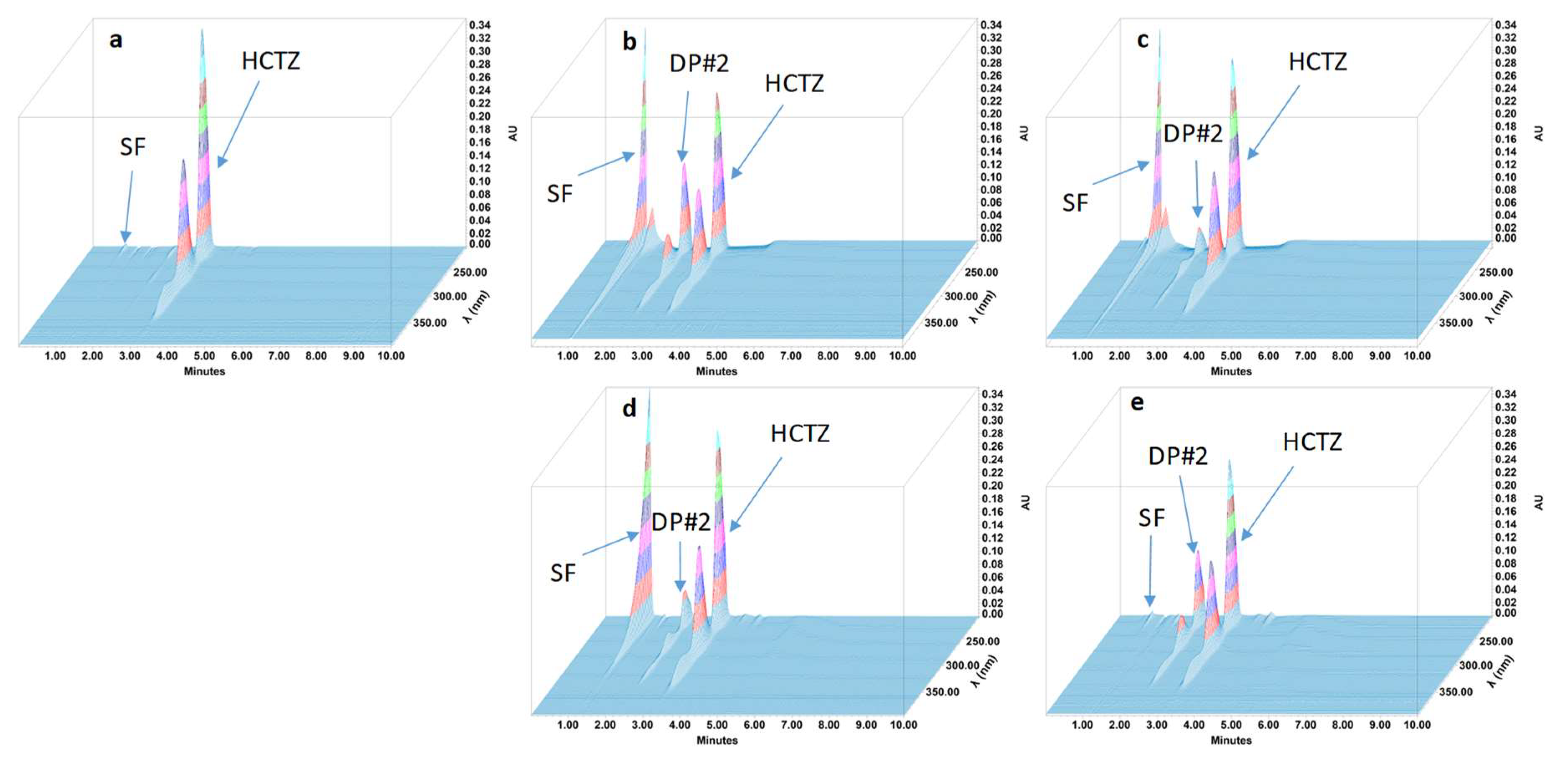
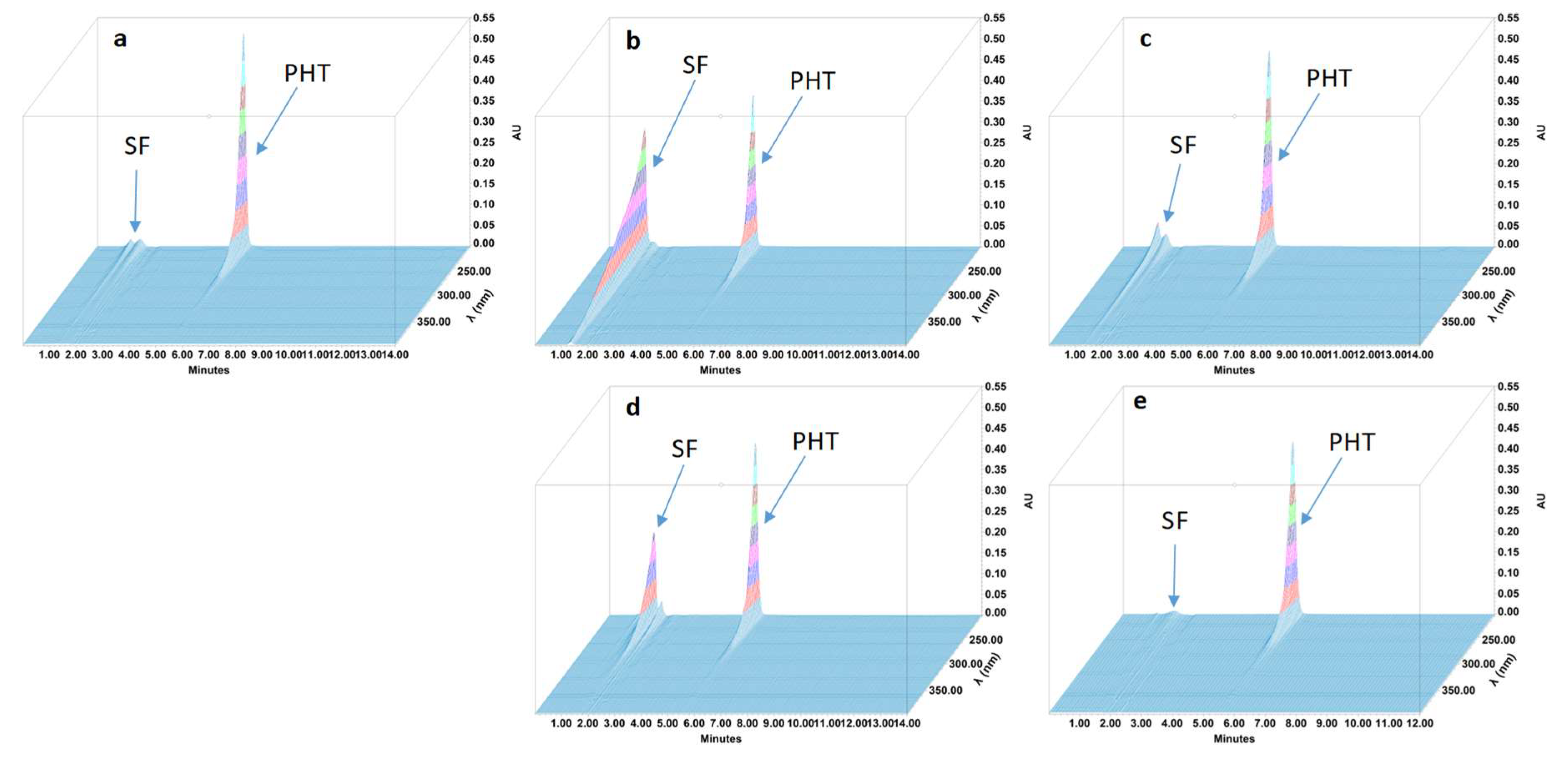
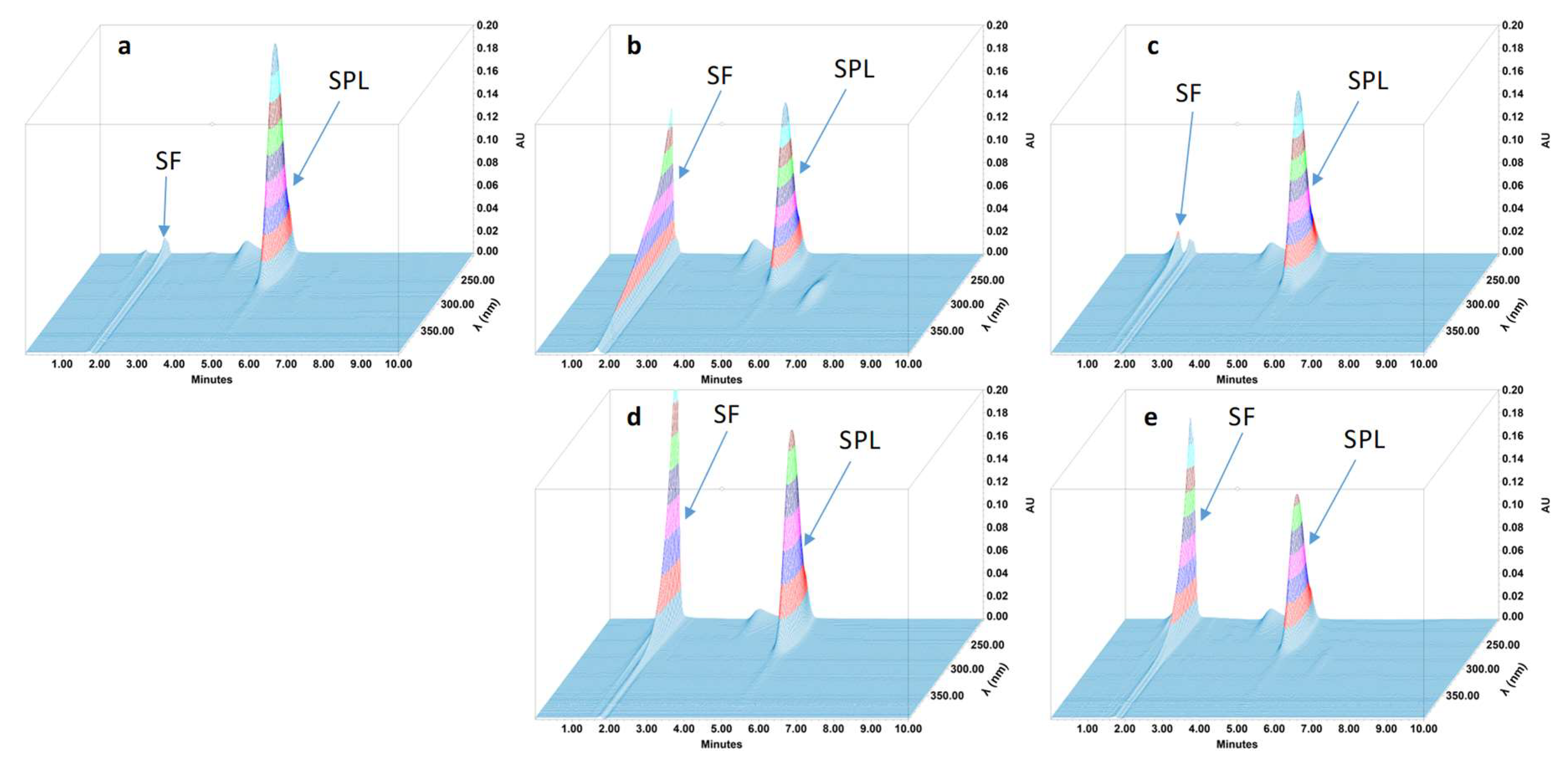
2.4.4. Specificity and Stability-Indicating Performance
3. Results
3.1. Method Validation
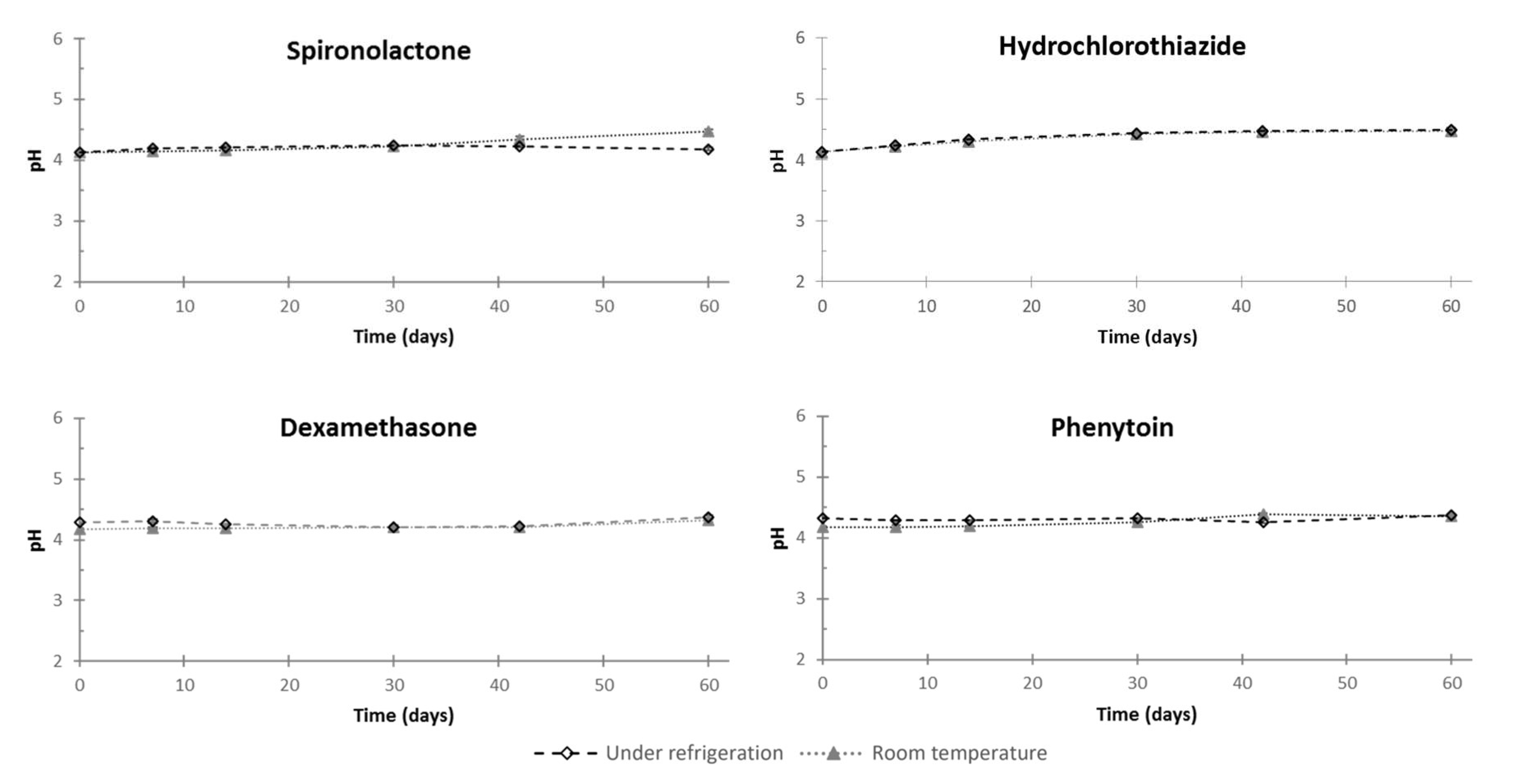
3.2. Feasibility and Stability Studies
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Nahata, M.C. Lack of Pediatric Drug Formulations. Pediatrics 1999, 104, 607–609. [Google Scholar]
- Mistry, P.; Batchelor, H. SPaeDD-UK project (Smart Paediatric Drug Development—UK) Evidence of acceptability of oral paediatric medicines: A review. J. Pharm. Pharmacol. 2017, 69, 361–376. [Google Scholar] [CrossRef]
- Conroy, S.; Choonara, I.; Impicciatore, P.; Mohn, A.; Arnell, H.; Rane, A.; Knoeppel, C.; Seyberth, H.; Pandolfini, C.; Raffaelli, M.P.; et al. Survey of unlicensed and off label drug use in paediatric wards in European countries. European Network for Drug Investigation in Children. BMJ 2000, 320, 79–82. [Google Scholar] [CrossRef]
- European Medicines Agency. Reflection Paper: Formulation of Choice for the Paediatric Population. 2006. Available online: https://www.ema.europa.eu/documents/scientific-guideline/reflection-paper-formulations-choice-paediatric-population_en.pdf (accessed on 14 March 2019).
- Richey, R.H.; Shah, U.U.; Peak, M.; Craig, J.V.; Ford, J.L.; Barker, C.E.; Nunn, A.J.; Turner, M.A. Manipulation of drugs to achieve the required dose is intrinsic to paediatric practice but is not supported by guidelines or evidence. BMC Pediatr. 2013, 13, 81. [Google Scholar] [CrossRef] [PubMed]
- Donovan, M.D.; Griffin, B.T.; Kharoshankaya, L.; Cryan, J.F.; Boylan, G.B. Pharmacotherapy for Neonatal Seizures: Current Knowledge and Future Perspectives. Drugs 2016, 76, 647–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitchell, C.D.; Richards, S.M.; Kinsey, S.E.; Lilleyman, J.; Vora, A.; Eden, T.O.B. Medical Research Council Childhood Leukaemia Working Party Benefit of dexamethasone compared with prednisolone for childhood acute lymphoblastic leukaemia: Results of the UK Medical Research Council ALL97 randomized trial. Br. J. Haematol. 2005, 129, 734–745. [Google Scholar] [CrossRef] [PubMed]
- Segar, J.L. Neonatal Diuretic Therapy: Furosemide, Thiazides, and Spironolactone. Clin. Perinatol. 2012, 39, 209–220. [Google Scholar] [CrossRef] [PubMed]
- Allen, L.V.; Erickson, M.A. Stability of ketoconazole, metolazone, metronidazole, procainamide hydrochloride, and spironolactone in extemporaneously compounded oral liquids. Am. J. Health-Syst. Pharm. AJHP Off. J. Am. Soc. Health-Syst. Pharm. 1996, 53, 2073–2078. [Google Scholar] [CrossRef] [PubMed]
- Binson, G.; Venisse, N.; Bacle, A.; Beuzit, K.; Dupuis, A. Preparation and Physico-Chemical Stability of Dexamethasone Oral Suspension. Pharm. Technol. Hosp. Pharm. 2017, 2, 193–201. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, A.O.; Polonini, H.C.; Silva, S.L.; Patrício, F.B.; Brandão, M.A.F.; Raposo, N.R.B. Feasibility of amlodipine besylate, chloroquine phosphate, dapsone, phenytoin, pyridoxine hydrochloride, sulfadiazine, sulfasalazine, tetracycline hydrochloride, trimethoprim and zonisamide in SyrSpend ® SF PH4 oral suspensions. J. Pharm. Biomed. Anal. 2016, 118, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Glass, B.D.; Haywood, A. Stability considerations in liquid dosage forms extemporaneously prepared from commercially available products. J. Pharm. Pharm. Sci. 2006, 9, 398–426. [Google Scholar]
- Mendes, C.; Costa, A.P.; Oliveira, P.R.; Tagliari, M.P.; Silva, M.A.S. Physicochemical and microbiological stability studies of extemporaneous antihypertensive pediatric suspensions for hospital use. Pharm. Dev. Technol. 2013, 18, 813–820. [Google Scholar] [CrossRef]
- Polonini, H.C.; Loures, S.; Lima, L.C.; Ferreira, A.O.; Brandão, M.A.F. Stability of Atenolol, Clonazepam, Dexamethasone, Diclofenac Sodium, Diltiazem, Enalapril Maleate, Ketoprofen, Lamotrigine, Penicillamine-D, and Thiamine in SyrSpend SF PH4 Oral Suspensions. Int. J. Pharm. Compd. 2016, 20, 167–174. [Google Scholar]
- Polonini, H.C.; Silva, S.L.; de Almeida, T.R.; Brandão, M.A.F.; Ferreira, A.O. Compatibility of caffeine, carvedilol, clomipramine hydrochloride, folic acid, hydrochlorothiazide, loperamide hydrochloride, methotrexate, nadolol, naltrexone hydrochloride and pentoxifylline in SyrSpend SF PH4 oral suspensions. Eur. J. Hosp. Pharm. 2016, 23, 352–358. [Google Scholar] [CrossRef]
- Fabiano, V.; Mameli, C.; Zuccotti, G.V. Paediatric pharmacology: Remember the excipients. Pharmacol. Res. 2011, 63, 362–365. [Google Scholar] [CrossRef]
- Golightly, L.K.; Smolinske, S.S.; Bennett, M.L.; Sutherland, E.W.; Rumack, B.H. Pharmaceutical excipients. Adverse effects associated with inactive ingredients in drug products (Part I). Med. Toxicol. Advers. Drug Exp. 1988, 3, 128–165. [Google Scholar]
- European Medicines Agency. Questions and Answers on Benzoic Acid and Benzoates Used as Excipients in Medicinal Products for Human Use. 2017. Available online: https://www.ema.europa.eu/documents/scientific-guideline/questions-answers-benzoic-acid-benzoates-used-excipients-medicinal-products-human-use_en.pdf (accessed on 14 March 2019).
- Nowak, K.; Ratajczak–Wrona, W.; Górska, M.; Jabłońska, E. Parabens and their effects on the endocrine system. Mol. Cell. Endocrinol. 2018, 474, 238–251. [Google Scholar] [CrossRef]
- Barouki, R.; Gluckman, P.D.; Grandjean, P.; Hanson, M.; Heindel, J.J. Developmental origins of non-communicable disease: Implications for research and public health. Environ. Health 2012, 11, 42. [Google Scholar] [CrossRef]
- Bergman, Å.; Heindel, J.; Jobling, S.; Kidd, K.; Zoeller, R.T. State of the science of endocrine disrupting chemicals. Toxicol. Lett. 2012, 211S, S3. [Google Scholar] [CrossRef]
- European Medicines Agency. Reflection Paper on the Use of Methyl-and Propylparaben as Excipients in Human Medicinal Products for Oral Use. 2013. Available online: https://www.ema.europa.eu/documents/scientific-guideline/reflection-paper-use-methyl-propylparaben-excipients-human-medicinal-products-oral-use_en.pdf (accessed on 14 March 2019).
- Korotkova, E.I.; Avramchik, O.A.; Angelov, T.M.; Karbainov, Y.A. Investigation of antioxidant activity and lipophilicity parameters of some preservatives. Electrochim. Acta 2005, 51, 324–332. [Google Scholar] [CrossRef]
- Yalkowsky, S.H.; He, Y.; Jain, P. Handbook of Aqueous Solubility Data. 2010. Available online: https://www.crcpress.com/Handbook-of-Aqueous-Solubility-Data/Yalkowsky-He-Jain/p/book/9781439802458 (accessed on 14 March 2019).
- Roulet, L.; Maillard, N.; Dupuis, A. Bonnes pratiques de préparations: Le projet de l’Afssaps. Actual. Pharm. Hosp. 2007, 3, 39–43. [Google Scholar] [CrossRef]
- Allen, L.V., Jr.; Bassani, G.S.; Elder, E.J.; Parr, A.F. Strength and Stability Testing for Compounded Preparations. US Pharmacop. 2014, pp. 1–7. Available online: https://www.usp.org/sites/default/files/usp/document/FAQs/strength-stability-testing-compounded-preparations.pdf (accessed on 14 March 2019).
- Abraham, J. International Conference on Harmonisation of Technical Requirements For Registration of Pharmaceuticals for Human Use. In Handbook of Transnational Economic Governance Regimes; Brouder, A., Tietje, C., Eds.; Brill: Leiden, The Netherlands, 2009; pp. 1041–1054. ISBN 978-90-04-16330-0. [Google Scholar]
- Larsen, E.C.; Devidas, M.; Chen, S.; Salzer, W.L.; Raetz, E.A.; Loh, M.L.; Mattano, L.A.; Cole, C.; Eicher, A.; Haugan, M.; et al. Dexamethasone and High-Dose Methotrexate Improve Outcome for Children and Young Adults with High-Risk B-Acute Lymphoblastic Leukemia: A Report from Children’s Oncology Group Study AALL0232. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2016, 34, 2380–2388. [Google Scholar] [CrossRef]
- Mutz, A.E.; Obladen, M.W. Hyperosmolar oral medication and necrotizing enterocolitis. Pediatrics 1985, 75, 371–372. [Google Scholar]
- SyrSpend® SF PH4 Dry | Fagron. Available online: https://fagron.com/en/product/syrspendr-sf-ph4-dry (accessed on 14 March 2019).
- European Medicines Agency. Guideline on the Investigation of Medicinal Products in the Term and Preterm Neonate. 2009. Available online: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003754.pdf (accessed on 14 March 2019).
- Warner, A. Drug use in the neonate: Interrelationships of pharmacokinetics, toxicity, and biochemical maturity. Clin. Chem. 1986, 32, 721–727. [Google Scholar]
- Nellis, G.; Metsvaht, T.; Varendi, H.; Lass, J.; Duncan, J.; Nunn, A.J.; Turner, M.A.; Lutsar, I. Product Substitution as a Way Forward in Avoiding Potentially Harmful Excipients in Neonates. Pediatr. Drugs 2016, 18, 221–230. [Google Scholar] [CrossRef]
- Nellis, G.; Metsvaht, T.; Varendi, H.; Toompere, K.; Lass, J.; Mesek, I.; Nunn, A.J.; Turner, M.A.; Lutsar, I. Potentially harmful excipients in neonatal medicines: A pan-European observational study. Arch. Dis. Child. 2015, 100, 694–699. [Google Scholar] [CrossRef]
- Soni, M.G.; Carabin, I.G.; Burdock, G.A. Safety assessment of esters of p-hydroxybenzoic acid (parabens). Food Chem. Toxicol. 2005, 43, 985–1015. [Google Scholar] [CrossRef]
- Karpuzoglu, E.; Holladay, S.D.; Gogal, R.M. Parabens: Potential impact of low-affinity estrogen receptor binding chemicals on human health. J. Toxicol. Environ. Health B Crit. Rev. 2013, 16, 321–335. [Google Scholar] [CrossRef]
- Skakkebaek, N.E.; Toppari, J.; Söder, O.; Gordon, C.M.; Divall, S.; Draznin, M. The Exposure of Fetuses and Children to Endocrine Disrupting Chemicals: A European Society for Paediatric Endocrinology (ESPE) and Pediatric Endocrine Society (PES) Call to Action Statement. J. Clin. Endocrinol. Metab. 2011, 96, 3056–3058. [Google Scholar] [CrossRef] [Green Version]
- WHO | Endocrine Disrupters and Child Health. Available online: http://www.who.int/ceh/publications/endocrine_disrupters_child/en/ (accessed on 14 March 2019).







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dexamethasone | Hydrochlorothiazide | Phenytoin | Spironolactone | |
|---|---|---|---|---|
| Mobile Phase | Acetonitrile and water (50:50, v:v) | Methanol and water (20:80, v:v) pH adjusted to 4.5 using acetic acid | Acetonitrile and water (60:40, v:v) | Methanol and water (70:30, v:v) |
| Flow rate (mL/min) | 1.0 | 1.5 | 1.0 | 1.0 |
| Column | Purospher® STAR RP-18 endcapped (5 µm) 150 × 4.6 mm | |||
| λ (nm) | 238 | 224 | 238 | 238 |
| Dexamethasone | Hydrochlorothiazide | Phenytoin | Spironolactone | |
|---|---|---|---|---|
| Calibration range (mg/mL) | 1.25–10 | 12–28 | 1.25–10 | 1.25–10 |
| r2 | 0.9996 | 0.9996 | 0.9998 | 0.9992 |
| Maximum residual value | 5.0% | 2.4% | 3.5% | 5.0% |
| S/N | 438 | 138 | 349 | 164 |
| Colonne1 | Spironolactone | Dexamethasone | Hydrochlorothiazide | Phenytoin |
|---|---|---|---|---|
| Target concentration (mg/mL) | 5.0 | 5.0 | 2.0 | 5.0 |
| Mean concentration (mg/mL) | Repeatability | |||
| 4.9 | 5.0 | 2.0 | 5.0 | |
| RSD (%) | 4.4 | 1.3 | 3.6 | 4.0 |
| CI 95% (%) | [4.2 ; 4.7] | [1.3 ; 1.4] | [3.5 ; 3.6] | [1.9 ; 6.0] |
| Mean concentration (mg/mL) | Intermediate precision | |||
| 5.0 | 4.9 | 2.0 | 5.0 | |
| RSD (%) | 3.0 | 3.2 | 4.0 | 3.1 |
| CI 95% (%) | [2.9 ; 3.1] | [3.1 ; 3.3] | [3.9 ; 4.0] | [2.3 ; 3.8] |
| Recovery rate (%) | Trueness | |||
| 100.5 | 98.8 | 99.8 | 99.7 | |
| 95% CI (%) | [99.0 ; 102.0] | [97.3 ; 100.4] | [97.8 ; 101.7] | [98.2 ; 101.2] |
| Retention Time (min) | RRT | Tailing Factor | Resolution | Theoritical Plates | |
|---|---|---|---|---|---|
| Dexamethasone | 6.2 | NA | 1.03 | NA | 6797 |
| Degradation product #1 | 3.2 | 0.52 | 1.12 | 12.74 | 6018 |
| Hydrochlorothiazide | 3.12 | NA | 1.16 | NA | 5399 |
| Degradation product #2 | 2.25 | 0.72 | 1.18 | 6.06 | 5713 |
| Phénytoïne | 5.44 | NA | 1.17 | NA | 6240 |
| All degradation products were in solvent front | |||||
| Spironolactone | 5.23 | NA | 1.16 | NA | 2547 |
| All degradation products were in solvent front | |||||
| Color | Precipitate | Osmolality (mOsm/kg) | |||
|---|---|---|---|---|---|
| Day 0 | Day 60 | Day 60 | Day 0 | Day 60 | |
| Dexamethasone | white opalescent | white opalescent | no | <LOQ | <LOQ |
| Hydrochlorothiazide | off-white opalescent | off-white opalescent | no | <LOQ | <LOQ |
| Phenytoin | white opalescent | white opalescent | no | <LOQ | <LOQ |
| Spironolactone | white opalescent | white opalescent | no | <LOQ | <LOQ |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Binson, G.; Beuzit, K.; Migeot, V.; Marco, L.; Troussier, B.; Venisse, N.; Dupuis, A. Preparation and Physicochemical Stability of Liquid Oral Dosage Forms Free of Potentially Harmful Excipient Designed for Pediatric Patients. Pharmaceutics 2019, 11, 190. https://doi.org/10.3390/pharmaceutics11040190
Binson G, Beuzit K, Migeot V, Marco L, Troussier B, Venisse N, Dupuis A. Preparation and Physicochemical Stability of Liquid Oral Dosage Forms Free of Potentially Harmful Excipient Designed for Pediatric Patients. Pharmaceutics. 2019; 11(4):190. https://doi.org/10.3390/pharmaceutics11040190
Chicago/Turabian StyleBinson, Guillaume, Karine Beuzit, Virginie Migeot, Léa Marco, Barbara Troussier, Nicolas Venisse, and Antoine Dupuis. 2019. "Preparation and Physicochemical Stability of Liquid Oral Dosage Forms Free of Potentially Harmful Excipient Designed for Pediatric Patients" Pharmaceutics 11, no. 4: 190. https://doi.org/10.3390/pharmaceutics11040190