Mathematical Modeling of Release Kinetics from Supramolecular Drug Delivery Systems


 , ,
, ,
Abstract
:1. Introduction
- -
- diffusion phenomena, when the support is represented by molecules;
- -
- convective transport by currents in fluids, described by fluid mechanics;
- -
- radiative transport of elementary particles.
2. Mathematical Methods for Solving Transfer Equations in Initial and Boundary Conditions Imposed by Particular Systems
2.1. Diffusion Equation
2.2. Initial and Boundary Conditions
- Different combinations of phenomenological conditions can lead to the same initial and boundary conditions and, consequently, to the same mathematical solutions. It frequently happens that experimentally determined release kinetics to fit a theoretical law are deduced in completely different phenomenological conditions.
- Derivation of solutions essentially implies the initial and boundary conditions, such that the in-depth analysis of phenomena and prediction possibilities are best achieved in connection with understanding of the mathematical aspects.
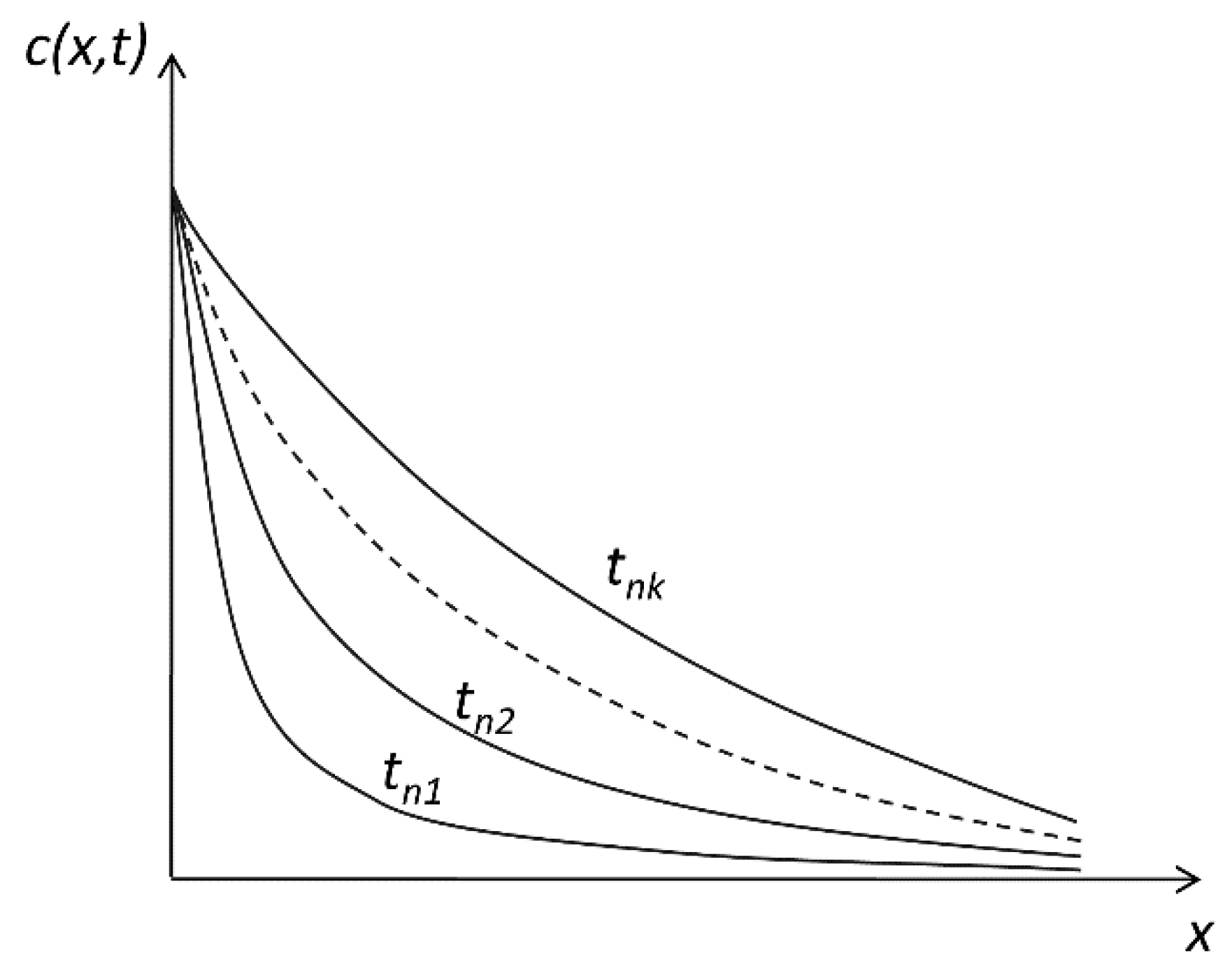
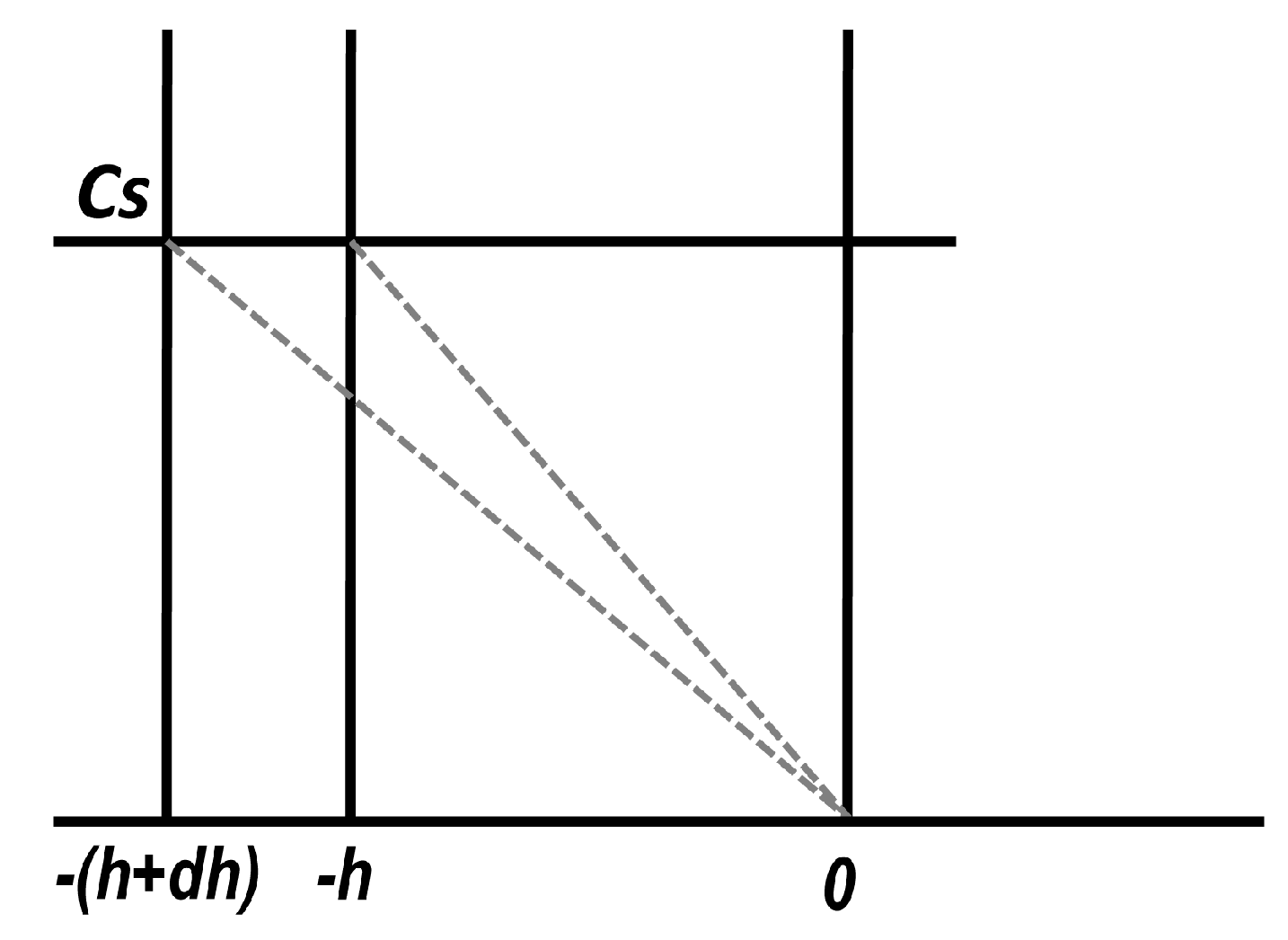
2.3. Release in an Infinite Medium from an Interface where Concentration Is Kept Constant: Laplace Transform Method
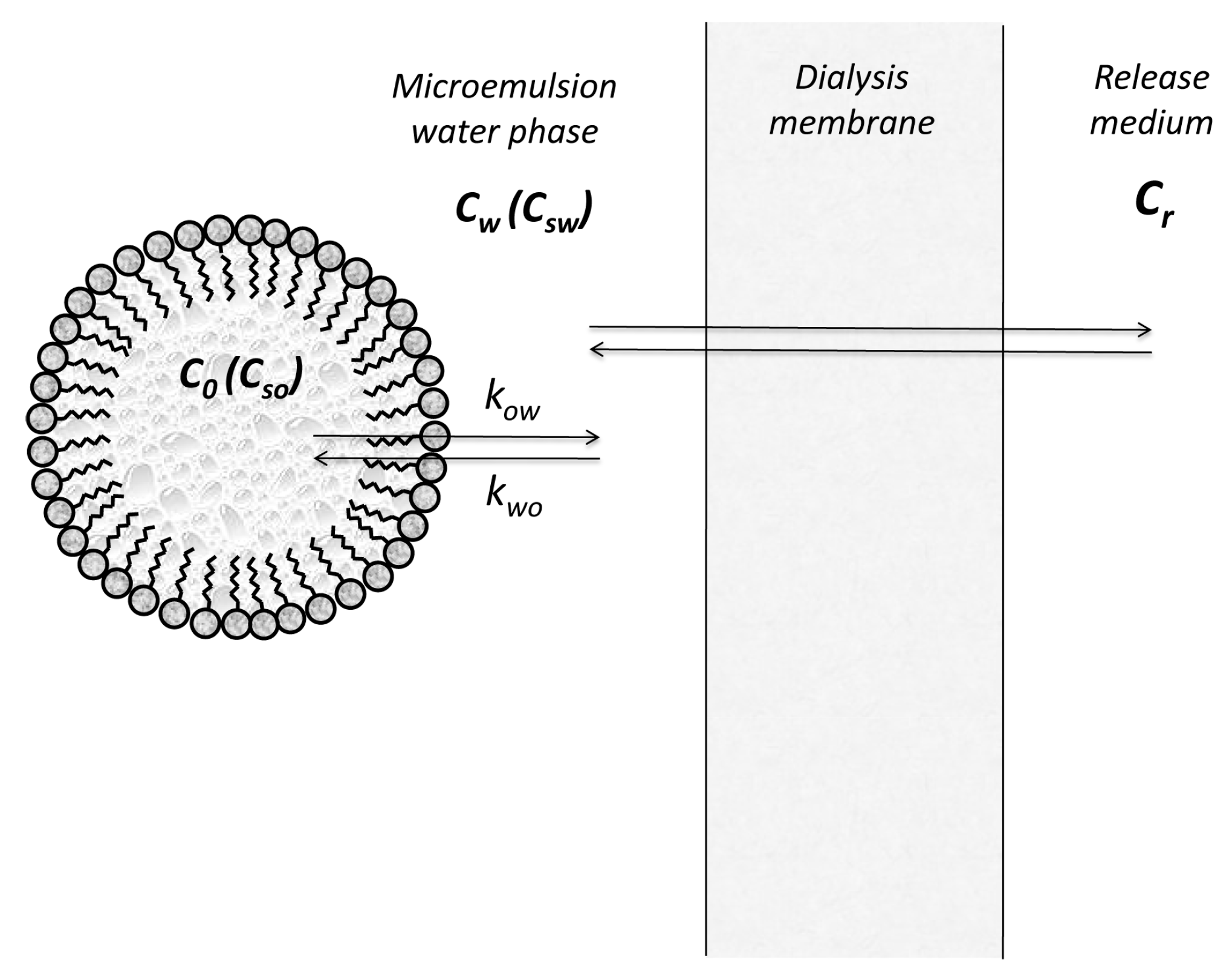
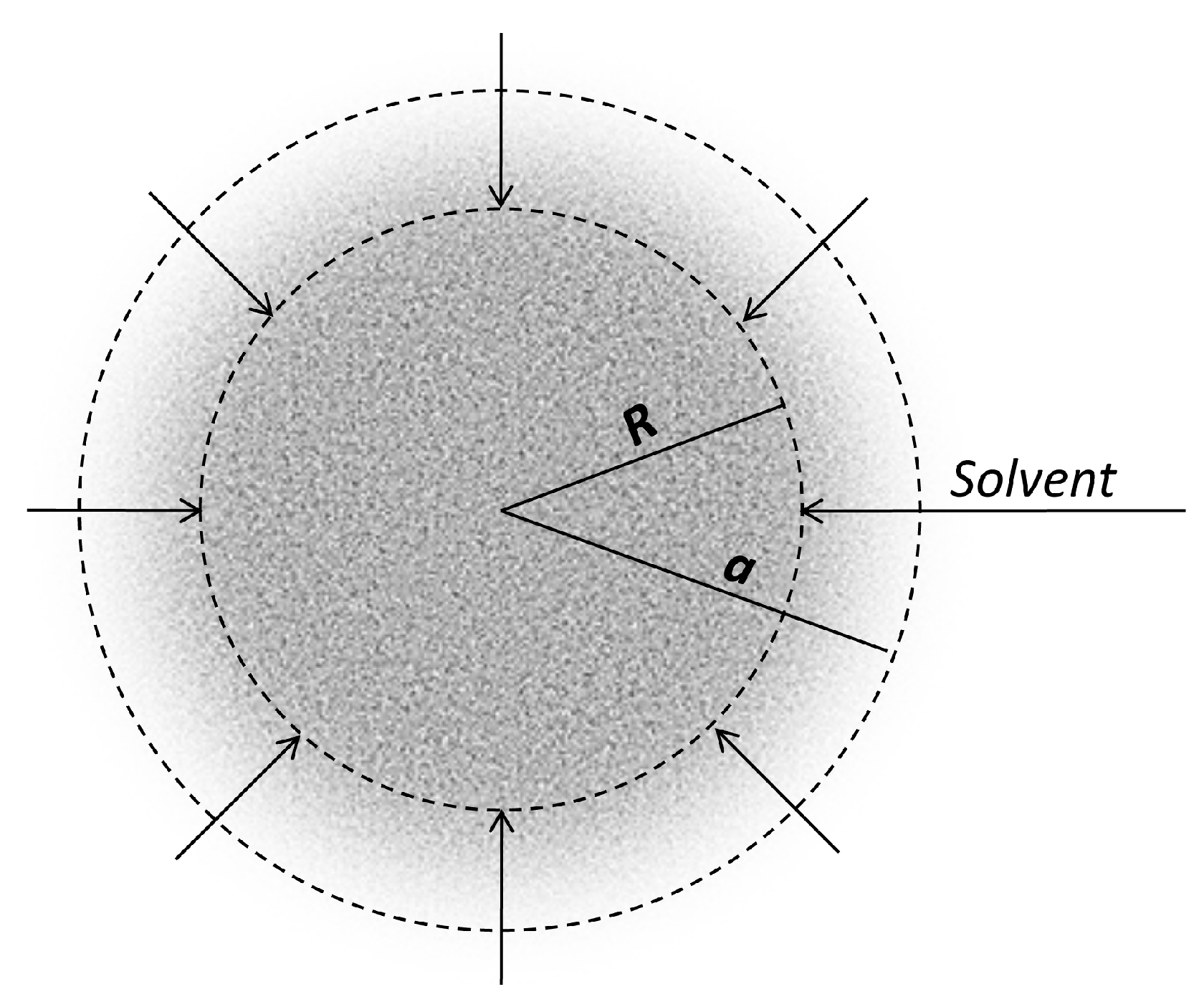
2.4. Transfer at Liquid/Liquid Interfaces: Release from Microemulsions
2.4.1. Stationary State Models
2.4.2. Compartmental Models
2.5. Diffusion in Membranes: Method of Separation of Variables
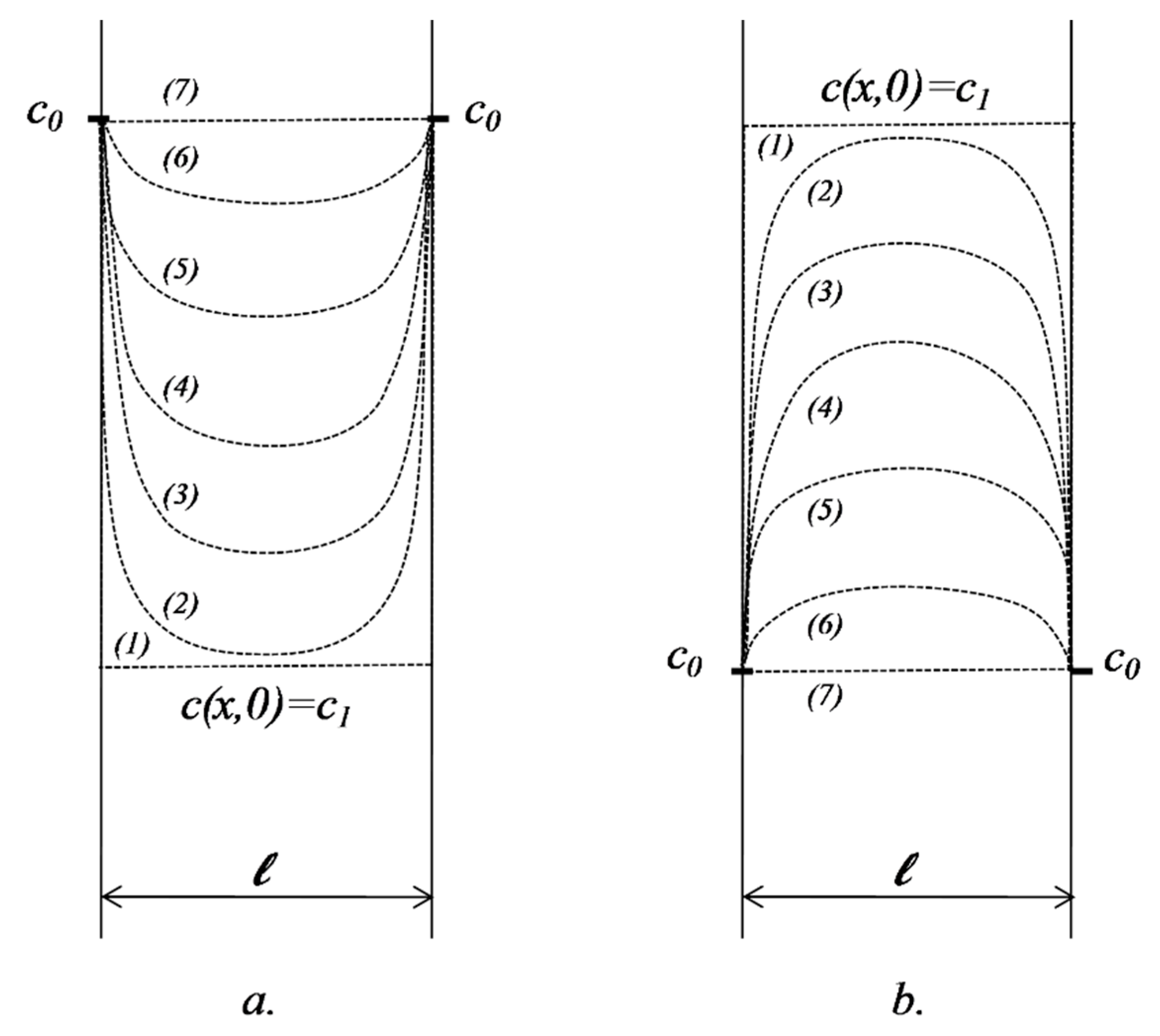
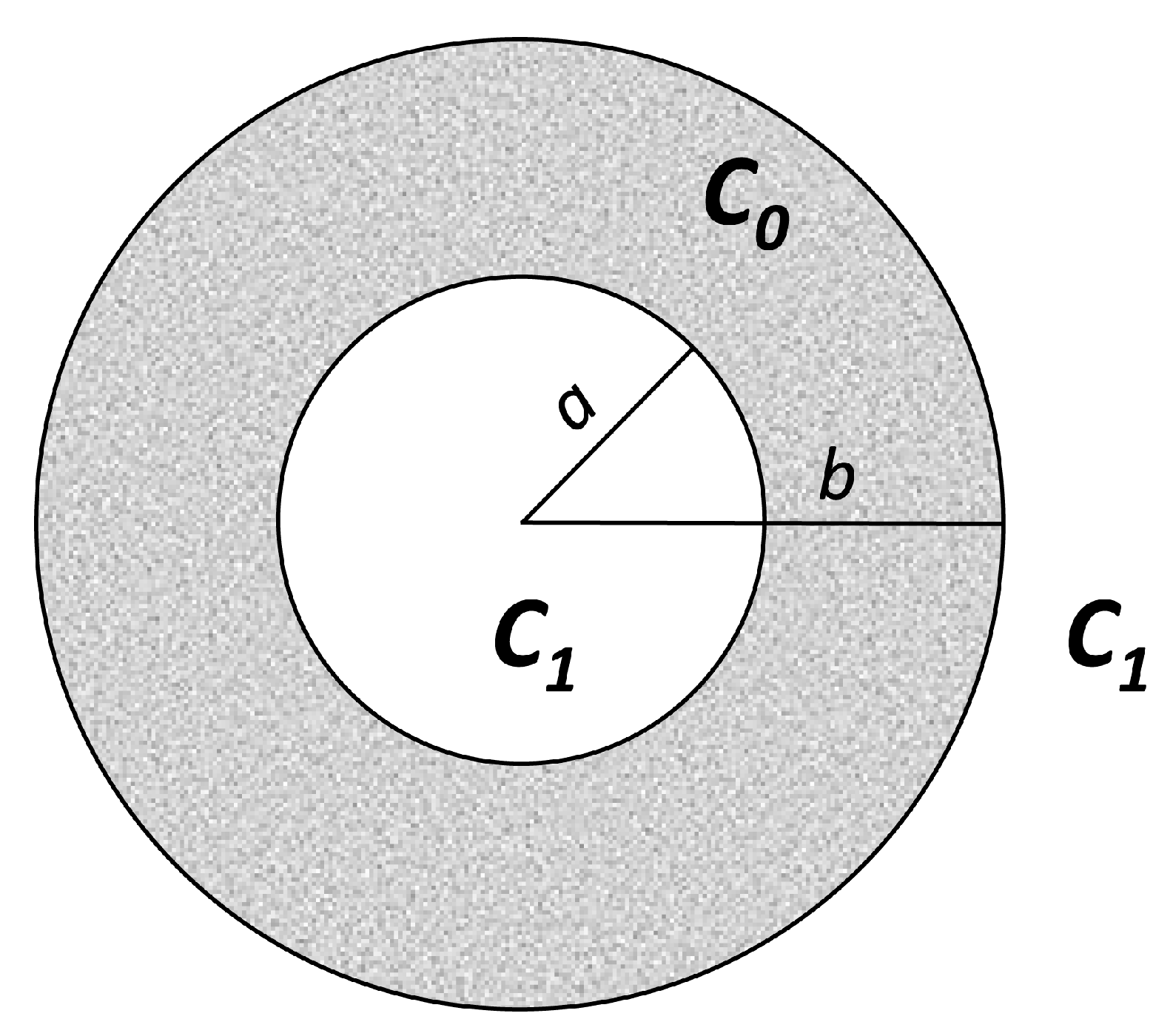
2.5.1. Diffusion in a Domain Bordered by Two Interfaces where Concentration Is Kept Constant
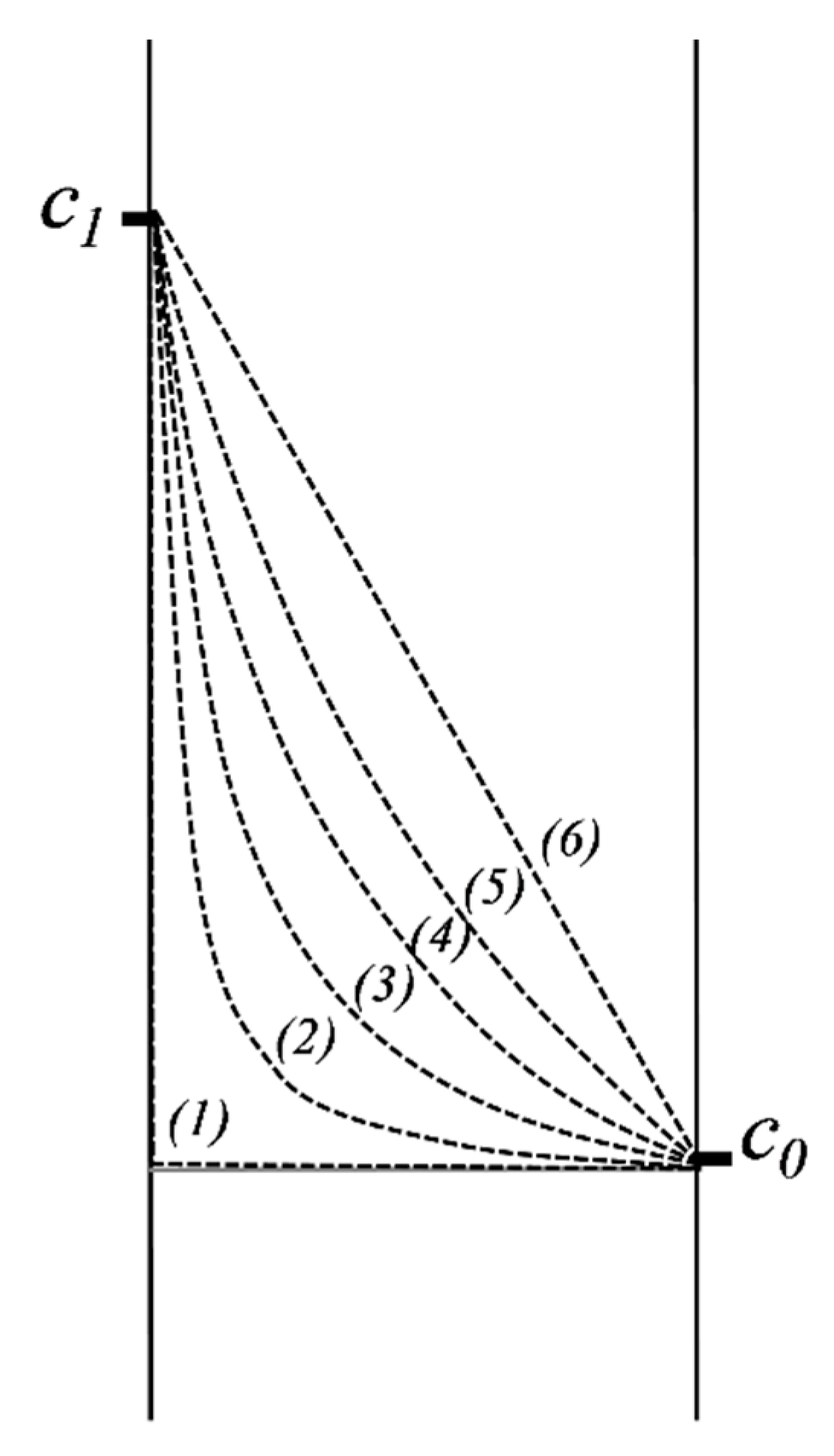
2.5.2. Diffusion in a Domain Bordered by Two Interfaces of Constant but Different Concentrations
2.6. Diffusion Equation in Spherical and Cylindrical Coordinates
2.6.1. Solutions of Diffusion Equation in Spherical Coordinates
2.6.2. Release from a Non-Degrading Polymer
2.6.3. Release from Lipid Dosage Forms
- The beads do not significantly swell or erode during drug release.
- The beads are spherical in shape.
- The drug is initially homogeneously distributed within the spheres.
- Perfect sink conditions are provided throughout the experiments.
- Mass transfer resistance due to liquid unstirred boundary layers at the surface of the spheres is negligible compared to mass transfer resistance due to diffusion within the systems.
- Drug dissolution is rapid and complete upon exposure to the release medium.
- Diffusion with time- and position-independent diffusion coefficients is the release rate-limiting mass transfer step.
2.6.4. Release from Lipid Implants with Cylindrical Geometry
- The implants do not significantly swell or erode during drug release.
- The implants are cylindrical in shape.
- Diffusional mass transport occurs in radial and axial direction, with the same diffusivities.
- The drug is initially homogeneously distributed within the implants.
- Perfect sink conditions are provided throughout the experiments.
- Mass transfer resistance due to liquid unstirred boundary layers at the surface of the implants is negligible compared to mass transfer resistance due to diffusion within the systems.
- Drug dissolution is rapid and complete upon exposure to the release medium.
- Diffusion with time- and position-independent diffusion coefficients is the release rate-limiting mass transfer step.
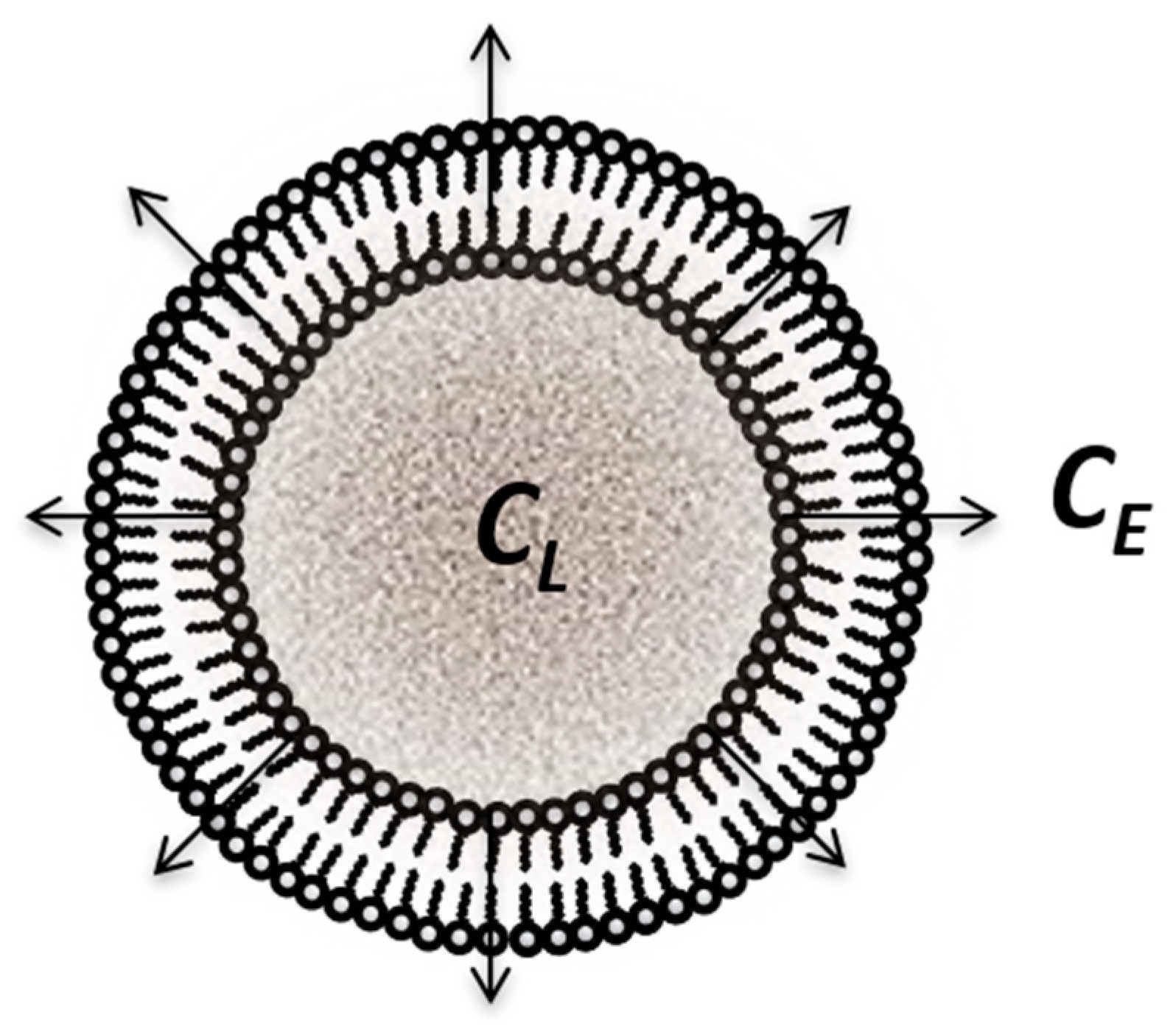
2.7. Release Controlled by Transfer across Membranes, Considered as Coupled Interfaces: Release from Liposomes
3. Mechanistic and Empirical Models in Systems with Moving Boundaries
3.1. Matrix Systems
3.1.1. Stefan’s Problem
3.1.2. Steady-State Higuchi’s Moving Boundary Model
3.1.3. Release from a Spherical Matrix
3.1.4. Boundary Layer Effect
3.2. Swellable Polymers
3.2.1. Intrusion of Water into Matrix
3.2.2. Swelling Component of Release from Polymers
3.3. Erodible Polymers
3.3.1. Kinetics of Release from a Sheet of Thickness 2l
3.3.2. Kinetics of Release from a Sphere of Initial Radius
3.3.3. Kinetics of Release from a Cylinder of Radius and Height 2
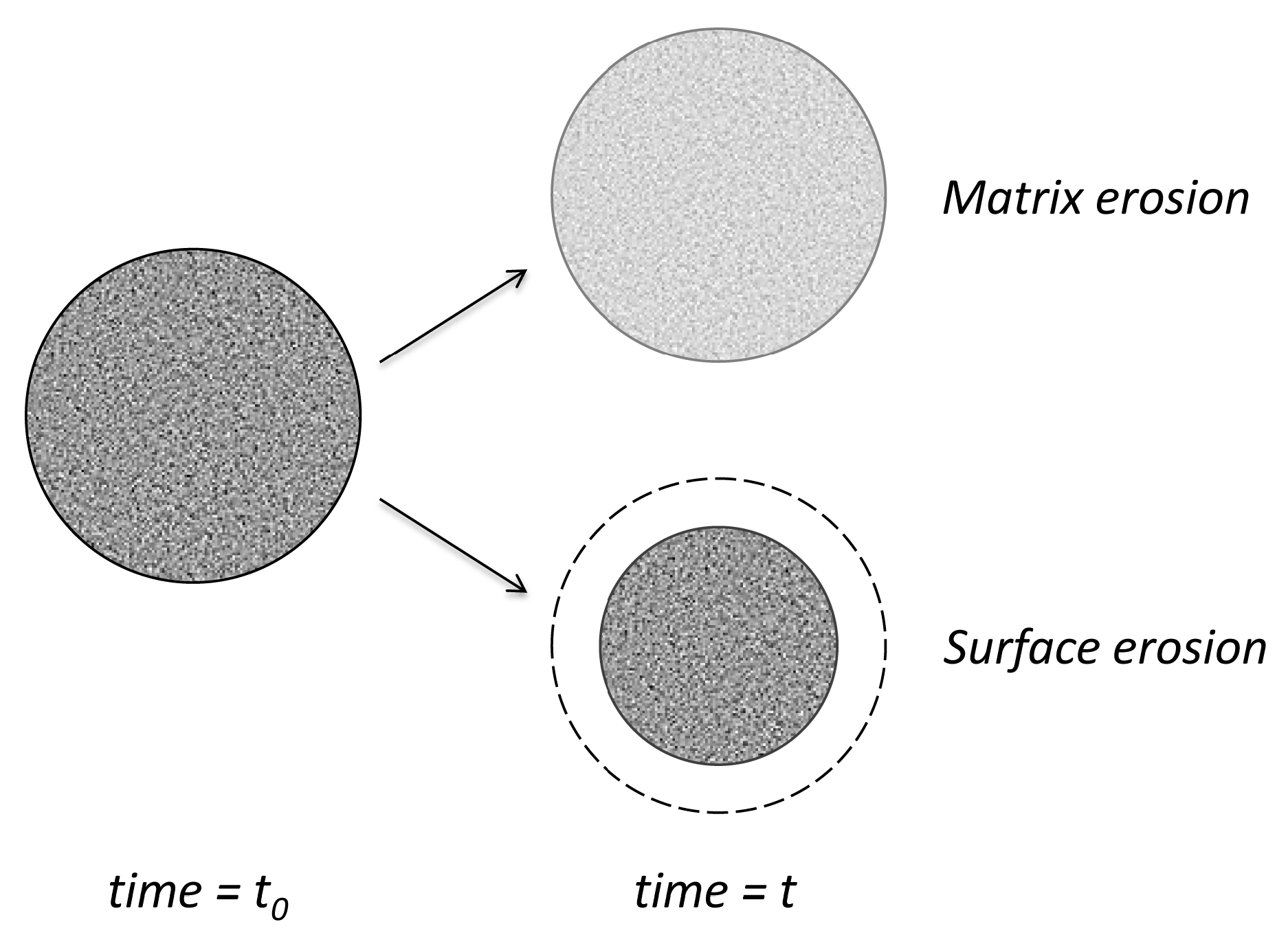
3.3.4. Empirical Surface Erosion Models
3.3.5. Mechanistic Surface Erosion Models
3.4. Complex, Multiparameter Release Models
3.4.1. Concomitant Depolymerization, Erosion, and Diffusion
3.4.2. Monte Carlo Simulation Models
3.4.3. Artificial Neural Network Models
4. Release Models Based on Fick’s First Law
4.1. Noyes–Whitney Model
4.2. “Empirical” Extensions
4.3. Applications of “Empirical” Models in Describing Release from Micro- and Nanostructured Carriers
4.3.1. Micro-Sized Polymeric Carriers
4.3.2. Nano-Sized Polymeric Carriers
- -
- the release models developed for transfer across plane surfaces are no longer applicable;
- -
- their curvature implies specific properties, primarily high free energy and aggregation tendency;
- -
- continuum models lack the ability to describe the kinetics of drug release as the concentration of the drug in the nanosystems fluctuates and the notion of concentration profile becomes meaningless.
4.3.3. Liquid Crystals
4.3.4. Liposomes
4.3.5. Solid Lipid Nanoparticles and Lipid Dosage Forms
4.4. Selection of the Mathematical Release Model
5. Conclusions
Funding
Conflicts of Interest
Appendix A
Diffusion in a Domain Bordered by Two Interfaces where Concentration is Kept Constant
References
- Crank, J. The Mathematics of Diffusion, 2nd ed.; Oxford Clarendon Press: Oxford, UK, 1975. [Google Scholar]
- Carslaw, H.S.; Jaeger, J.C. Conduction of Heat in Solids, 2nd ed.; Oxford University Press: Oxford, UK, 1959. [Google Scholar]
- Voicu, V.A.; Mircioiu, C. Mecanisme Farmacologice la Interfete Membranare [Farmacological Mechanisms at Membranar Interfaces]; Ed Academiei: Bucharest, Romania, 1994. [Google Scholar]
- Bolisetti, S.S.; Reddy, M.S. Formulation and in-vitro evaluation of gastro retentive in-situ floating gels of repaglinide cubosomes. J. Pharm. Res. 2013, 6, 787. [Google Scholar]
- Al-Kady, A.S.; Gaber, M.; Hussein, M.M.; Ebeid, E.Z. Nanostructure-loaded mesoporous silica for controlled release of coumarin derivatives: A novel testing of the hyperthermia effect. Eur. J. Pharm. Biopharm. 2011, 77, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Anuta, V.; Nitulescu, G.M.; Dinu-Pîrvu, C.E.; Olaru, O.T. Biopharmaceutical profiling of new antitumor pyrazole derivatives. Molecules 2014, 19, 16381–16401. [Google Scholar] [CrossRef] [PubMed]
- Avrămescu, R.-E.; Ghica, M.; Dinu-Pîrvu, C.; Prisada, R.; Popa, L. Superhydrophobic Natural and Artificial Surfaces—A Structural Approach. Materials 2018, 11, 866. [Google Scholar] [CrossRef] [PubMed]
- Larsen, A.T.; Ohlsson, A.G.; Polentarutti, B.; Barker, R.A.; Phillips, A.R.; Abu-Rmaileh, R.; Dickinson, P.A.; Abrahamsson, B.; Ostergaard, J.; Müllertz, A. Oral bioavailability of cinnarizine in dogs: Relation to SNEDDS droplet size, drug solubility and in vitro precipitation. Eur. J. Pharm. Sci. 2013, 48, 339–350. [Google Scholar] [CrossRef]
- Martins, S.; Sarmento, B.; Ferreira, D.C.; Souto, E.B. Lipid-based colloidal carriers for peptide and protein delivery--liposomes versus lipid nanoparticles. Int. J. Nanomed. 2007, 2, 595–607. [Google Scholar]
- Onoue, S.; Uchida, A.; Kuriyama, K.; Nakamura, T.; Seto, Y.; Kato, M.; Hatanaka, J.; Tanaka, T.; Miyoshi, H.; Yamada, S. Novel solid self-emulsifying drug delivery system of coenzyme Q10 with improved photochemical and pharmacokinetic behaviors. Eur. J. Pharm. Sci. 2012, 46, 492–499. [Google Scholar] [CrossRef] [PubMed]
- Shegokar, R.; Muller, R.H. Nanocrystals: Industrially feasible multifunctional formulation technology for poorly soluble actives. Int. J. Pharm. 2010, 399, 129–139. [Google Scholar] [CrossRef]
- Thomas, N.; Holm, R.; Garmer, M.; Karlsson, J.; Mullertz, A.; Rades, T. Supersaturated self-nanoemulsifying drug delivery systems (Super-SNEDDS) enhance the bioavailability of thepoorly water-soluble drug simvastatin in dogs. AAPS J. 2013, 15, 219–227. [Google Scholar] [CrossRef]
- Grassi, M.; Grassi, G.; Lapasin, L.; Colombo, I. Understanding Drug Release and Absorption Mechanisms. A Physical and Mathematical Approach; CRC Press, Taylor & Francis Group: Boca Raton, FL, USA, 2007. [Google Scholar]
- Washington, C. Drug release from monodisperse systems: A critical review. Int. J. Pharm. 1990, 58, 1–12. [Google Scholar] [CrossRef]
- Yotsuyanagi, T.; Higuchi, W.I.; Ghanem, A.H. Theoretical treatment of diffusional transport into and through an oil-water emulsion with an interfacial barrier at the oil-water interface. J. Pharm. Sci. 1973, 62, 40–43. [Google Scholar] [CrossRef] [PubMed]
- Friedman, D.; Benita, S. A mathematical model for drug release from 0/W emulsions: Application to controlled release morphine emulsions. Drug Dev. Ind. Pharm. 1987, 13, 2067–2085. [Google Scholar] [CrossRef]
- Grassi, M.; Coceani, N.; Magarotto, L. Mathematical modeling of drug release from microemulsions: Theory in comparison with experiments. J. Colloid Interface Sci. 2000, 228, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Sirotti, C.; Coceani, N.; Colombo, I.; Lapasin, R.; Grassi, M. Modeling of drug release from microemulsions: A peculiar case. J. Membr. Sci. 2002, 204, 401–412. [Google Scholar] [CrossRef]
- Grassi, M.; Lamberti, G.; Cascone, S.; Grassi, G. Mathematical modeling of simultaneous drug release and in vivo absorption. Int. J. Pharm. 2011, 418, 130–141. [Google Scholar] [CrossRef] [PubMed]
- Mircioiu, C.; Voicu, V.A.; Ionescu, M.; Miron, D.S.; Radulescu, F.S.; Nicolescu, A.C. Evaluation of in vitro absorption, decontamination and desorption of organophosphorous compounds from skin and synthetic membranes. Toxicol. Lett. 2013, 219, 99–106. [Google Scholar] [CrossRef]
- Mircioiu, C.; Perju, A.; Neagu, A.; Griu, E.; Calin, G.; Miron, D.S. Pharmacokinetics of progesterone in postmenopausal women: 1. Pharmacokinetics following intravaginal administration. Eur. J. Drug Metab. Pharm. 1998, 23, 391–396. [Google Scholar] [CrossRef]
- Tvrdonova, M.; Dedik, L.; Mircioiu, C.; Miklovicova, D.; Durisova, M. Physiologically motivated time-delay model to account for mechanisms underlying enterohepatic circulation of piroxicam in human beings. Basic Clin. Pharm. Toxicol. 2009, 104, 35–42. [Google Scholar] [CrossRef]
- Mehta, S.K.; Kaur, G.; Bhasin, K.K. Tween-embedded microemulsions—Physicochemical and spectroscopic analysis for antitubercular drugs. AAPS PharmSciTech 2010, 11, 143–153. [Google Scholar] [CrossRef]
- Sandulovici, R.; Prasacu, I.; Mircioiu, C.; Voicu, V.; Medvedovici, A.; Anuta, V. Mathematical and phenomenological criteria in selection of pharmacokinetic model for M1 metabolite of pentoxyphylline. Farmacia 2009, 57, 235–246. [Google Scholar]
- D’Aurizio, E.; Sozio, P.; Cerasa, L.S.; Vacca, M.; Brunetti, L.; Orlando, G.; Chiavaroli, A.; Kok, R.J.; Hennink, W.E.; Di Stefano, A. Biodegradable microspheres loaded with an anti-parkinson prodrug: An in vivo pharmacokinetic study. Mol. Pharm. 2011, 8, 2408–2415. [Google Scholar] [CrossRef] [PubMed]
- Bidah, D.; Ouriemchi, E.M.; Vergnaud, J.M. Diffusional process of drug delivery from a dosage form with a Gelucire matrix. Int. J. Pharm. 1992, 80, 145–149. [Google Scholar] [CrossRef]
- Siepmann, J.; Siepmann, F. Mathematical modeling of drug release from lipid dosage forms. Int. J. Pharm. 2011, 418, 42–53. [Google Scholar] [CrossRef] [PubMed]
- Guse, C.; Koennings, S.; Kreye, F.; Siepmann, F.; Goepferich, A.; Siepmann, J. Drug release from lipid-based implants: Elucidation of the underlying mass transport mechanisms. Int. J. Pharm. 2006, 314, 137–144. [Google Scholar] [CrossRef]
- Guse, C.; Koennings, S.; Maschke, A.; Hacker, M.; Becker, C.; Schreiner, S.; Blunk, T.; Spruss, T.; Goepferich, A. Biocompatibility and erosion behavior of implants made of triglycerides and blends with cholesterol and phospholipids. Int. J. Pharm. 2006, 314, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Vergaud, J.M. Controlled Drug Release of Oral Dosage Forms; Hellis Horwood: Chichester, UK, 1993. [Google Scholar]
- Fugit, K.D. Quantification of Factors Governing Drug Release Kinetics from Nanoparticles: A Combined Experimental and Mechanistic Modeling Approach. Ph.D. Thesis, University of Kentucky, Kentucky, UK, 2014. [Google Scholar]
- Tsuchiya, H.; Mizogami, M. Interaction of local anesthetics with biomembranes consisting of phospholipids and cholesterol: Mechanistic and clinical implications for anesthetic and cardiotoxic effects. Anesthesiol. Res. Pract. 2013, 2013, 297141. [Google Scholar] [CrossRef] [PubMed]
- Butu, A.; Rodino, S.; Golea, D.; Butu, M.; Butnariu, M.; Negoescu, C.; DinuPirvu, C.-E. Liposomal nanodelivery system for proteasome inhibitor anticancer drug bortezomib. Farmacia 2015, 63, 224–229. [Google Scholar]
- Enden, G.; Schroeder, A. A mathematical model of drug release from liposomes by low frequency ultrasound. Ann. Biomed. Eng. 2009, 37, 2640–2645. [Google Scholar] [CrossRef]
- Diamond, J.M.; Katz, Y. Interpretation of nonelectrolyte partition coefficients between dimyristoyl lecithin and water. J. Membr. Biol. 1974, 17, 121–154. [Google Scholar] [CrossRef]
- Xiang, T.X.; Anderson, B.D. The relationship between permeant size and permeability in lipid bilayer membranes. J. Membr. Biol. 1994, 140, 111–122. [Google Scholar] [CrossRef]
- Mayer, P.T.; Anderson, B.D. Transport across 1,9-decadiene precisely mimics the chemical selectivity of the barrier domain in egg lecithin bilayers. J. Pharm. Sci. 2002, 91, 640–646. [Google Scholar] [CrossRef]
- Xiang, T.X.; Anderson, B.D. Permeability of acetic acid across gel and liquid-crystalline lipid bilayers conforms to free-surface-area theory. Biophys. J. 1997, 72, 223–237. [Google Scholar] [CrossRef]
- Xiang, T.X.; Anderson, B.D. Influence of chain ordering on the selectivity of dipalmitoylphosphatidylcholine bilayer membranes for permeant size and shape. Biophys. J. 1998, 75, 2658–2671. [Google Scholar] [CrossRef]
- Xiang, T.X.; Xu, Y.H.; Anderson, B.D. The barrier domain for solute permeation varies with lipid bilayer phase structure. J. Membr. Biol. 1998, 165, 77–90. [Google Scholar] [CrossRef]
- Drug Delivery Mechanism and Efficiency of Liposomes into Skin. Available online: https://ecommons.cornell.edu/bitstream/handle/1813/2612/grp2.doc;sequence=1 (accessed on 10 December 2018).
- Lee, P.I. Modeling of drug release from matrix systems involving moving boundaries: Approximate analytical solutions. Int. J. Pharm. 2011, 418, 18–27. [Google Scholar] [CrossRef]
- Higuchi, T. Rate of release of medicaments from ointment bases containing drugs in suspension. J. Pharm. Sci. 1961, 50, 874–875. [Google Scholar] [CrossRef]
- Higuchi, T. Mechanism of sustained-action medication. Theoretical analysis of rate of release of solid drugs dispersed in solid matrices. J. Pharm. Sci. 1963, 52, 1145–1149. [Google Scholar] [CrossRef]
- Koizumi, T.; Ueda, M.; Kakemi, M.; Kameda, H. Rate of release of medicaments from ointment bases containing drugs in suspension. Chem. Pharm. Bull. (Tokyo) 1975, 23, 3288–3292. [Google Scholar] [CrossRef]
- Koizumi, T.; Panomsuk, T. Release of medicaments from spherical matrices containing drug in suspension—Theoretical aspects. Int. J. Pharm. 1995, 116, 45–49. [Google Scholar] [CrossRef]
- Roseman, T.J.; Higuchi, W.I. Release of medroxyprogesterone acetate from a silicone polymer. J. Pharm. Sci. 1970, 59, 353–357. [Google Scholar] [CrossRef]
- Koennings, S.; Tessmar, J.; Blunk, T.; Gopferich, A. Confocal microscopy for the elucidation of mass transport mechanisms involved in protein release from lipid-based matrices. Pharm. Res. 2007, 24, 1325–1335. [Google Scholar] [CrossRef]
- Vergaud, J.M.; Rosca, I.D. Assessing Bioavailablility of Drug Delivery Systems: Mathematical Modeling; CRC Press: Boca Raton, FL, USA, 2005. [Google Scholar]
- Koennings, S.; Beri, A.; Tessmar, J.; Blunk, T.; Goepferich, A. Influence of wettability and surface activity on release behavior of hydrophilic substances from lipid matrices. J. Control. Release 2007, 119, 173–181. [Google Scholar] [CrossRef]
- Dinu-Pîrvu, C.; Ivana, S. A study of the influence of crosslinking degree on the physicochemical properties of gelatin microparticles. Cellul. Chem. Technol. 2013, 47, 721–726. [Google Scholar]
- Zaky, A.; Elbakry, A.; Ehmer, A.; Breunig, M.; Goepferich, A. The mechanism of protein release from triglyceride microspheres. J. Control. Release 2010, 147, 202–210. [Google Scholar] [CrossRef]
- Efentakis, M.; Al-Hmoud, H.; Buckton, G.; Rajan, Z. The influence of surfactants on drug release from a hydrophobic matrix. Int. J. Pharm. 1991, 70, 153–158. [Google Scholar] [CrossRef]
- Vergaud, J.M. Liquid Transport Processes in Polymeric Materials; Prentice Hall: Englewood Cliffs, NJ, USA, 1991. [Google Scholar]
- Hopfenberg, H.B.; Hsu, K.C. Swelling-controlled, constant rate delivery systems. Polym. Eng. Sci. 1978, 18, 1186–1191. [Google Scholar] [CrossRef]
- Peppas, N.A. Release of bioactive agents from swellable polymers: Theory and experiments. In Recent Advances in Drug Delivery Systems; Anderson, J., Kim, S., Eds.; Springer: New York, NY, USA, 1984; pp. 279–289. [Google Scholar]
- Bakhouya-Sabbahi, N.; Bouzon, J.; Vergnaud, J.M. Absorption of liquid by a sphere with radial diffusion and finite surface coefficient of matter transfer and subsequent change in dimension. Polym. Compos. 1994, 2. [Google Scholar]
- Peppas, N.A. Analysis of Fickian and non-Fickian drug release from polymers. Pharm. Acta Helv. 1985, 60, 110–111. [Google Scholar]
- Bakhouya, N.; Sabbahi, A. Determination of diffusion parameters for polymer spheres undergoing high volume liquid transfer. Plast. Rubber Compos. 1999, 28, 271–276. [Google Scholar] [CrossRef]
- Azaar, K.; Rosca, I.D.; Vergnaud, J.M. Anisotropic behaviour of thin EPDM rubber discs towards absorption of toluene. Plast. Rubber Compos. 2002, 31, 220–225. [Google Scholar] [CrossRef]
- Brazel, C.S.; Peppas, N.A. Mechanisms of solute and drug transport in relaxing, swellable, hydrophilic glassy polymers. Polymer 1999, 40, 3383–3398. [Google Scholar] [CrossRef]
- Brazel, C.S.; Peppas, N.A. Modeling of drug release from swellable polymers. Eur. J. Pharm. Biopharm. 2000, 49, 47–58. [Google Scholar] [CrossRef]
- Alfrey, T.; Gurnee, E.F.; Lloyd, W.G. Diffusion in glassy polymers. J. Polym. Sci. C Polym. Symp. 1966, 12, 261. [Google Scholar] [CrossRef]
- Davidson, G.W.R., III; Peppas, N.A. Solute and penetrant diffusion in swellable polymers: V. Relaxation-controlled transport in P(HEMA-co-MMA) copolymers. J. Control. Release 1986, 3, 243–258. [Google Scholar] [CrossRef]
- Davidson, G.W.R., III; Peppas, N.A. Solute and penetrant diffusion in swellable polymers: VI. The Deborah and swelling interface numbers as indicators of the order of biomolecular release. J. Control. Release 1986, 3, 259–271. [Google Scholar] [CrossRef]
- Klier, J.; Peppas, N.A. Solute and penetrant diffusion in swellable polymers: VIII. Influence of the swelling interface number on solute concentration profiles and release. J. Control. Release 1988, 7, 61–68. [Google Scholar] [CrossRef]
- Korsmeyer, R.W.; Lustig, S.R.; Peppas, N.A. Solute and penetrant diffusion in swellable polymers. I. Mathematical modeling. J. Polym. Sci. B Polym. Phys. 1986, 24, 395–408. [Google Scholar] [CrossRef]
- Korsmeyer, R.W.; Von Meerwall, E.; Peppas, N.A. Solute and penetrant diffusion in swellable polymers. II. Verification of theoretical models. J. Polym. Sci. B Polym. Phys. 1986, 24, 409–434. [Google Scholar] [CrossRef]
- Lustig, S.R.; Peppas, N.A. Solute and penetrant diffusion in swellable polymers. VII. A free volume-based model with mechanical relaxation. J. Appl. Polym. Sci. 1987, 33, 533–549. [Google Scholar] [CrossRef]
- Peppas, N.A.; Korsmeyer, R.W. Dynamically swelling hydrogels in controlled release applications. In Hydrogels in Medicine and Pharmacy, vol. 3. Properties and Applications; Peppas, N.A., Ed.; CRC Press: Boca Raton, FL, USA, 1987; pp. 109–136. [Google Scholar]
- Bettini, R.; Catellani, P.L.; Santi, P.; Massimo, G.; Peppas, N.A.; Colombo, P. Translocation of drug particles in HPMC matrix gel layer: Effect of drug solubility and influence on release rate. J. Control. Release 2001, 70, 383–391. [Google Scholar] [CrossRef]
- Colombo, P.; Bettini, R.; Massimo, G.; Catellani, P.L.; Santi, P.; Peppas, N.A. Drug diffusion front movement is important in drug release control from swellable matrix tablets. J. Pharm. Sci. 1995, 84, 991–997. [Google Scholar] [CrossRef]
- Ferrero, C.; Munoz-Ruiz, A.; Jimenez-Castellanos, M.R. Fronts movement as a useful tool for hydrophilic matrix release mechanism elucidation. Int. J. Pharm. 2000, 202, 21–28. [Google Scholar] [CrossRef]
- Ghica, M.V.; Hîrjău, M.; Lupuleasa, D.; Dinu-Pîrvu, C.-E. Flow and thixotropic parameters for rheological characterization of hydrogels. Molecules 2016, 21, 786. [Google Scholar] [CrossRef]
- Toderescu, C.D.; Dinu-Pîrvu, C.; Ghica, M.V.; Anuța, V.; Popa, D.E.; Vlaia, L.; Lupuliasa, D. Influence of formulation variables on ketoprofen diffusion profiles from hydroalcoholic gels. Farmacia 2016, 64, 728–735. [Google Scholar]
- Rinaki, E.; Valsami, G.; Macheras, P. The power law can describe the ‘entire’ drug release curve from HPMC-based matrix tablets: A hypothesis. Int. J. Pharm. 2003, 255, 199–207. [Google Scholar] [CrossRef]
- Bunde, A.; Havlin, S.; Nossal, R.; Stanley, H.E.; Weiss, G.H. On controlled diffusion-limited drug release from a leaky matrix. J. Chem. Phys. 1985, 83, 5909–5913. [Google Scholar] [CrossRef]
- Leuenberger, H.; Rohera, B.D.; Haas, C. Percolation theory—A novel approach to solid dosage form design. Int. J. Pharm. 1987, 38, 109–115. [Google Scholar] [CrossRef]
- Ghica, M.V.; Kaya, M.G.A.; Dinu-Pîrvu, C.E.; Lupuleasa, D.; Udeanu, D.I. Development, optimization and in vitro/in vivo characterization of collagen-dextran spongious wound dressings loaded with flufenamic acid. Molecules 2017, 22, 1552. [Google Scholar] [CrossRef]
- Irimia, T.; Ghica, M.V.; Popa, L.; Anuţa, V.; Arsene, A.L.; Dinu-Pîrvu, C.E. Strategies for improving ocular drug bioavailability and cornealwound healing with chitosan-based delivery systems. Polymers 2018, 10, 1221. [Google Scholar] [CrossRef]
- Riley, R.G.; Smart, J.D.; Tsibouklis, J.; Dettmar, P.W.; Hampson, F.; Davis, J.A.; Kelly, G.; Wilber, W.R. An investigation of mucus/polymer rheological synergism using synthesised and characterised poly(acrylic acid)s. Int. J. Pharm. 2001, 217, 87–100. [Google Scholar] [CrossRef]
- Riley, R.G.; Smart, J.D.; Tsibouklis, J.; Young, S.A.; Hampson, F.; Davis, A.; Kelly, G.; Dettmar, P.W.; Wilber, W.R. An in vitro model for investigating the gastric mucosal retention of 14C-labelled poly(acrylic acid) dispersions. Int. J. Pharm. 2002, 236, 87–96. [Google Scholar] [CrossRef]
- Rossi, S.; Bonferoni, M.C.; Caramella, C.; Ironi, L.; Tentoni, S. Model-based interpretation of creep profiles for the assessment of polymer-mucin interaction. Pharm. Res. 1999, 16, 1456–1463. [Google Scholar] [CrossRef] [PubMed]
- Ritger, P.L.; Peppas, N.A. A simple equation for description of solute release I. Fickian and non-fickian release from non-swellable devices in the form of slabs, spheres, cylinders or discs. J. Control. Release 1987, 5, 23–36. [Google Scholar] [CrossRef]
- Siepmann, J.; Kranz, H.; Bodmeier, R.; Peppas, N.A. HPMC-matrices for controlled drug delivery: A new model combining diffusion, swelling, and dissolution mechanisms and predicting the release kinetics. Pharm. Res. 1999, 16, 1748–1756. [Google Scholar] [CrossRef] [PubMed]
- Siepmann, J.; Podual, K.; Sriwongjanya, M.; Peppas, N.A.; Bodmeier, R. A new model describing the swelling and drug release kinetics from hydroxypropyl methylcellulose tablets. J. Pharm. Sci. 1999, 88, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Arifin, D.Y.; Lee, L.Y.; Wang, C.H. Mathematical modeling and simulation of drug release from microspheres: Implications to drug delivery systems. Adv. Drug Deliv. Rev. 2006, 58, 1274–1325. [Google Scholar] [CrossRef] [PubMed]
- Siepmann, J.; Gopferich, A. Mathematical modeling of bioerodible, polymeric drug delivery systems. Adv. Drug Deliv. Rev. 2001, 48, 229–247. [Google Scholar] [CrossRef]
- Siepmann, J.; Peppas, N.A. Hydrophilic matrices for controlled drug delivery: An improved mathematical model to predict the resulting drug release kinetics (the “sequential layer” model). Pharm. Res. 2000, 17, 1290–1298. [Google Scholar] [CrossRef]
- Siepmann, J.; Peppas, N.A. Mathematical modeling of controlled drug delivery. Adv. Drug Deliv. Rev. 2001, 48, 137–138. [Google Scholar] [CrossRef]
- Langer, R.; Peppas, N.A. Chemical and physical structure of polymers as carriers for controlled release of bioactive agents: A review. J. Macromol. Sci. C 1983, 23, 61–126. [Google Scholar] [CrossRef]
- Allen, C.; Maysinger, D.; Eisenberg, A. Nano-engineering block copolymer aggregates for drug delivery. Colloid Surf. B 1999, 16, 3–27. [Google Scholar] [CrossRef]
- Kwon, G.S. Polymeric micelles for delivery of poorly water-soluble compounds. Crit. Rev. Ther. Drug Carr. Syst. 2003, 20, 357–403. [Google Scholar] [CrossRef]
- Kataoka, K.; Harada, A.; Nagasaki, Y. Block copolymer micelles for drug delivery: Design, characterization and biological significance. Adv. Drug Deliv. Rev. 2001, 47, 113–131. [Google Scholar] [CrossRef]
- Hopfenberg, H.B. Controlled release from erodible slabs, cylinders, and spheres. Am. Chem. Soc. Div. Org. Coat. Plast. Chem. Prepr. 1976, 36, 229–234. [Google Scholar]
- Hixson, A.W.; Crowell, J.H. Dependence of reaction velocity upon surface and agitation. Ind. Eng. Chem. 1931, 23, 923–931. [Google Scholar] [CrossRef]
- El-Arini, S.K.; Leuenberger, H. Dissolution properties of praziquantel-PVP systems. Pharm. Acta Helv. 1998, 73, 89–94. [Google Scholar] [CrossRef]
- Cooney, D.O. Effect of geometry on the dissolution of pharmaceutical tablets and other solids: Surface detachment kinetics controlling. AIChE J. 1972, 18, 446–449. [Google Scholar] [CrossRef]
- Heller, J.; Baker, R.W. Theory and practice of controlled Drug Delivery from bioerodible polymers. In Controlled Release of Bioactive Materials; Baker, W., Ed.; Academic Press: New York, NY, USA, 1980; pp. 1–18. [Google Scholar]
- Harland, R.S.; Dubernet, C.; Benoit, J.P.; Peppas, N.A. A model of dissolution-controlled, diffusional drug release from non-swellable polymeric microspheres. J. Control. Release 1988, 7, 207–215. [Google Scholar] [CrossRef]
- Kosmidis, K.; Rinaki, E.; Argyrakis, P.; Macheras, P. Analysis of Case II drug transport with radial and axial release from cylinders. Int. J. Pharm. 2003, 254, 183–188. [Google Scholar] [CrossRef]
- Satterfield, C.N.; Colton, C.K.; Pitcher, W.H. Restricted diffusion in liquids within fine pores. AIChE J. 1973, 19, 628–635. [Google Scholar] [CrossRef]
- Joshi, A.; Himmelstein, K.J. Dynamics of controlled release from bioerodible matrices. J. Control. Release 1991, 15, 95–104. [Google Scholar] [CrossRef]
- Thombre, A.G. Theoretical aspects of polymer biodegradation: Mathematical modeling of drug release and acid-catalyzed poly(ortho-ester) biodegradation. In Biodegradable Polymers and Plastics; Vert, M., Feijen, J., Albertsson, A.C., Scott, G., Chellini, E., Eds.; The Royal Society of Chemisty: Cambridge, UK, 1992; pp. 215–225. [Google Scholar]
- Thombre, A.G.; Himmelstein, K.J. A simultaneous transport-reaction model for controlled drug delivery from catalyzed bioerodible polymer matrices. AIChE J. 1985, 31, 759–766. [Google Scholar] [CrossRef]
- Charlier, A.; Leclerc, B.; Couarraze, G. Release of mifepristone from biodegradable matrices: Experimental and theoretical evaluations. Int. J. Pharm. 2000, 200, 115–120. [Google Scholar] [CrossRef]
- Lee, P.I. Diffusional release of a solute from a polymeric matrix—Approximate analytical solutions. J. Membr. Sci. 1980, 7, 255–275. [Google Scholar]
- Lee, P.I. Interpretation of drug-release kinetics from hydrogel matrices in terms of time-dependent diffusion coefficients. In Controlled-Release Technology; American Chemical Society: Washington, DC, USA, 1987; pp. 71–83. [Google Scholar]
- Raman, C.; Berkland, C.; Kyekyoon, K.; Pack, D.W. Modeling small-molecule release from PLG microspheres: Effects of polymer degradation and nonuniform drug distribution. J. Control. Release 2005, 103, 149–158. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Zhong, C.; Mi, J. Modeling of drug release from bioerodible polymer matrices. Drug Deliv. 2005, 12, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Yang, Z.; Chow, L.L.; Wang, C.H. Simulation of drug release from biodegradable polymeric microspheres with bulk and surface erosions. J. Pharm. Sci. 2003, 92, 2040–2056. [Google Scholar] [CrossRef]
- Siepmann, J.; Faisant, N.; Akiki, J.; Richard, J.; Benoit, J.P. Effect of the size of biodegradable microparticles on drug release: Experiment and theory. J. Control. Release 2004, 96, 123–134. [Google Scholar] [CrossRef]
- Wada, R.; Hyon, S.H.; Ikada, Y. Kinetics of diffusion-mediated drug release enhanced by matrix degradation. J. Control. Release 1995, 37, 151–160. [Google Scholar] [CrossRef]
- Siepmann, J.; Elkharraz, K.; Siepmann, F.; Klose, D. How autocatalysis accelerates drug release from plga-based microparticles: A quantitative treatment. Biomacromolecules 2005, 6, 2312–2319. [Google Scholar] [CrossRef]
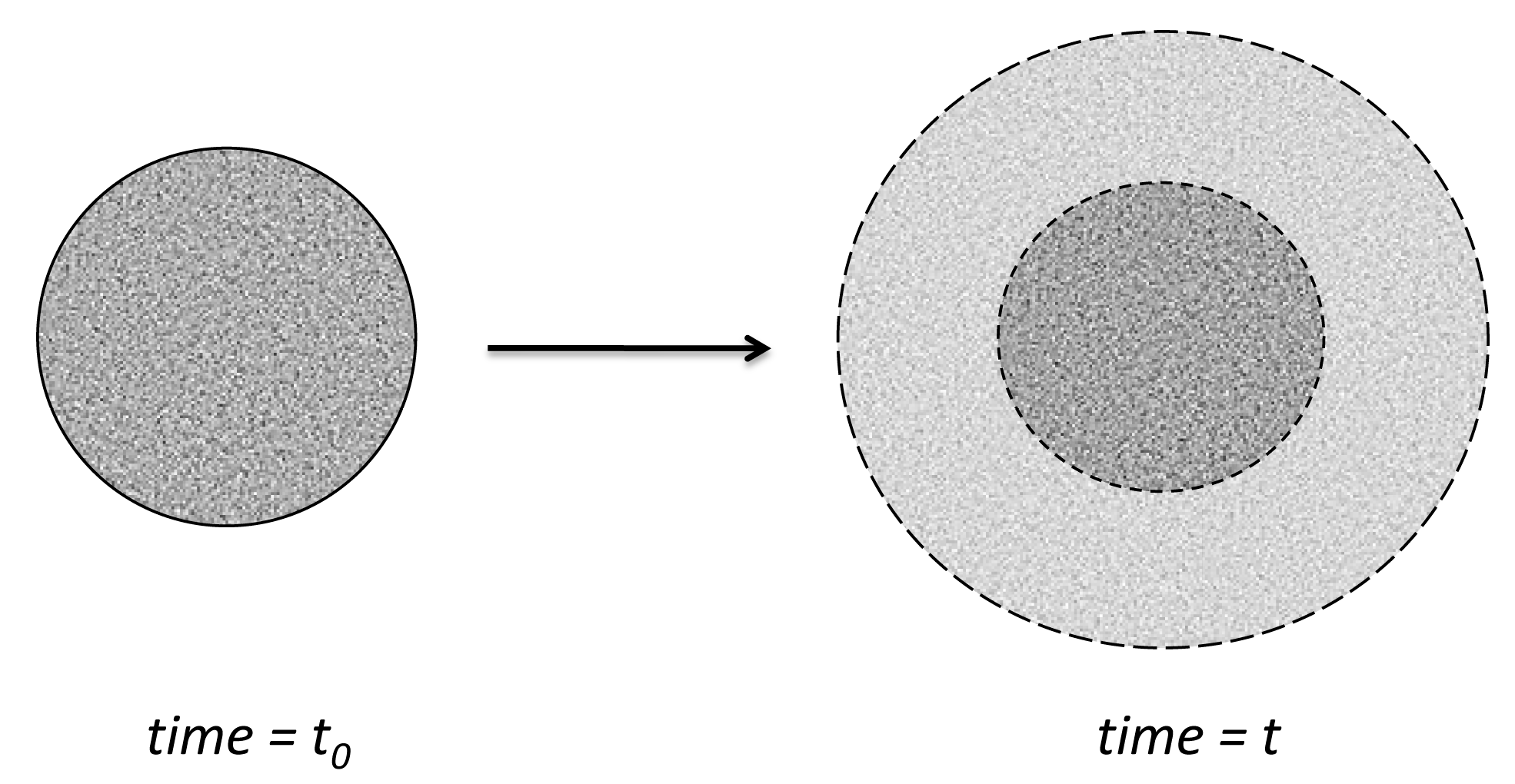
- Zygourakis, K. Development and temporal evolution of erosion fronts in bioerodible controlled release devices. Chem. Eng. Sci. 1990, 45, 2359–2366. [Google Scholar] [CrossRef]
- Zygourakis, K.; Markenscoff, P.A. Computer-aided design of bioerodible devices with optimal release characteristics: A cellular automata approach. Biomaterials 1996, 17, 125–135. [Google Scholar] [CrossRef]
- Gopferich, A. Mechanisms of polymer degradation and erosion. Biomaterials 1996, 17, 103–114. [Google Scholar] [CrossRef]
- Gopferich, A.; Langer, R. Modeling of polymer erosion. Macromolecules 1993, 26, 4105–4112. [Google Scholar] [CrossRef]
- Gopferich, A.; Langer, R. The influence of microstructure and monomer properties on the erosion mechanism of a class of polyanhydrides. J. Polym. Sci. A Polym. Chem. 1993, 31, 2445–2458. [Google Scholar] [CrossRef]
- Gopferich, A.; Langer, R. Modeling of polymer erosion in three dimensions: Rotationally symmetric devices. AIChE J. 1995, 41, 2292–2299. [Google Scholar] [CrossRef]
- Gopferich, A.; Langer, R. Modeling monomer release from bioerodible polymers. J. Control. Release 1995, 33, 55–69. [Google Scholar] [CrossRef]
- Kosmidis, K.; Macheras, P. Monte Carlo simulations for the study of drug release from matrices with high and low diffusivity areas. Int. J. Pharm. 2007, 343, 166–172. [Google Scholar] [CrossRef]
- Kosmidis, K.; Argyrakis, P.; Macheras, P. A reappraisal of drug release laws using Monte Carlo simulations: The prevalence of the Weibull function. Pharm. Res. 2003, 20, 988–995. [Google Scholar] [CrossRef]
- Kosmidis, K.; Argyrakis, P.; Macheras, P. Fractal kinetics in drug release from finite fractal matrices. J. Chem. Phys. 2003, 119, 6373–6377. [Google Scholar] [CrossRef]
- Landau, D.P.; Binder, K. Monte Carlo Simulations in Statistical Physics; Cambridge University Press: Cambridge, UK, 2000. [Google Scholar]
- Papadopoulou, V.; Kosmidis, K.; Vlachou, M.; Macheras, P. On the use of the Weibull function for the discernment of drug release mechanisms. Int. J. Pharm. 2006, 309, 44–50. [Google Scholar] [CrossRef]
- Chen, Y.; McCall, T.W.; Baichwal, A.R.; Meyer, M.C. The application of an artificial neural network and pharmacokinetic simulations in the design of controlled-release dosage forms. J. Control. Release 1999, 59, 33–41. [Google Scholar] [CrossRef]
- Takahara, J.; Takayama, K.; Nagai, T. Multi-objective simultaneous optimization technique based on an artificial neural network in sustained release formulations. J. Control. Release 1997, 49, 11–20. [Google Scholar] [CrossRef]
- Takayama, K.; Fujikawa, M.; Nagai, T. Artificial neural network as a novel method to optimize pharmaceutical formulations. Pharm. Res. 1999, 16, 1–6. [Google Scholar] [CrossRef]
- Wu, T.; Pan, W.; Chen, J.; Zhang, R. Formulation optimization technique based on artificial neural network in salbutamol sulfate osmotic pump tablets. Drug Dev. Ind. Pharm. 2000, 26, 211–215. [Google Scholar] [CrossRef]
- Mircioiu, C.; Borisova, S.A.; Voicu, V.A. Biopharmaceutic metrics applied in comparison of clusters of time courses of effect in clinical trials. J. Appl. Biopharm. Pharmacokinet. 2013, 1, 37–44. [Google Scholar] [CrossRef]
- Ibriç, S.; Jovanoviç, M.; Djuriç, Z.; Parojìiç, J.; Solomun, L. The application of generalized regression neural network in the modeling and optimization of aspirin extended release tablets with Eudragit-RS PO as matrix substance. J. Control. Release 2002, 82, 213–222. [Google Scholar] [CrossRef]
- Ghaffari, A.; Abdollahi, H.; Khoshayand, M.R.; Bozchalooi, I.S.; Dadgar, A.; Rafiee-Tehrani, M. Performance comparison of neural network training algorithms in modeling of bimodal drug delivery. Int. J. Pharm. 2006, 327, 126–138. [Google Scholar] [CrossRef]
- Nernst, W. Theorie der Reaktionsgeschwindigkeit in heterogenen Systemen. Z. Phys. Chem. 1904, 47, 52–55. [Google Scholar] [CrossRef]
- Noyes, A.A.; Whitney, W.R. The rate of solution of solid substances in their own solutions. J. Am. Chem. Soc. 1897, 19, 930–934. [Google Scholar] [CrossRef]
- Langenbucher, F. Linearization of dissolution rate curves by the Weibull distribution. J. Pharm. Pharm. 1972, 24, 979–981. [Google Scholar] [CrossRef]
- Marin, M.M.; Ignat, M.; Ghica, M.V.; Kaya, M.A.; Pirvu, C.D.; Anuta, V.; Popa, L. Collagen—Lidocaine microcapsules with controlled release for tooth extraction pain. Rev. Chim. 2018, 69, 1213–1215. [Google Scholar]
- Zambrano-Zaragoza, M.L.; Quintanar-Guerrero, D.; Del Real, A.; Piñon-Segundo, E.; Zambrano-Zaragoza, J.F. The release kinetics of β-carotene nanocapsules/xanthan gum coating and quality changes in fresh-cut melon (cantaloupe). Carbohydr. Polym. 2017, 157, 1874–1882. [Google Scholar] [CrossRef] [PubMed]
- Adibkia, K.; Siahi Shadbad, M.R.; Nokhodchi, A.; Javadzedeh, A.; Barzegar-Jalali, M.; Barar, J.; Mohammadi, G.; Omidi, Y. Piroxicam nanoparticles for ocular delivery: Physicochemical characterization and implementation in endotoxin-induced uveitis. J. Drug Target. 2007, 15, 407–416. [Google Scholar] [CrossRef] [PubMed]
- Vilaca, N.; Amorim, R.; Machado, A.F.; Parpot, P.; Pereira, M.F.; Sardo, M.; Rocha, J.; Fonseca, A.M.; Neves, I.C.; Baltazar, F. Potentiation of 5-fluorouracil encapsulated in zeolites as drug delivery systems for in vitro models of colorectal carcinoma. Colloids Surf. B Biointerfaces 2013, 112, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Ignjatovic, N.L.; Ninkov, P.; Sabetrasekh, R.; Uskokovic, D.P. A novel nano drug delivery system based on tigecycline-loaded calciumphosphate coated with poly-DL-lactide-co-glycolide. J. Mater. Sci. Mater. Med. 2010, 21, 231–239. [Google Scholar] [CrossRef]
- Fierăscu, R.C.; Dinu-Pîrvu, C.E.; Fierăscu, I.; Țărmure, V.; Stanică, N.; Nicolae, C.A.; Somoghi, R.; Trică, B.; Anuța, V. Inorganic/organic core-shell magnetic materials for removal of endocrine disrupting pharmaceuticals from water. Farmacia 2018, 66, 316–322. [Google Scholar]
- Ortan, A.; Fierascu, I.; Ungureanu, C.; Fierascu, R.C.; Avramescu, S.M.; Dumitrescu, O.; Dinu-Pirvu, C.E. Innovative phytosynthesized silver nanoarchitectures with enhanced antifungal and antioxidant properties. Appl. Surf. Sci. 2015, 358, 540–548. [Google Scholar] [CrossRef]
- Bhaskar, K.; Krishna, M.C.; Lingam, M.; Prabhakar, R.V.; Venkateswarlu, V.; Madhusudan, R.Y. Development of nitrendipine controlled release formulations based on SLN and NLC for topical delivery: In vitro and ex vivo characterization. Drug Dev. Ind. Pharm 2008, 34, 719–725. [Google Scholar] [CrossRef]
- Pi, F.M.; Tu, X.D.; Wu, Y. [Preparation of ATP-2Na loaded liposome and its effect on tissues energy state in myocardial ischemic mice]. Yao Xue Xue Bao 2010, 45, 1322–1326. [Google Scholar]
- Pippa, N.; Dokoumetzidis, A.; Pispas, S.; Demetzos, C. The interplay between the rate of release from polymer grafted liposomes and their fractal morphology. Int. J. Pharm. 2014, 465, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Guo, J.; Zheng, X.; Wu, J.; Zhou, Y.; Yu, Y.; Ye, Y.; Zhang, L.; Zhao, L. Preparation, pharmacokinetics and biodistribution of baicalin-loaded liposomes. Int. J. Nanomed. 2014, 9, 3623–3630. [Google Scholar] [CrossRef]
- Zhang, X.; Sun, P.; Bi, R.; Wang, J.; Zhang, N.; Huang, G. Targeted delivery of levofloxacin-liposomes for the treatment of pulmonary inflammation. J. Drug Target. 2009, 17, 399–407. [Google Scholar] [CrossRef] [PubMed]
- Macheras, P.; Dokoumetzidis, A. On the heterogeneity of drug dissolution and release. Pharm. Res. 2000, 17, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Dokoumetzidis, A.; Karalis, V.; Iliadis, A.; Macheras, P. The heterogeneous course of drug transit through the body. Trends Pharmacol. Sci. 2004, 25, 140–146. [Google Scholar] [CrossRef]
- Dokoumetzidis, A.; Macheras, P. The changing face of the rate concept in biopharmaceutical sciences: From Classical to fractal and finally to fractional. Pharm. Res. 2011, 28, 1229–1232. [Google Scholar] [CrossRef] [PubMed]
- Dokoumetzidis, A.; Papadopoulou, V.; Valsami, G.; Macheras, P. Development of a reaction-limited model of dissolution: Application to official dissolution tests experiments. Int. J. Pharm. 2008, 355, 114–125. [Google Scholar] [CrossRef]
- Pereira, L.M. Fractal pharmacokinetics. Comput. Math. Methods Med. 2010, 11, 161–184. [Google Scholar] [CrossRef]
- Pippa, N.; Dokoumetzidis, A.; Demetzos, C.; Macheras, P. On the ubiquitous presence of fractals and fractal concepts in pharmaceutical sciences: A review. Int. J. Pharm. 2013, 456, 340–352. [Google Scholar] [CrossRef]
- Pathak, Y.; Thassu, D. (Eds.) Drug Delivery Nanoparticles Formulation and Characterization; Informa Healthcare: London, UK, 2009. [Google Scholar]
- Huang, Y.; Li, Y.; Li, X.Z.; Liu, S.; Lei, P.; Xiao, J. [Study on the release of oleanolic acid loaded nanocapsules in vitro]. Zhong Yao Cai 2008, 31, 283–285. [Google Scholar]
- Bege, N.; Renette, T.; Endres, T.; Beck-Broichsitter, M.; Hanggi, D.; Kissel, T. In situ forming nimodipine depot system based on microparticles for the treatment of posthemorrhagic cerebral vasospasm. Eur. J. Pharm. Biopharm. 2013, 84, 99–105. [Google Scholar] [CrossRef]
- Nippe, S.; General, S. Combination of injectable ethinyl estradiol and drospirenone drug-delivery systems and characterization of their in vitro release. Eur. J. Pharm. Sci. 2012, 47, 790–800. [Google Scholar] [CrossRef]
- Tawfeek, H.; Khidr, S.; Samy, E.; Ahmed, S.; Murphy, M.; Mohammed, A.; Shabir, A.; Hutcheon, G.; Saleem, I. Poly(glycerol adipate-co-omega-pentadecalactone) spray-dried microparticles as sustained release carriers for pulmonary delivery. Pharm. Res. 2011, 28, 2086–2097. [Google Scholar] [CrossRef]
- Puthli, S.; Vavia, P. Formulation and performance characterization of radio-sterilized progestin-only microparticles intended for contraception. AAPS PharmSciTech 2009, 10, 443–452. [Google Scholar] [CrossRef]
- Zidan, A.S.; Sammour, O.A.; Hammad, M.A.; Megrab, N.A.; Hussain, M.D.; Khan, M.A.; Habib, M.J. Formulation of anastrozole microparticles as biodegradable anticancer drug carriers. AAPS PharmSciTech 2006, 7, 61. [Google Scholar] [CrossRef]
- Gupta, K.C.; Jabrail, F.H. Glutaraldehyde and glyoxal cross-linked chitosan microspheres for controlled delivery of centchroman. Carbohydr. Res. 2006, 341, 744–756. [Google Scholar] [CrossRef]
- Glavas-Dodov, M.; Fredro-Kumbaradzi, E.; Goracinova, K.; Simonoska, M.; Calis, S.; Trajkovic-Jolevska, S.; Hincal, A.A. The effects of lyophilization on the stability of liposomes containing 5-FU. Int. J. Pharm. 2005, 291, 79–86. [Google Scholar] [CrossRef]
- Dubey, R.R.; Parikh, R.H. Two-stage optimization process for formulation of chitosan microspheres. AAPS PharmSciTech 2004, 5, E5. [Google Scholar] [CrossRef]
- Sitta, D.L.; Guilherme, M.R.; da Silva, E.P.; Valente, A.J.; Muniz, E.C.; Rubira, A.F. Drug release mechanisms of chemically cross-linked albumin microparticles: Effect of the matrix erosion. Colloids Surf. B Biointerfaces 2014, 122C, 404–413. [Google Scholar] [CrossRef]
- Dash, V.; Mishra, S.K.; Singh, M.; Goyal, A.K.; Rath, G. Release kinetic studies of aspirin microcapsules from ethyl cellulose, cellulose acetate phthalate and their mixtures by emulsion solvent evaporation method. Sci. Pharm. 2010, 78, 93–101. [Google Scholar] [CrossRef]
- Jain, A.; Thakur, K.; Kush, P.; Jain, U.K. Docetaxel loaded chitosan nanoparticles: Formulation, characterization and cytotoxicity studies. Int. J. Biol. Macromol. 2014, 69, 546–553. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Ahuja, M. Carboxymethyl gum kondagogu-chitosan polyelectrolyte complex nanoparticles: Preparation and characterization. Int. J. Biol. Macromol. 2013, 62, 80–84. [Google Scholar] [CrossRef] [PubMed]
- Katara, R.; Majumdar, D.K. Eudragit RL 100-based nanoparticulate system of aceclofenac for ocular delivery. Colloids Surf. B Biointerfaces 2013, 103, 455–462. [Google Scholar] [CrossRef]
- Sonaje, K.; Italia, J.L.; Sharma, G.; Bhardwaj, V.; Tikoo, K.; Kumar, M.N. Development of biodegradable nanoparticles for oral delivery of ellagic acid and evaluation of their antioxidant efficacy against cyclosporine A-induced nephrotoxicity in rats. Pharm. Res. 2007, 24, 899–908. [Google Scholar] [CrossRef] [PubMed]
- Mittal, G.; Sahana, D.K.; Bhardwaj, V.; Ravi Kumar, M.N. Estradiol loaded PLGA nanoparticles for oral administration: Effect of polymer molecular weight and copolymer composition on release behavior in vitro and in vivo. J. Control. Release 2007, 119, 77–85. [Google Scholar] [CrossRef]
- Choubey, J.; Bajpai, A.K. Investigation on magnetically controlled delivery of doxorubicin from superparamagnetic nanocarriers of gelatin crosslinked with genipin. J. Mater. Sci. Mater. Med. 2010, 21, 1573–1586. [Google Scholar] [CrossRef]
- Bajpai, A.K.; Choubey, J. Design of gelatin nanoparticles as swelling controlled delivery system for chloroquine phosphate. J. Mater. Sci. Mater. Med. 2006, 17, 345–358. [Google Scholar] [CrossRef]
- Zhang, T.; Murowchick, J.; Youan, B.B. Optimization of formulation variables affecting spray-dried oily core nanocapsules by response surface methodology. J. Pharm. Sci. 2011, 100, 1031–1044. [Google Scholar] [CrossRef]
- Mudgil, M.; Pawar, P.K. Preparation and in vitro/ex vivo evaluation of moxifloxacin-loaded PLGA nanosuspensions for ophthalmic application. Sci. Pharm. 2013, 81, 591–606. [Google Scholar] [CrossRef]
- Lee, K.W.Y.; Nguyen, T.H.; Hanley, T.; Boyd, B.J. Nanostructure of liquid crystalline matrix determines in vitro sustained release and in vivo oral absorption kinetics for hydrophilic model drugs. Int. J. Pharm. 2009, 365, 190–199. [Google Scholar] [CrossRef]
- Tilley, A.; Morton, D.A.; Hanley, T.; Boyd, B.J. Liquid crystalline coated drug particles as a potential route to long acting intravitreal steroids. Curr. Drug Deliv. 2009, 6, 322–331. [Google Scholar] [CrossRef] [PubMed]
- Rosenbaum, E.; Tavelin, S.; Johansson, L.B. A characterisation study on the application of inverted lyotropic phases for subcutaneous drug release. Int. J. Pharm. 2010, 388, 52–57. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.; Kim, J.; Um, J.Y.; Kwon, I.C.; Jeong, S.Y. Self-assembled “nanocubicle” as a carrier for peroral insulin delivery. Diabetologia 2002, 45, 448–451. [Google Scholar] [CrossRef] [PubMed]
- Esposito, E.; Cortesi, R.; Drechsler, M.; Paccamiccio, L.; Mariani, P.; Contado, C.; Stellin, E.; Menegatti, E.; Bonina, F.; Puglia, C. Cubosome dispersions as delivery systems for percutaneous administration of indomethacin. Pharm. Res. 2005, 22, 2163–2173. [Google Scholar] [CrossRef]
- Han, S.; Shen, J.Q.; Gan, Y.; Geng, H.M.; Zhang, X.X.; Zhu, C.L.; Gan, L. Novel vehicle based on cubosomes for ophthalmic delivery of flurbiprofen with low irritancy and high bioavailability. Acta Pharm. Sin. 2010, 31, 990–998. [Google Scholar] [CrossRef] [PubMed]
- Malmsten, M. Soft drug delivery systems. Soft Matter 2006, 2, 760–769. [Google Scholar] [CrossRef]
- Ibrahim, H.K.; El-Leithy, I.S.; Makky, A.A. Mucoadhesive nanoparticles as carrier systems for prolonged ocular delivery of gatifloxacin/prednisolone bitherapy. Mol. Pharm. 2010, 7, 576–585. [Google Scholar] [CrossRef]
- Larsson, K. Cubic lipid-water phases: Structures and biomembrane aspects. J. Phys. Chem. 1989, 93, 7304–7314. [Google Scholar] [CrossRef]
- Rizwan, S.B.; Hanley, T.; Boyd, B.J.; Rades, T.; Hook, S. Liquid crystalline systems of phytantriol and glyceryl monooleate containing a hydrophilic protein: Characterisation, swelling and release kinetics. J. Pharm. Sci. 2009, 98, 4191–4204. [Google Scholar] [CrossRef]
- Zhao, X.Y.; Zhang, J.; Zheng, L.Q.; Li, D.H. Studies of cubosomes as a sustained drug delivery system. J. Dispers. Sci. Technol. 2005, 25, 795–799. [Google Scholar] [CrossRef]
- Boyd, B.J. Characterisation of drug release from cubosomes using the pressure ultrafiltration method. Int. J. Pharm. 2003, 260, 239–247. [Google Scholar] [CrossRef]
- Ahmed, A.R.; Dashevsky, A.; Bodmeier, R. Drug release from and sterilization of in situ cubic phase forming monoglyceride drug delivery systems. Eur. J. Pharm. Biopharm. 2010, 75, 375–380. [Google Scholar] [CrossRef] [PubMed]
- Allababidi, S.; Shah, J.C. Kinetics and mechanism of release from glyceryl monostearate-based implants: Evaluation of release in a gel simulating in vivo implantation. J. Pharm. Sci. 1998, 87, 738–744. [Google Scholar] [CrossRef] [PubMed]
- Clogston, J.; Craciun, G.; Hart, D.J.; Caffrey, M. Controlling release from the lipidic cubic phase by selective alkylation. J. Control. Release 2005, 102, 441–461. [Google Scholar] [CrossRef] [PubMed]
- Shah, M.H.; Paradkar, A. Effect of HLB of additives on the properties and drug release from the glyceryl monooleate matrices. Eur. J. Pharm. Biopharm. 2007, 67, 166–174. [Google Scholar] [CrossRef] [PubMed]
- Sherif, S.; Bendas, E.R.; Badawy, S. The clinical efficacy of cosmeceutical application of liquid crystalline nanostructured dispersions of alpha lipoic acid as anti-wrinkle. Eur. J. Pharm. Biopharm. 2014, 86, 251–259. [Google Scholar] [CrossRef]
- Nazaruk, E.; Szlezak, M.; Gorecka, E.; Bilewicz, R.; Osornio, Y.M.; Uebelhart, P.; Landau, E.M. Design and assembly of pH-sensitive lipidic cubic phase matrices for drug release. Langmuir 2014, 30, 1383–1390. [Google Scholar] [CrossRef]
- Peng, X.; Wen, X.; Pan, X.; Wang, R.; Chen, B.; Wu, C. Design and in vitro evaluation of capsaicin transdermal controlled release cubic phase gels. AAPS PharmSciTech 2010, 11, 1405–1410. [Google Scholar] [CrossRef]
- Lara, M.G.; Bentley, M.V.r.; Collett, J.H. In vitro drug release mechanism and drug loading studies of cubic phase gels. Int. J. Pharm. 2005, 293, 241–250. [Google Scholar] [CrossRef]
- Ahmed, A.R.; Dashevsky, A.; Bodmeier, R. Reduction in burst release of PLGA microparticles by incorporation into cubic phase-forming systems. Eur. J. Pharm. Biopharm. 2008, 70, 765–769. [Google Scholar] [CrossRef]
- Kelmann, R.G.; Kuminek, G.; Teixeira, H.F.; Koester, L.S. Carbamazepine parenteral nanoemulsions prepared by spontaneous emulsification process. Int. J. Pharm. 2007, 342, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Wen, J.; Du, Y.; Li, D.; Alany, R. Development of water-in-oil microemulsions with the potential of prolonged release for oral delivery of L-glutathione. Pharm. Dev. Technol. 2013, 18, 1424–1429. [Google Scholar] [CrossRef]
- Ozyazici, M.; Gökçe, E.H.; Ertan, G. Release and diffusional modeling of metronidazole lipid matrices. Eur. J. Pharm. Biopharm. 2006, 63, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Karasulu, E.; Karasulu, H.Y.; Ertan, G.; Kirilmaz, L.; Gueneri, T. Extended release lipophilic indomethacin microspheres: Formulation factors and mathematical equations fitted drug release rates. Eur. J. Pharm. Sci. 2003, 19, 99–104. [Google Scholar] [CrossRef]
- Yamagata, Y.; Iga, K.; Ogawa, Y. Novel sustained-release dosage forms of proteins using polyglycerol esters of fatty acids. J. Control. Release 2000, 63, 319–329. [Google Scholar] [CrossRef]
- Badilli, U.; Sengel-Turk, C.T.; Onay-Besikci, A.; Tarimci, N. Development of etofenamate-loaded semisolid sln dispersions and evaluation of anti-inflammatory activity for topical application. Curr. Drug Deliv. 2015, 12, 200–209. [Google Scholar] [CrossRef]
- Tiyaboonchai, W.; Tungpradit, W.; Plianbangchang, P. Formulation and characterization of curcuminoids loaded solid lipid nanoparticles. Int. J. Pharm. 2007, 337, 299–306. [Google Scholar] [CrossRef]
- Rao, M.P.; Manjunath, K.; Bhagawati, S.T.; Thippeswami, B.S. Bixin loaded solid lipid nanoparticles for enhanced hepatoprotection—Preparation, characterisation and in vivo evaluation. Int. J. Pharm. 2014, 473, 485–492. [Google Scholar] [CrossRef]
- Abul, K.M.; Sultana, Y.; Ali, A.; Aqil, M.; Mishra, A.K.; Aljuffali, I.A.; Alshamsan, A. Part I: Development and optimization of solid-lipid nanoparticles using Box-Behnken statistical design for ocular delivery of gatifloxacin. J. Biomed. Mater. Res. A 2013, 101, 1813–1827. [Google Scholar] [CrossRef]
- Shi, X.; Peng, T.; Huang, Y.; Mei, L.; Gu, Y.; Huang, J.; Han, K.; Li, G.; Hu, C.; Pan, X. Comparative studies on glycerol monooleate-and phytantriol-based cubosomes containing oridonin in vitro and in vivo. Pharm. Dev. Technol. 2017, 22, 322–329. [Google Scholar] [CrossRef]
- Badie, H.; Abbas, H. Novel small self-assembled resveratrol-bearing cubosomes and hexosomes: Preparation, charachterization, and ex vivo permeation. Drug Dev. Ind. Pharm. 2018, 44, 2013–2025. [Google Scholar] [CrossRef] [PubMed]
- Paolino, D.; Tudose, A.; Celia, C.; Di Marzio, L.; Cilurzo, F.; Mircioiu, C. Mathematical Models as Tools to Predict the Release Kinetic of Fluorescein from Lyotropic Colloidal Liquid Crystals. Materials 2019, 12, 693. [Google Scholar] [CrossRef] [PubMed]
- Harrold, J.M.; Eiseman, J.L.; Joseph, E.; Strychor, S.; Zamboni, W.C.; Parker, R.S. Control-relevant modeling of the antitumor effects of 9-nitrocamptothecin in SCID mice bearing ht29 human colon xenografts. J. Pharmacokinet. Pharmacodyn. 2005, 32, 65–83. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, G. Estimating the dimension of a model. Ann. Stat. 1978, 6, 461–464. [Google Scholar] [CrossRef]
- Mircioiu, I.; Anuta, V.; Purcaru, S.-O.; Radulescu, F.; Miron, D.; Dumitrescu, I.-B.; Ibrahim, N.; Mircioiu, C. In vitro dissolution of poorly soluble drugs in the presence of surface active agents-in vivo pharmacokinetics correlations. II. Nimesulide. Farmacia 2013, 61, 88–102. [Google Scholar]
- Pahomi, G.; Corlan, G.; Anuta, V.; Sandulovici, R.; Mircioiu, I. Study of the influence of bile salts and lecithin on distribution of ketoconazole between plasma and methylene chloride. Farmacia 2012, 60, 809–821. [Google Scholar]
- Purcaru, S.-O.; Ionescu, M.; Raneti, C.; Anuta, V.; Mircioiu, I.; Belu, I. Study of nimesulide release from solid pharmaceutical formulations in tween 80 solutions. Curr. Health Sci. J. 2010, 36, 42–49. [Google Scholar]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | Supramolecular System | Main Excipients | Release Experiment | Empirical Model | Reference |
|---|---|---|---|---|---|
| Cefpodoxime proxetil | Micro-balloons (hollow microspheres) | Hydroxypropylmethyl cellulose (HPMC) ethyl cellulose (EC) | Method (M): United States Pharmacopoeia (USP) paddle apparatus Dissolution medium (DM): 0.1 N HCl (pH 1.2) | Higher values of correlation coefficients were obtained in the case of Higuchi’s square root of time kinetic treatment; diffusion was the predominant mechanism of drug release. | [156] |
| Nimodipine Coumarin | Microparticles | PLGA | DM: 50/50 (w/w) mixture of phosphate-buffered saline (PBS), pH 7.4 and ethanol | Higuchi model | [157] |
| Ethinyl estradiol (EE) Drospirenone (DRSP) | Microparticles | PLGA | M: dialysis sac method DM: USP phosphate buffer pH 7.4 + 8% 2-Hydroxypropyl--β-cyclodextrin | EE release from PLGA microparticles was faster than DRSP release; EE release is assumed to be primarily controlled by drug diffusion. | [158] |
| Sodium fluorescein (hydrophilic compound) | Spray-dried microparticle | Poly(glycerol adipate-co-ω-pentadecalactone), l-arginine, l-leucine | DM: PBS, pH 7.4 (n = 3) | Higuchi model | [159] |
| Levonorgestrel | Microparticles | PLGA; Methocel Polyvinyl alcohol | DM: 0.9% sodium chloride + 0.5% sodium dodecyl sulfate | Release kinetics followed predominantly a zero-order release profile. | [160] |
| Anastrozole | Microparticles | PLGA | M: modified dialysis method DM: 0.1 N HCl (pH 1.2) and phosphate buffer (pH 7.4). | An initial burst release phase was followed by a gradual release phase with good correlation coefficients for the Higuchi model. | [161] |
| Centchroman | Microparticles | Glutaraldehyde Glyoxal | NA | A burst release of 29% centchroman within an initial period of 40 h was seen, and the remaining 70% was released in the next 60 h following zero-order release kinetics. | [162] |
| 5-fluorouracil (5-FU) | Microspheres | Bovine serum albumin Galactosylated chitosan (coating) | M: dynamic dialysis DM: phosphate buffered saline (pH 7.4, PBS) | Attenuated burst release in comparison with uncoated microspheres. Release followed Higuchi’s square root model. | [163] |
| Methotrexate (MTX) 5-fluorouracil (5-FU) | Microspheres | Chitosan | DM: PBS, pH 7.4 | Biphasic release (more prominent for MTX microspheres). 5-FU release followed Higuchi’s model, whereas MTX was released more slowly with a combination of first-order kinetics and Higuchi’s square-root model | [164] |
| Vitamin B12 | Microparticles | Bovine serum albumin (BSA) | M: dialysis technique DM: pH 2, pH 6 and pH 10 buffers | First stage: power law and Weibull equations. The second stage: super case II transport mechanism, as a result of diffusion, relaxation, and erosion. Application of Hixson–Crowell model confirmed the erosion mechanism. | [165] |
| Aspirin | Microcapsules | Ethyl cellulose, Cellulose Acetate Phthalate | M: USP apparatus 2 DM: pH-1.2 for 2 h followed by acetate buffer at pH 6.0 for 7 h | The best fit was the Higuchi model, indicating diffusion-controlled release. The n in Korsemeyer–Peppas model varied between 0.5 and 0.7, suggesting a diffusion-controlled release. | [166] |
| Drug | Supramolecular System | Main Excipients | Release Experiment | Empirical Model | Reference |
|---|---|---|---|---|---|
| Docetaxel | Nanoparticles | Chitosan | Method (M): dialysis sac method Dissolution medium (DM): PBS pH 7.4 | Higuchi’s square-root and Korsmeyer–Peppas; 0.45 ≤ n ≤ 0.89 indicates a combination of both diffusion of drug through the polymer and dissolution of the polymer. | [167] |
| Ofloxacin | Nanoparticles | Carboxymethyl gum kondagogu; Chitosan | M: dialysis sac method DM: phosphate buffer solution pH 7.4 | Higuchi model; ‘n’ exponent of Peppas equation (n < 0.43) suggested diffusion-controlled mechanism. | [168] |
| Aceclofenac | Nanoparticles | Eudragit RL 100- | M: dialysis sac method DM: Sorenson’s phosphate buffer | Higuchi model (0.43 < n < 0.85) | [169] |
| Ellagic Acid | Biodegradable nanoparticles | PLGA polycaprolactone (PCL) | M: dialysis technique DM: phosphate buffer pH 7.4 | An initial burst release was followed by Higuchi’s square-root pattern in the case of PLGA and PCL nanoparticles. | [170] |
| Estradiol | Nanoparticles | PLGA | M: dialysis technique DM: phosphate buffer pH 7.4 | Zero order for low-molecular-weight nanoparticles; it was considered that degradation plays a dominant role and controls the release rate. High-molecular-weight nanoparticles showed the best fit into the Higuchi’s model. | [171] |
| Doxorubicin | Nanoparticles | Gelatin cross-linked with genipin Fe3O4 | DM: PBS pH 7.4 | A correlation between the quantity of released drug and swelling of the nanoparticles was established using a power-law model. | [172] |
| Chloroquine phosphate | Nanoparticles | Gelatin | DM: PBS pH 7.4 and distilled water | Fick’s power law allowed establishing a correlation between the quantity of released drug and swelling of the nanoparticles. | [173] |
| Indomethacin | Nanocapsules | Pluronic F127 Polylactide (PLA) Labrafac CC | M: dialysis technique DM: PBS pH 7.4 | The release pattern was found to follow a power-law model, with n values ranging between 0.35 and 1.03 (depending on the preparation method). | [174] |
| Tigecycline | Nanoparticles | Calcium phosphate (CP) PLGA | DM: physiological solution at 37 °C under static conditions | The tigecycline content was released within a 35-day period. The in vitro data were best fitted with the Weibull model, and the release was defined as non-Fickian transport. | [141] |
| Moxifloxacin | Nanosuspensions | PLGA | M: USP apparatus 1 DM: simulated tear fluid (pH 7.4) | All formulations followed Korsemeyer–Peppas release kinetics with n values between 0.45 and 0.89 (anomalous behavior). | [175] |
| Drug | Supramolecular System | Main Excipients | Release Experiment | Empirical Model | Reference |
|---|---|---|---|---|---|
| Alpha lipoic acid (ALA) | Cubosomes loaded gel | Glycerol monooleate (GMO) Poloxamer P407 | M: USP Apparatus 5, paddle over disk assembly DM: hydro-alcoholic solution (1:1), 700 mL | Higuchi model ALA release from cubosomes in gels was shown to be primarily controlled by diffusion through the matrix. | [192] |
| Doxorubicin | Bicontinuous lipidic cubic phases (LCPs) | GMO Phytantriol (PT) | DM: pH 7.4 and pH 5.8 buffer | Higuchi model was n > 0.5 in all cases, indicating non-Fickian anomalous transport in which both diffusion and matrix effects. | [193] |
| Capsaicin | Cubic phase gels | GMO: propylene glycol (1,2-propanediol, PG): water | DM: isotonic phosphate buffered solution (PBS) | Release kinetics were determined to fit Higuchi’s square-root equation indicating that the release was under diffusion control. The calculated diffusion exponent showed the release from cubic phase gels was anomalous transport (n = 0.57–0.60) | [194] |
| Salicylic acid | Cubic phase gels | GMO Myverol 18–99® distilled monoglycerides | M: USP app I DM: Isotonic phosphate buffer | Release mechanism could be fitted to both Higuchi and first-order models. | [195] |
| 2-pyrrolidone (model) | In situ cubic phase forming monoglyceride drug delivery systems | Monoglyceride (GMO or glycerol monolinoleate) Cosolvents (ethanol, PEG 300, 2-pyrrolidone, DMSO) | DM: 0.1 M phosphate buffer, pH 7.4, with 0.1% sodium azide as preservative | The release of oligonucleotide from the fully swollen cubic phase matrix followed a diffusion-controlled release mechanism square-root Higuchi model in 24-h intervals for all formulations. | [196] |
| Carbamazepine | Nanoemulsion | Castor oil; Lipophilic emulsifier (lecithin or polyoxyl 35 castor oil); Tween 80 | M: dialysis technique DM: phosphate buffer pH 7.4 | Higuchi model best characterized the release profiles for the nanoemulsions and for the free drug, and drug release was described as a diffusion process based on Fick’s law. | [197] |
| l-glutathione | Microemulsions Liquid crystal systems | - | NA | Higuchi model | [198] |
| Drug | Supramolecular System | Main Excipients | Release Experiment | Empirical Model | Reference |
|---|---|---|---|---|---|
| Etofenamate | Solid Lipid Nanoparticles (SLN) | Compritol 888 ATO Precirol ATO 5 | NA | Higuchi model for Compritol 888 ATO SLNs; Zero-order release for Precirol ATO 5 SLNs | [202] |
| Curcuminoids | SLN | Poloxamer 188 Dioctyl sodium sulfosuccinate Stearic acid Glyceryl monostearate | M; vertical Franz diffusion cells DM: 50% (v/v) ethanol | 25% burst release of the curcuminoids within 10 min followed by controlled release pattern following Higuchi’s square-root model for 12 h | [203] |
| Bixin | SLN | Trimyristin Glycerol monostearate | M: diffusion using Franz diffusion cells Receptor medium: Sorensen buffer pH 7.7 | The release was first-order diffusion-controlled. The n-values obtained from the Korsmeyer–Peppas model (n = 0.697) indicated the release mechanism was non-Fickian type. | [204] |
| Gatifloxacin | SLN | Stearic acid (SA)/ Compritol/Gelucire Poloxamer-188 Sodium taurocholate | M: Automated transdermal diffusion cells Receptor medium: phosphate buffer (pH 7.4) | The release pattern was found to follow Korsmeyer–Peppas model (n = 0.15). | [205] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mircioiu, C.; Voicu, V.; Anuta, V.; Tudose, A.; Celia, C.; Paolino, D.; Fresta, M.; Sandulovici, R.; Mircioiu, I. Mathematical Modeling of Release Kinetics from Supramolecular Drug Delivery Systems. Pharmaceutics 2019, 11, 140. https://doi.org/10.3390/pharmaceutics11030140
Mircioiu C, Voicu V, Anuta V, Tudose A, Celia C, Paolino D, Fresta M, Sandulovici R, Mircioiu I. Mathematical Modeling of Release Kinetics from Supramolecular Drug Delivery Systems. Pharmaceutics. 2019; 11(3):140. https://doi.org/10.3390/pharmaceutics11030140
Chicago/Turabian StyleMircioiu, Constantin, Victor Voicu, Valentina Anuta, Andra Tudose, Christian Celia, Donatella Paolino, Massimo Fresta, Roxana Sandulovici, and Ion Mircioiu. 2019. "Mathematical Modeling of Release Kinetics from Supramolecular Drug Delivery Systems" Pharmaceutics 11, no. 3: 140. https://doi.org/10.3390/pharmaceutics11030140