Drug Transport across Porcine Intestine Using an Ussing Chamber System: Regional Differences and the Effect of P-Glycoprotein and CYP3A4 Activity on Drug Absorption


Abstract
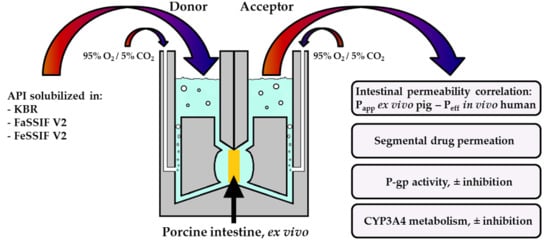
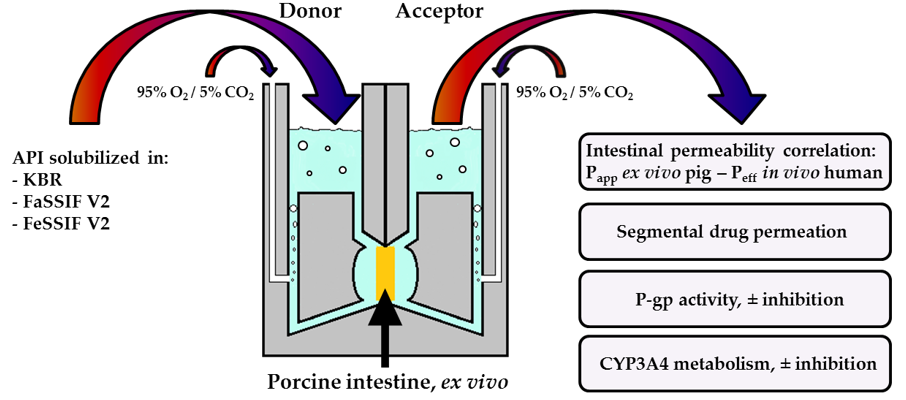
:
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Porcine Intestinal Tissue
2.3. Ussing Chamber Setup and Procedures for Intestinal Absorption Experiments
2.4. Analytical Methods
2.5. Data Analysis
2.5.1. Permeability Calculations
2.5.2. Data Fitting
2.5.3. Evaluation of the Relative Contributions of Drug Deposition and Permeation During Intestinal Transport: The Transport Index (TI)
2.6. Statistical Analysis
3. Results and Discussion
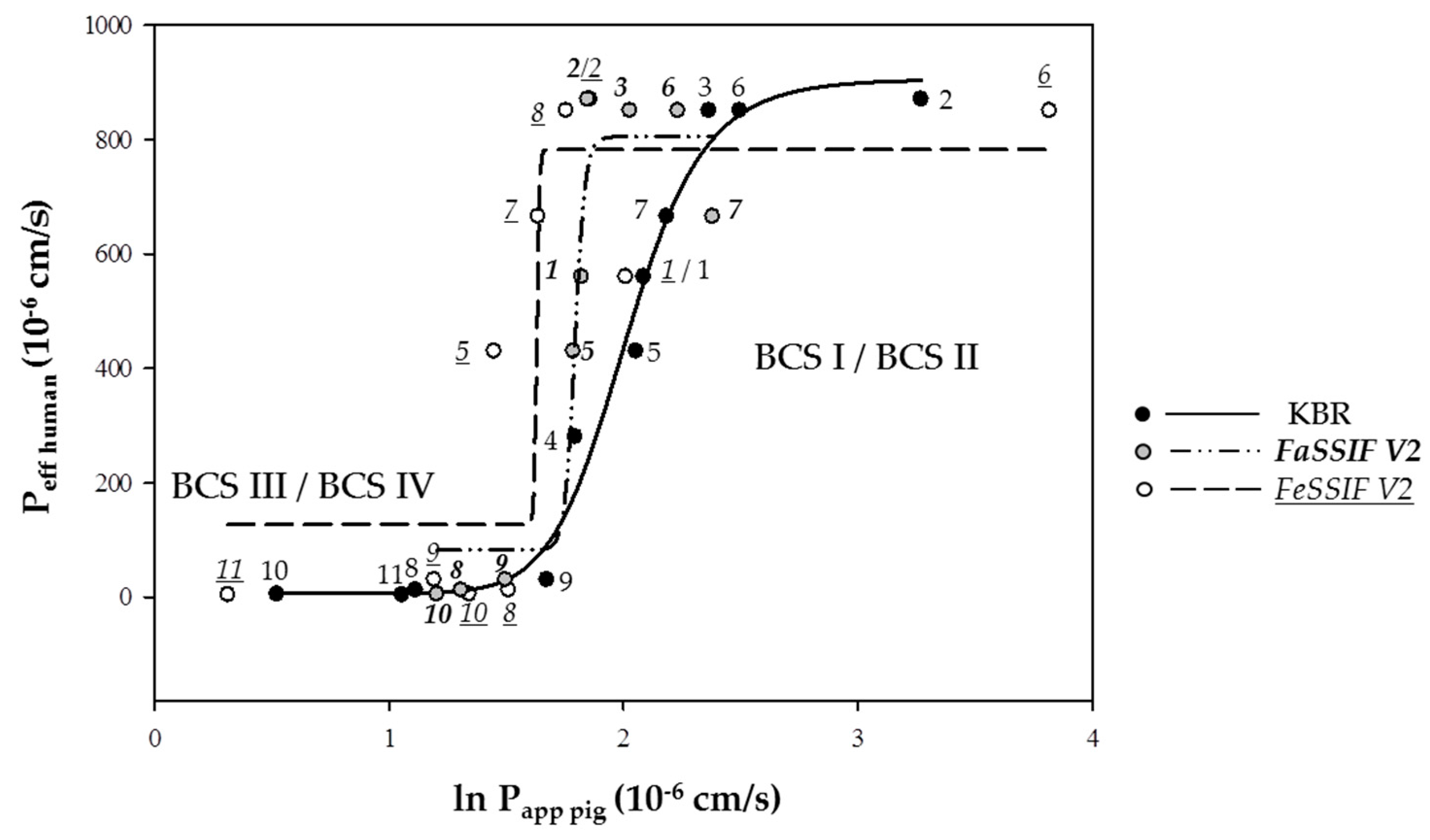
3.1. Intestinal Absorption of Drugs from KBR and FaSSIF V2 and FeSSIF V2
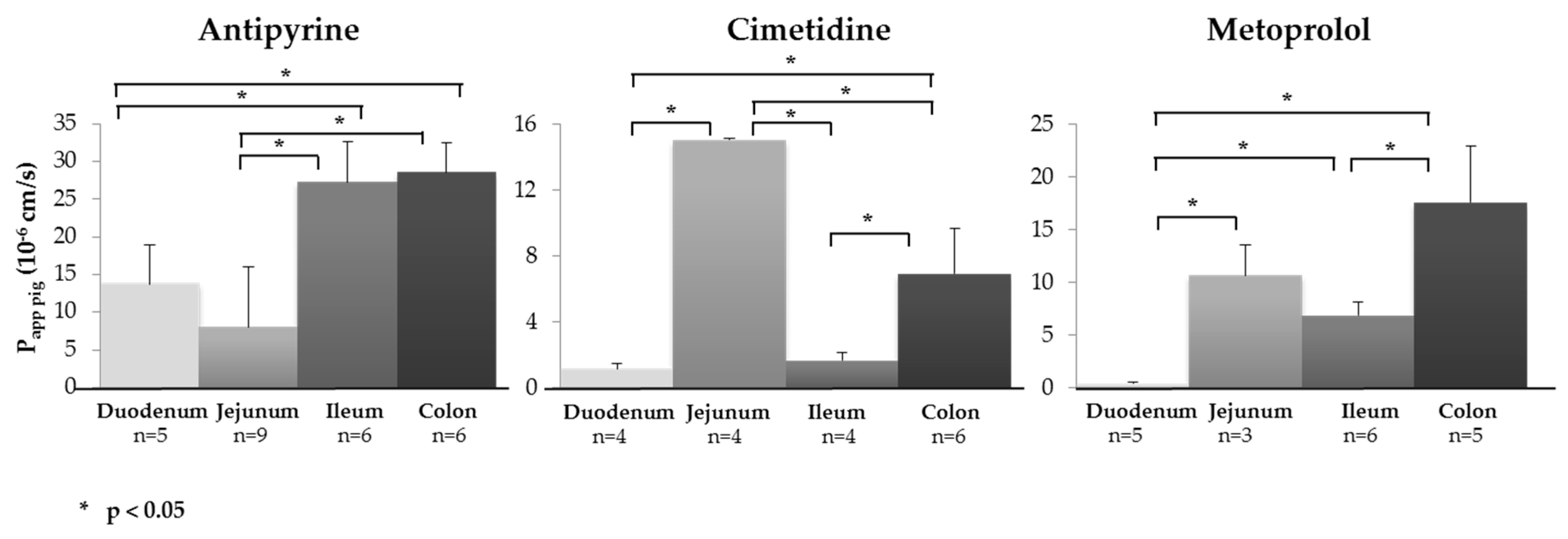
3.2. Segmental Intestinal Drug Permeation: Regional Variations in Drug Uptake
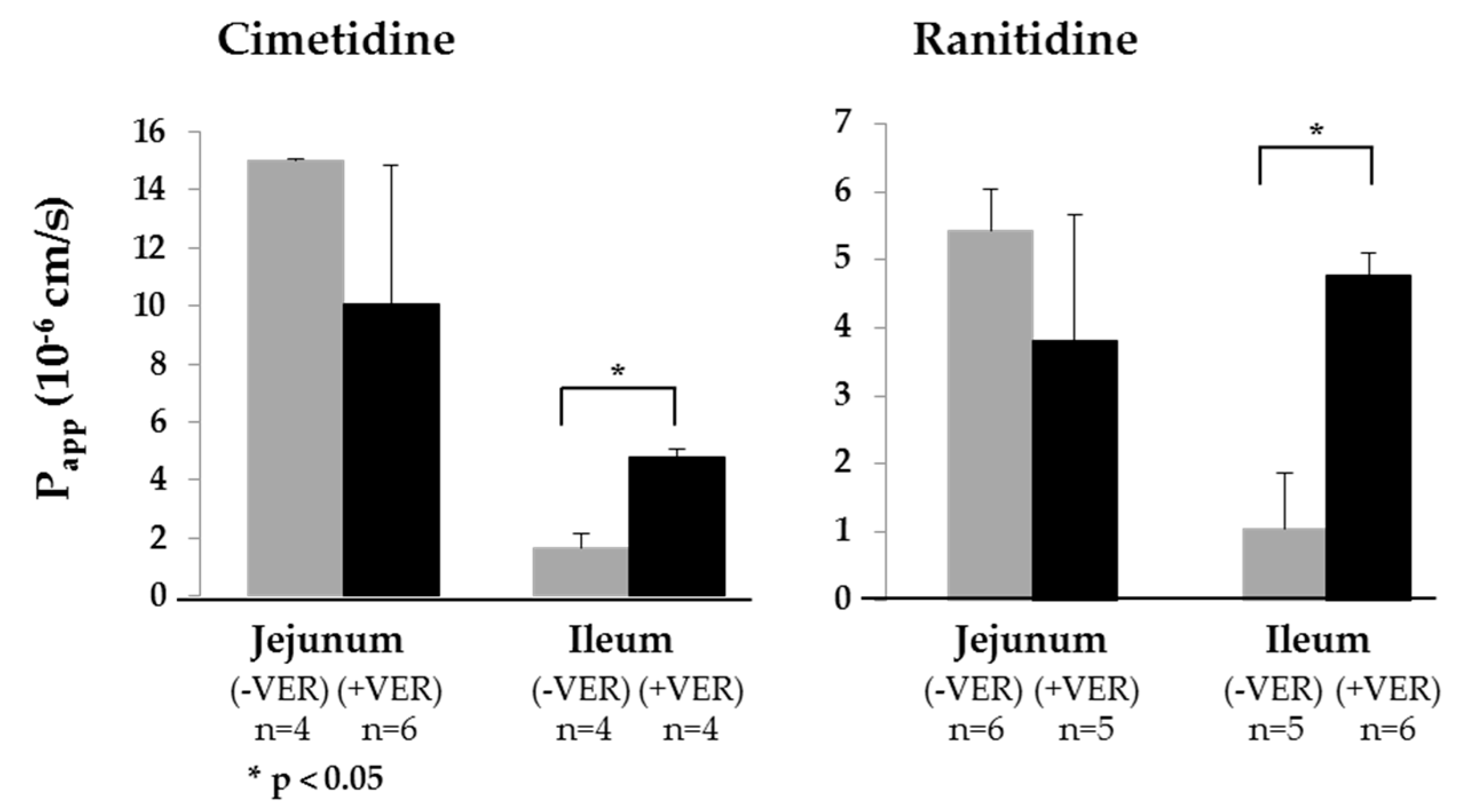
3.3. Demonstrating Activity of the P-gp Efflux Transporter
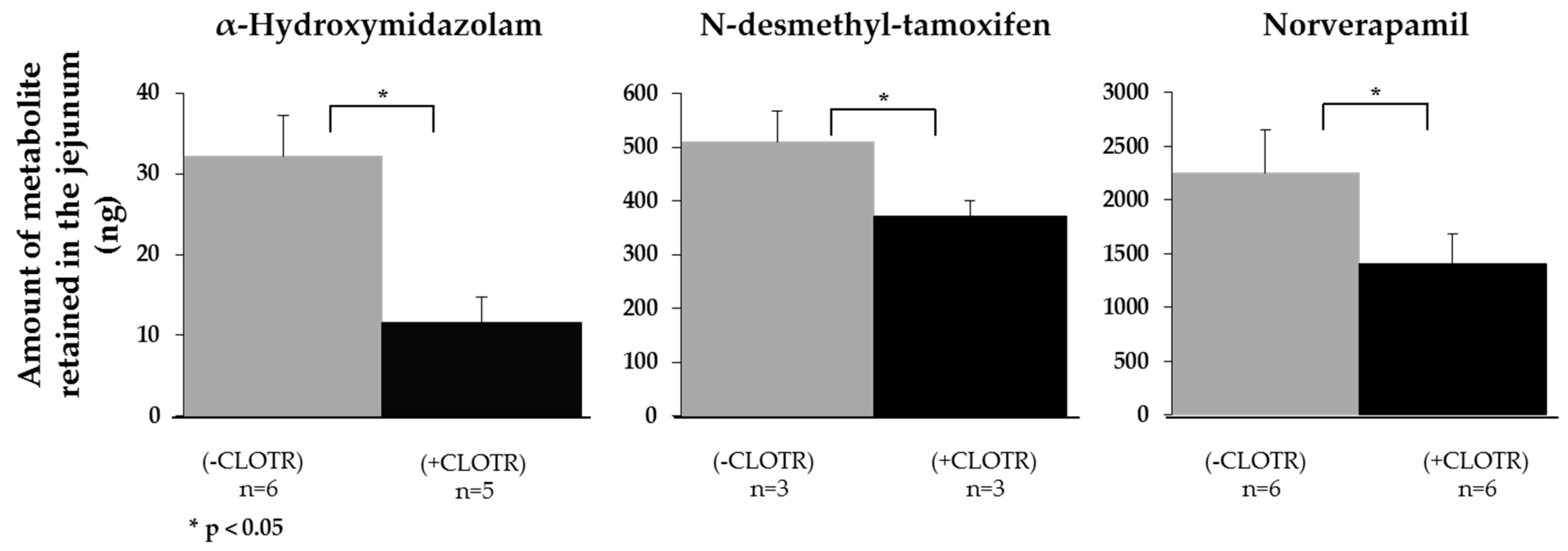
3.4. Demonstrating that CYP3A4 Retains Activity in the Porcine Intestine Ex Vivo
3.5. Relative Contribution of Drug Deposition (QDEP) and Drug Permeation (QPERM) to the Transport Index (TI)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rogers, S.M.; Back, D.J.; Orme, M.L.E. Intestinal metabolism of ethinyloestradiol and paracetamol in vitro: Studies using Ussing chambers. Br. J. Clin. Pharmacol. 1987, 23, 727–734. [Google Scholar] [CrossRef] [PubMed]
- Arnold, Y.E.; Imanidis, G.; Kuentz, M.T. Advancing in-vitro drug precipitation testing: New process monitoring tools and a kinetic nucleation and growth model. J. Pharm. Pharmacol. 2011, 63, 333–341. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.L.; Hubert, B.B.; Leeson, L.J.; Rhodes, C.T.; Robinson, J.R.; Roseman, T.J.; Shefter, E. The development of USP dissolution and drug release standards. Pharm. Res. 1990, 7, 983–987. [Google Scholar] [CrossRef]
- Minekus, M.; Marteau, P.; Havenaar, R.; Huis, J.H.J. A multicompartmental dynamic computer-controlled model simulating the stomach and small intestine. Altern. Lab. Anim. ATLA 1995, 23, 197–209. [Google Scholar]
- Barker, R.; Abrahamsson, B.; Kruusmaegi, M. Application and validation of an advanced gastrointestinal in vitro model for the evaluation of drug product performance in pharmaceutical development. J. Pharm. Sci. 2014, 103, 3704–3712. [Google Scholar] [CrossRef]
- Blanquet, S.; Zeijdner, E.; Beyssac, E.; Meunier, J.-P.; Denis, S.; Havenaar, R.; Alric, M. A dynamic artificial gastrointestinal system for studying the behavior of orally administered drug dosage forms under various physiological conditions. Pharm. Res. 2004, 21, 585–591. [Google Scholar] [CrossRef] [PubMed]
- Arnold, Y.; Bravo Gonzalez, R.; Versace, H.; Kuentz, M. Comparison of different in vitro tests to assess oral lipid-based formulations using a poorly soluble acidic drug. J. Drug Del. Sci. Technol. 2010, 20, 143–148. [Google Scholar] [CrossRef]
- Walter, E.; Janich, S.; Roessler, J.; Hilfinger, J.; Amidon, G. H29-MTX/Caco-2 cocultures as an in vitro model for the intestinal epithelium: In vitro-in vivo correlation with permeabiltiy data from rats and humans. J. Pharm. Sci. 1996, 85, 1070–1076. [Google Scholar] [CrossRef]
- Hilgendorf, C.; Spahn-Langguth, H.; Regardh, C.; Lipka, E.; Amidon, G.; Langguth, P. Caco-2 versus Caco-2/HT29-MTX co-cultured cell lines: Permeabilities via diffusion, inside- and outside-directed carrier-mediated transport. J. Pharm. Sci. 2000, 89, 63–75. [Google Scholar] [CrossRef]
- Araujo, F.; Sarmento, B. Towards the characterization of an in vitro triple co-culture intestine cell model for permeability studies. Int. J. Pharm. 2013, 458, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Lozoya-Agullo, I.; Araujo, F.; Gonzalez-Alvarez, I.; Merino-Sanjuan, M.; Gonzalez-Alvarez, M.; Bermejo, M.; Sarmento, B. Usefulness of Caco-2/HT29-MTX and Caco-2/HT29-MTX/Raji B coculture models to predict intestinal and colonic permeability compared to Caco-2 monoculture. Mol. Pharm. 2017, 14, 1264–1270. [Google Scholar] [CrossRef] [PubMed]
- Wuyts, B.; Riethorst, D.; Brouwers, J.; Tack, J.; Annaert, P.; Augustijns, P. Evaluation of fasted satte human intestinal fluid as apical solvent system in the Caco-2 absoprtion model and comparison with FaSSIF. Eur. J. Pharm. Sci. 2015, 67, 126–135. [Google Scholar] [CrossRef]
- Antoine, D.; Pellequer, Y.; Tempesta, C.; Lorscheidt, S.; Kettel, B.; Tamaddon, L.; Jannin, V.; Demarne, F.; Lamprecht, A.; Béduneau, A. Biorelevant media resistant co-culture model mimicking permeability of human intestine. Int. J. Pharm. 2015, 481, 27–36. [Google Scholar] [CrossRef]
- Lozoya-Agullo, I.; Gonzalez-Alvarez, I.; Zur, M.; Fine-Shamir, N.; Cohen, Y.; Markovic, M.; Garrigues, T.M.; Dahan, A.; Gonzalez-Alvarez, M.; Merino-Sanjuan, M.; et al. Closed-loop Doluisio (colon, small intestine) and single-pass intestinal perfusion (colon, jejunum) in rat-biophysical model and predictions based on Caco-2. Pharm. Res. 2018, 2018, 2–23. [Google Scholar] [CrossRef]
- Mani, V.; Hollis, J.H.; Gabler, N.K. Dietary oil composition differentially modulates intestinal endotoxin transport and postprandial endotoxemia. Nutr. Metab. 2013, 10, 1–9. [Google Scholar] [CrossRef]
- Kansara, V.; Mitra, A.K. Evaluation of an ex vivo model implication for carrier-mediated retinal drug delivery. Curr. Eye Res. 2006, 31, 415–426. [Google Scholar] [CrossRef]
- Eisenhut, M. Changes in ion transport in inflammatory disease. J. Inflamm. 2006, 3. [Google Scholar] [CrossRef]
- Burshtein, G.; Friedman, M.; Greenberg, S.; Amnon, H. Transepithelial transport of a natural cholinesterase inhibitor, huperzine A, along the gastrointestinal tract: The role of ionization on absorption mechanism. Planta Med. 2013, 79, 259–265. [Google Scholar] [CrossRef]
- Jin, L.; Boyd, B.J.; White, P.J.; Pennington, M.W.; Norton, R.S.; Nicolazzo, J.A. Buccal mucosal delivery of a potent peptide leads to therapeutically-relevant plasma concentrations for the treatment of autoimmune diseases. J. Control. Release 2015, 199, 37–44. [Google Scholar] [CrossRef]
- Boudry, G. The Ussing chamber technique to evaluate alternatives to in-feed antibiotics for young pigs. Anim. Res. 2005, 54, 219–230. [Google Scholar] [CrossRef] [Green Version]
- Ussing, H.H.; Zerahn, K. Active transport of sodium as the source of electric current in the short-circuited isolated frog skin. Acta Physiol. Scand. 1951, 23, 110–127. [Google Scholar] [CrossRef] [PubMed]
- Wu-Pong, S.; Livesay, V.; Dvorchik, B.; Barr, W.H. Oligonucleotide transport in rat and human intestine Ussing chamber models. Biopharm. Drug Dispos. 1999, 20, 411–416. [Google Scholar] [CrossRef]
- Nejdfors, P.; Ekelund, M.; Jeppson, B.; Weström, B.R. Mucosal in vitro permeability in the intestinal tract of the pig, the rat, and man: Species- and region-related differences. Scand. J. Gastroenterol. 2000, 35, 501–507. [Google Scholar] [PubMed]
- Ungell, A.-L.; Nylander, S.; Bergstrand, S.; Sjöberg, A.; Lennernäs, H. Membrane transport of drugs in different regions of the intestinal tract of the rat. J. Pharm. Sci. 1998, 87, 360–366. [Google Scholar] [CrossRef]
- Haslam, I.S.; O’Reilly, D.A.; Sherlock, D.J.; Kauser, A.; Womack, C.; Coleman, T. Pancreatoduodenectomy as a source of human small intestine for Ussing chamber investigations and comparative studies with rat tissue. Biopharm. Drug Dispos. 2011, 32, 210–221. [Google Scholar] [CrossRef] [Green Version]
- Rozehnal, V.; Nakai, D.; Hoepner, U.; Fischer, T.; Kamiyama, E.; Takahashi, M.; Mueller, J. Human small intestinal and colonic tissue mounted in the Ussing chamber as a tool for characterizing the intestinal absorption of drugs. Eur. J. Pharm. Sci. 2012, 46, 367–373. [Google Scholar] [CrossRef] [PubMed]
- Miyake, M.; Toguchi, H.; Nishibayashi, T.; Higaki, K.; Sugita, A.; Koganei, K.; Kamada, N.; Kitazume, M.T.; Hisamatsu, T.; Sato, T.; et al. Establishment of novel prediction system of intestinal absorption in humans using human intestinal tissue. J. Pharm. Sci. 2013, 102, 2564–2571. [Google Scholar] [CrossRef]
- Sjöberg, A.; Lutz, M.; Tannergren, C.; Wingolf, C.; Borde, A.; Ungell, A.-L. Comprehensive study on regional human intestinal permeability and prediction of fraction absorbed of drugs using the Ussing chamber technique. Eur. J. Pharm. Sci. 2013, 48, 166–180. [Google Scholar] [CrossRef]
- Söderholm, J.D.; Hedman, L.; Artursson, P.; Franzén, L.; Larsson, J.; Pantzar, N.; Permert, J.; Olaison, G. Integrity and metabolism of human ileal mucosa in vitro in the Ussing chamber. Acta Physiol. Scand. 1998, 162, 47–56. [Google Scholar] [CrossRef]
- Menon, R.M.; Barr, W.H. Comparison of ceftibuten transport across Caco-2 cells and rat jejunum mounted on modified Ussing chambers. Biopharm. Drug Dispos. 2003, 24, 299–308. [Google Scholar] [CrossRef]
- Kim, J.-S.; Mitchell, S.; Kijek, P.; Tsume, Y.; Hilfinger, J.; Amidon, G.L. The suitability of an in situ perfusion model for permeability determinations: Utility for BCS Class I biowaiver requests. Mol. Pharm. 2006, 3, 686–694. [Google Scholar] [CrossRef] [PubMed]
- Lennernäs, H. Animal data: The contributions of the Ussing chamber and perfusion systems to predicting human oral drug delivery in vivo. Adv. Drug Deliver. Rev. 2007, 59, 1103–1120. [Google Scholar] [CrossRef]
- Kararli, T.T. Comparison of the gastrointestinal anatomy, physiology, and biochemistry of humans and commonly used laboratory animals. Biopharm. Drug Dispos. 1995, 16, 351–380. [Google Scholar] [CrossRef] [PubMed]
- Sjoegren, E.; Abrahamsson, B.; Augustijns, P.; Becker, D.; Bolger, M.B.; Brewster, M.; Brouwers, J.; Flanagan, T.; Harwood, M.; Heinen, C.; et al. In vivo methods for drug absorption—Comparative physiologies, model selection, correlation with in vitro methods (IVIC), and applications for formulation/API/excipient characterization including food effects. Eur. J. Pharm. Sci. 2014, 57, 99–151. [Google Scholar] [CrossRef] [PubMed]
- Patterson, J.K.; Lei, X.G.; Miller, D.D. The pig as an experimental model for elucidation the mechanisms governing dietary influence on mineral absorption. Exp. Bio. Med. 2008, 233, 651–664. [Google Scholar] [CrossRef] [PubMed]
- Musther, H.; Olivares-Morales, A.; Hatley, O.J.D.; Liu, B.; Hodjegan, A.R. Animal versus human oral drug bioavailability: Do they correlate? Eur. J. Pharm. Sci. 2014, 57, 280–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, E.; Ullrey, D. The pig as a model for human nutrition. Ann. Rev. Nutr. 1987, 7, 361–382. [Google Scholar] [CrossRef]
- Witkamp, R.; Monshouwer, M. Pharmacokinetics in vivo and in vitro in swine. Scan. J. Lab. Anim. Sci. 1998, 25, 45–56. [Google Scholar]
- Skaanild, M. Porcine cytochrome P450 and metabolism. Curr. Pharm. Des. 2006, 12, 1421–1427. [Google Scholar] [CrossRef] [PubMed]
- Schrickx, J. ABC-Transporters in the Pig; Faculty of Veterinary Medicine, Utrecht University: Utrecht, The Netherlands, 2006. [Google Scholar]
- Mouly, S.; Paine, M.F. P-glyocportein increases from proximal to distal regions of human small intestine. Pharm. Res. 2003, 20, 1595–1599. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Pak, Y.; Mayersohn, M. Protein expression pattern of p-glycoprotein along the gastrointestinal tract of the yucatan micropig. J. Biochem. Mol. Toxicol. 2004, 18, 18–22. [Google Scholar] [CrossRef] [PubMed]
- Deusser, H.; Rogoll, D.; Scheppach, W.; Volk, A.; Melcher, R.; Richling, E. Gastrointestinal absorption and metabolism of apple polyphenols ex vivo by the pig intestinal mucosa in the Ussing chamber. Biotechnol. J. 2013, 8, 363–370. [Google Scholar] [CrossRef]
- Erk, T.; Hauser, J.; Williamson, G.; Renouf, M.; Steiling, H.; Dionisi, F.; Richling, E. Structure- and dose-absorption relationships of coffee polyphenols. Biofactors 2013, 40, 103–112. [Google Scholar] [CrossRef]
- Westerhout, J.; van de Steeg, A.; Grossouw, D.; Zeijdner, E.E.; Krul, C.A.M.; Verwei, M.; Wortelboer, H.M. A new approach to predict human intestinal absorption using porcine intestinal tissue and biorelevant matrices. Eur. J. Pharm. Sci. 2014, 63, 167–177. [Google Scholar] [CrossRef]
- Neirinckx, E.; Vervaet, C.; Michiels, J.; De Smet, S.; Van den Broeck, W.; Remon, J.P.; De Backer, P.; Croubels, S. Feasibility of the Ussing chamber technique for the determination of in vitro jejunal permeability of passively absorbed compounds in different animal species. J. Vet. Pharmacol. Therap. 2010, 34, 290–297. [Google Scholar] [CrossRef]
- Herrmann, J.; Hermes, R.; Breves, G. Transepithelial transport and intraepithelial metabolism of short-chain fatty acids (SCFA) in the porcine proximal colon are influenced by SCFA concentration and luminal pH. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 2011, 158, 169–176. [Google Scholar] [CrossRef]
- Lampen, A.; Zhang, Y.; Hackbarth, I.; Benet, L.Z.; Sewig, K.-F.; Christians, U. Metabolism and transport of the macrolide immunosuppressant sirolimus in the small intestine. J. Pharmcol. Exp. Ther. 1998, 285, 1104–1112. [Google Scholar]
- Lampen, A.; Christians, U.; Gonschior, A.-K.; Bader, A.; Hackbarth, I.; von Engelhardt, W.; Sewing, K.-F. Metabolism of the macrolide immunosuppressant, tacrolimus, by the pig gut mucosa in the Ussing chamber. Br. J. Pharmacol. 1996, 117, 1730–1734. [Google Scholar] [CrossRef] [Green Version]
- Aucamp, M.; Odendaal, R.; Wilna, L.; Hamman, J. Amorphous azithromycin with improved aqueous solubility and intestinal membrane permeability. Drug Dev. Ind. Pharm. 2015, 41, 1100–1108. [Google Scholar] [CrossRef]
- Atlabachew, M.; Combrinck, S.; Viljoen, A.M.; Hamman, J.H.; Gouws, C. Isolation and in vitro permeation of phenylpropylamino alkaloids from Khat (Catha edulis) across oral and intestinal mucosal tissues. J. Ethnopharmacol. 2016, 194, 307–315. [Google Scholar] [CrossRef]
- De Bruyn, S.; Willers, C.; Steyn, D.; Steenekamp, J.; Hamman, J. Development and evaluation of double-phase multiple-unit dosage form for enhanced insulin intestinal delivery. Drug Deliv. Lett. 2018, 8, 52–60. [Google Scholar] [CrossRef]
- Gerber, W.; Hamman, J.H.; Steyn, J.D. Excipient-drug pharmacokinetic interactions: Effect of disintegrants on efflux across excised pig intestinal tissues. J. Food Drug Anal. 2018, 26, S115–S124. [Google Scholar] [CrossRef] [PubMed]
- Pietzonka, P.; Walter, E.; Duda-Johner, S.; Langguth, P.; Merkle, H.P. Compromised integrity of excised porcine intestinal epithelium obtained from the abattoir affects the outcome of in vitro particle uptake studies. Eur. J. Pharm. Sci. 2002, 15, 39–47. [Google Scholar] [CrossRef]
- Shikanga, E.A.; Hamman, J.H.; Chen, W.; Combrinck, S.; Gericke, N.; Viljoen, A.M. In vitro permeation of mesembrine alkaloids from Sceletium tortuosum across porcine buccal, sublingual, and intestinal mucosa. Planta Med. 2012, 78, 260–268. [Google Scholar] [CrossRef] [PubMed]
- Hoegman, M.; Moerk, A.-C.; Roomans, G.M. Hypertonic saline increases tight junction permeability in airway epithelium. Eur. Respir. J. 2002, 20, 1444–1448. [Google Scholar] [CrossRef] [Green Version]
- Clark, L.L. A guide to Ussing chamber studies of mouse intestine. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 296, G1151–G1166. [Google Scholar] [CrossRef] [PubMed]
- Jantratid, E.; Niels, J.; Reppas, C.; Dressman, J.B. Dissolution media simulating conditions in the proximal human gastrointestinal tract: An update. Pharm. Res. 2008, 25, 1663–1676. [Google Scholar] [CrossRef] [PubMed]
- Winiwarter, S.; Bonham, N.M.; Ax, F.; Hallberg, A.; Lennernäs, H.; Karlén, A. Correlation of human jejunal permeability (in vivo) of drugs with experimentally and theoretically derived parameters. A multivariate data analysis approach. J. Med. Chem. 1998, 41, 4934–4949. [Google Scholar] [CrossRef]
- Sarti, F.; Müller, C.; Iqbal, J.; Perera, G.; Laffleur, F.; Bernkop-Schnürch, A. Development and in vivo evaluation of an oral vitamin B12 delivery system. Eur. J. Pharm. Biopharm. 2013, 84, 132–137. [Google Scholar] [CrossRef]
- Zhang, W.; Ramamoorthy, Y.; Kilicarslan, T.; Nolte, H.; Tyndale, R.F.; Sellers, E.M. Inhibition of cytochromes P450 by antifungal imidazole derivatives. Drug Metab. Dispos. 2002, 30, 314–318. [Google Scholar] [CrossRef]
- Artursson, P. Epithelial transport of drugs in cell culture. I: A model for studying the passive diffusion of drugs over intestinal absorbtive (Caco-2) cells. J. Pharm. Sci. 1990, 79, 476–482. [Google Scholar] [CrossRef] [PubMed]
- Cazares-Delgadillo, J.; Naik, A.; Ganem-Rondero, A.; Quintanar-Guerrero, D.; Kalia, Y. Transdermal delivery of Cytochrome C—A 12.4 kDa protein-across intact skin by constant-current iontophoresis. Pharm. Res. 2007, 24, 1360–1368. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Kalaria, D.R.; Kalia, Y.N. Erbium: YAG fractional laser ablation for the percutaneous delivery of intact functional therapeutic antibodies. J. Control. Release 2011, 156, 53–59. [Google Scholar] [CrossRef]
- Lapteva, M.; Mondon, K.; Moller, M.; Gurny, R.; Kalia, Y.N. Polymeric micelle nanocarriers for the cutaneous delivery of tacrolimus: A targeted approach for the treatment of psoriasis. Mol. Pharm. 2014, 11, 2989–3001. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Kalia, Y.N. Short-duration ocular iontophoresis of ionaziable aciclovir prodrugs: A new approach to treat herpes simplex infections in the anterior and posterior segments of the eye. Int. J. Pharm. 2018, 536, 292–300. [Google Scholar] [CrossRef]
- Miyake, M.; Kondo, S.; Koga, T.; Yoda, N.; Nakazato, S.; Emoto, C.; Mukai, T.; Toguchi, H. Evaluation of intestinal metabolism and absorption using Ussing chamber system, equipped with intestinal tissue from rats and dogs. Eur. J. Pharm. Biopharm. 2018, 122, 49–53. [Google Scholar] [CrossRef]
- Bachhav, Y.G.; Heinrich, A.; Kalia, Y.N. Using laser microporation to improve transdermal deliveriy of diclofenac: Increasing bioavailability and the range of therapeutic applications. Eur. J. Pharm. Biopharm. 2011, 78, 408–414. [Google Scholar] [CrossRef]
- Kalaria, D.R.; Patel, P.; Merino, V.; Patravale, V.B.; Kalia, Y.N. Controlled iontophoretic transport of huperzine A across skin in vitro and in vivo: Effect of delivery conditions and comparison of pharmacokinetic models. Mol. Pharm. 2013, 10, 4322–4329. [Google Scholar] [CrossRef]
- Kalaria, D.R.; Patel, P.; Merino, V.; Patravale, V.B.; Kalia, Y.N. Controlled iontophoretic delivery of pramipexole: Electrotransport kinetics in vitro and in vivo. Eur. J. Pharm. Biopharm. 2014, 88, 56–63. [Google Scholar] [CrossRef]
- Dressman, J.B.; Amidon, G.L.; Reppas, C.; Shah, V.P. Dissolution testing as a prognostic tool for oral drug absorption: Immediate release dosage forms. Pharm. Res. 1998, 15, 11–22. [Google Scholar] [CrossRef]
- Ingels, F.; Deferme, S.; Destexhe, E.; Oth, M.; Van den Mooter, G.; Augustijns, P. Simulated intestinal fluid as transport medium in the Caco-2 cell culture model. Int. J. Pharm. 2002, 232, 183–192. [Google Scholar] [CrossRef]
- Patel, N.; Forbes, B.; Eskola, S.; Murray, J. Use of simulated intestinal fluids with Caco-2 cells. Drug Dev. Ind. Pharm. 2006, 32, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Lind, M.L.; Jacobsen, J.; Holm, R.; Müllertz, A. Development of simulated intestinal fluids containing nutrients as transport media in the Caco-2 cell culture model: Assessment of cell viability, monolayer integrity and transport of poorly aqueous soluble drug and a substrate of efflux mechanisms. Eur. J. Pharm. Sci. 2007, 32, 261–270. [Google Scholar] [CrossRef] [PubMed]
- Markopoulos, C.; Thoenen, F.; Preisig, D.; Symillides, M.; Vertzoni, M.; Parrott, N.; Reppas, C.; Imanidis, G. Biorelevant media for transport experiments in the Caco-2 model to evaluate drug absorption in the fasted and the fed state and their usefulness. Eur. J. Pharm. Biopharm. 2014, 86, 438–448. [Google Scholar] [CrossRef] [PubMed]
- Knutson, L.; Odlind, B.; Hällgren, R. A new technique for segmental jejunal perfusion in man. Am. J. Gastroenterol. 1989, 84, 1278–1284. [Google Scholar]
- Olivares-Morales, A.; Lennernaes, H.; Aarons, L.; Rostami-Hodjegan, A. Translating human effective jejunal intestinal permeability to surface-dependent intrinsic permeability: A pragmatic method for a more mechanistic prediction of regional oral drug absorption. AAPS J. 2015, 17, 1177–1192. [Google Scholar] [CrossRef]
- Lennernäs, H. Human intestinal permeability. J. Pharm. Sci. 1998, 87, 403–410. [Google Scholar] [CrossRef]
- Miller, J.M.; Beig, A.; Krieg, B.J.; Carr, R.A.; Borchardt, T.B.; Amidon, G.E.; Amidon, G.L.; Dahan, A. The solubility-permeability interplay: Mechanistic modeling and predicitive application of the impact of micellar solubilization on intestinal permeation. Mol. Pharm. 2011, 8, 1848–1856. [Google Scholar] [CrossRef]
- Yano, K.; Masaoka, Y.; Kataoka, M.; Sakuma, S.; Yamashita, S. Mechanisms of membrane transport of poorly soluble drugs: Role of micelles in oral absorption processes. J. Pharm. Sci. 2010, 99, 1336–1345. [Google Scholar] [CrossRef]
- Tannergren, C.; Bergendal, A.; Lennernäs, H.; Abrahamsson, B. Toward an increased understanding of the barriers to colonic drug absorption in humans: Implication for early controlled release candidate assessment. Mol. Pharm. 2009, 6, 60–73. [Google Scholar] [CrossRef]
- Lennernäs, H.; Palm, K.; Fagerholm, U.; Artursson, P. Comparison between active and passive drug transport in human intestinal epithelial (Caco-2) cells in vitro and human jejunum in vivo. Int. J. Pharm. 1996, 127, 103–107. [Google Scholar] [CrossRef]
- Collett, A.; Higgs, N.B.; Sims, M.; Rowland, M.; Warhurst, G. Modulation of the permeability of H2 receptor antagonists cimetidine and ranitidine by p-glycoprotein in rat intestine and the human colonic cell line Caco-2. J. Pharmacol. Exp. Ther. 1999, 288, 171–178. [Google Scholar] [PubMed]
- Dahlgren, D.; Roos, C.; Sjögren, E.; Lennernäs, H. Direct in vivo human intestinal permeability (Peff) determined with different clinical perfusion and intubation methods. J. Pharm. Sci. 2015, 104, 2702–2726. [Google Scholar] [CrossRef]
- Lennernäs, H. Regional intestinal drug permeation: Biopharmaceutics and drug development. Eur. J. Pharm. Sci. 2014, 57, 333–341. [Google Scholar] [CrossRef] [PubMed]
- Ashmawy, S.M.; El-Gizawy, S.A.; El Maghraby, G.M.; Osman, M.A. Regional difference in intestinal drug absorption as a measure for the potential effect of P-glycoprotein efflux transporters. J. Pharm. Pharmacol. 2019, 71, 362–370. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Gibbs, S.T.; Fang, L.; Miller, H.A.; Landowski, C.P.; Shin, H.-C.; Lennernas, H.; Zhing, Y.; Amidon, G.L.; Yu, L.X.; et al. Why is it challenging to predict intestinal drug absorption and oral bioavailability in human using rat model. Pharm. Res. 2006, 28, 1675–1686. [Google Scholar] [CrossRef] [PubMed]
- Salomon, J.J.; Hagos, Y.; Petzke, S.; Kühne, A.; Gausterer, J.C.; Hosoya, K.-I.; Ehrhardt, C. Beta-2 adrenergic agonists are substrates and inhibitors of human organic cation transporter 1. Mol. Pharm. 2015, 12, 2633–2641. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-H.; Jones, D.R.; Hall, S.D. Differential mechanism-based inhibition of CYP3A4 and CYP3A5 by verapamil. Drug Metab. Dispos. 2005, 33, 664–671. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| API | MW (g/mol) | log P | log D [59] Octanol/H2O pH 7.4 pH 6.5 pH 5.5 | Solubility (n = 3) (mg/mL) | Study | ||||
|---|---|---|---|---|---|---|---|---|---|
| KBR | FaSSIF V2 | FeSSIF V2 | |||||||
| BCS I | |||||||||
| Antipyrine (1) C11H12N2O | 188.23 | 0.38 | 0.6 | 0.6 | 0.6 | 103.00 ± 54.61 | 261.10 ± 132.29 | 564.44 ± 83.12 | Passive b Regional c |
| Ketoprofen (2) C16H14O3 | 254.28 | 3.12 | 0.1 | 0.8 | 1.8 | 2.82 ± 0.62 | 6.73 ± 2.70 | 6.37 ± 1.52 | Passive |
| (+/−)-Metoprolol (3) C15H25NO3 | 267.36 | 1.88 | 0.0 | −0.5 | −0.6 | 298.03 ± 13.86 | 43.85 ± 15.81 | 380.69 ± 33.79 | Passive Regional |
| Midazolam C18H13ClFN3 | 325.77 | 3.89 | 3.0 a | 3.6 a | 3.9 a | n.a. | n.a. | n.a. | CYP3A4 d |
| Propranolol (4) C16H21NO2 | 259.34 | 3.48 | 1.4 | 0.9 | 0.7 | 158.69 ± 4.34 | 247.53 ± 7.47 | 222.57 ± 2.00 | Passive |
| Verapamil C27H38N2O4 | 454.60 | 3.79 | 3.8a | 3.8 a | 3.8 a | 1.04 ± 0.09 | 5.60 ± 1.04 | 17.60 ± 0.98 | CYP3A4 |
| BCS II | |||||||||
| Carbamazepine (5) C15H12N2O | 236.27 | 2.1 | 2.45 | 2.45 | 2.45 | 0.20 ± 0.02 | 0.32 ± 0.01 | 0.73 ± 0.01 | Passive |
| Naproxen (6) C14H14O3 | 230 | 3.18 | 0.3 | 1.1 | 2.1 | 4.51 ± 0.04 | 14.59 ± 1.19 | 13.00 ± 2.37 | Passive |
| Piroxicam (7) C15H13N3O4S | 331.35 | 3.06 | 0.2 a | 1.1 a | 2.0 a | 0.43 ± 0.02 | 0.29 ± 0.02 | 0.06 ± 0.00 | Passive |
| Tamoxifen C26H29NO | 371.51 | 5.93 | 5.6 a | 5.9 a | 5.9 a | 23.32 ± 1.54 | 32.19 ± 0.42 | 27.87 ± 0.66 | CYP3A4 |
| BSC III | |||||||||
| Atenolol (8) C14H22N2O3 | 365.40 | 0.75 | −2.0 | −2.0 | −2.0 | 35.57 ± 7.29 | 33.73 ± 2.06 | 41.28 ± 1.16 | Passive |
| Cimetidine C14H22N2O3 | 252.34 | 0.40 | 0.4 a | 0.4 a | 0.4 a | 5.65 ± 0.13 | 5.88 ± 0.26 | 0.36 ± 0.26 | Regional P-gp e |
| Ranitidine C14H22N2O3 | 314.40 | 0.27 | 0.2 a | 0.3 a | 0.3 a | 19.64 ± 2.78 | 15.89 ± 1.47 | 807.51 ± 70.47 | P-gp |
| Terbutaline (9) C14H22N2O3 | 225.28 | 0.9 | −1.4 | −1.3 | −1.3 | 213.73 ± 15.18 | 372.59 ± 18.86 | 305.07 ± 124.48 | Passive |
| BCS IV | |||||||||
| Furosemide (10) C12H11ClN2O5S | 330.74 | 2.03 | −0.9 | −0.5 | 0.4 | 5.62 ± 0.10 | 24.90 ± 2.84 | 19.68 ± 2.59 | Passive |
| Hydrochlorothiazide (11) C7H8ClN3O4S2 | 297.74 | −0.16 | −0.2 | −0.2 | −0.2 | 0.81 ± 0.17 | 1.18 ± 0.14 | 1.04 ± 0.19 | Passive |
| Drug | Papp,pig Ex Vivo (10−6 cm/s) | Peff,human a | Peff,human b | QDEP (%) | QPERM (%) | (n) c | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| KBR (n) | FaSSIF V2 (n) | FeSSIF V2 (n) | In Vivo (10−6 cm/s) | In Vivo (10−6 cm/s) | KBR | FaSSIF V2 | FeSSIF V2 | |||||||
| BCS I | ||||||||||||||
| (1) Antipyrine | 8.06 ± 7.91 (10) | 6.18 ± 2.18 (4) | 7.47 ± 0.95 (3) | 560 ± n.a. [31] | 19–29 [77] | 0.72 ± 0.20 | 2.18 ± 0.53 | (6) | 1.07 ± 0.22 | 0.33 ± 0.16 | (4) | 2.90 ± 0.26 | 1.03 ± 0.28 | (3) |
| (2) Ketoprofen | 26.31 ± 1.49 (3) | 6.34 ± 2.63 (5) | 6.42 ± 2.44 (4) | 870 ± n.a. [31] | 29–45 [77] | 11.57 ± 2.65 | 6.25 ± 0.92 | (6) | 4.49 ± 1.61 | 0.90 ± 0.46 | (5) | 0.37 ± 0.08 | 0.97 ± 0.54 | (4) |
| (3) Metoprolol | 10.64 ± 2.92 (3) | 7.62 ± 1.41 (3) | 5.79 ± 1.38 (3) | 850 ± n.a. [31] | 5.2–7.9 [77] | 0.28 ± 0.04 | 0.18 ± 0.05 | (6) | 0.12 ± 0.03 | 0.89 ± 0.19 | (3) | 0.22 ± 0.01 | 0.74 ± 0.08 | (3) |
| (4) Propranolol | 6.01 ± 3.41(5) | 0.71 ± 0.24 (3) | 0.93 ± 0.55 (3) | 280 ± 130 [31] | 9.3–14 [77] | 0.35 ± 0.06 | 0.04 ± 0.05 | (6) | 4.17 ± 0.15 | 0.27 ± 0.03 | (3) | 1.61 ± 0.57 | 0.27 ± 0.30 | (3) |
| BCS II | ||||||||||||||
| (5) Carbamazepine | 7.81 ± 5.69 (16) | 5.97 ± 0.52 (4) | 4.26 ± 0.96 (6) | 430 ± n.a. [31] | - | 1.44 ± 0.16 | 2.39 ± 0.42 | (6) | n.a. | n.a. | - | 3.45 ± 0.52 | 0.58 ± 0.22 | (6) |
| (6) Naproxen | 12.14 ± 4.83 (3) | 9.33 ± 3.36 (3) | 45.47 ± 7.97 (5) | 850 ± n.a. [31] | 27–42 [77] | n.a. | n.a. | - | n.a. | n.a. | - | 4.74 ± 1.04 | 8.11 ± 1.22 | (5) |
| (7) Piroxicam | 8.93 ± 2.03 (3) | 10.83 ± 3.70 (3) | 5.14 ± 0.78 (4) | 665 ± n.a. [31] | - | 4.12 ± 0.15 | 2.52 ± 0.70 | (3) | 5.63 ± 0.26 | 2.32 ± 0.79 | (3) | 0.21 ± 0.04 | 1.13 ± 0.46 | (5) |
| BCS III | ||||||||||||||
| (8) Atenolol | 3.05 ± 0.73 (6) | 3.70 ± 1.28 (6) | 4.53 ± 1.71 (5) | 20 ± n.a. [31] | 1.8–2.8 [77] | 7.38 ± 3.11 | 0.67 ± 0.13 | (6) | 0.25 ± 0.06 | 0.48 ± 0.09 | (6) | 0.55 ± 0.11 | 0.61 ± 0.12 | (5) |
| (9) Terbutaline | 5.33 ± 1.95 (17) | 4.47 ± 0.62 (5) | 3.30 ± 0.68 (6) | 30 ± 30 [78] | 1.7–2.6 [77] | 0.52 ± 0.11 | 0.66 ± 0.22 | (6) | 39.47 ± 2.51 | 0.54 ± 0.29 | (5) | 0.04 ± 0.00 | 0.39 ± 0.10 | (6) |
| BCS IV | ||||||||||||||
| (10) Furosemide | 1.69 ± 0.89 (16) | 3.34 ± 1.46 (4) | 3.83 ± 0.77 (6) | 5 ± n.a. [31] | 1–1.6 [77] | n.a. | n.a. | - | 2.22 ± 0.17 | 0.43 ± 0.09 | (4) | 1.52 ± 0.24 | 1.38 ± 0.24 | (6) |
| (11) Hydrochlorothiazide | 2.88 ± 2.84 (17) | 0.2 ± 0.25 (3) | 1.37 ± 0.53 (6) | 4 ± n.a. [31] | - | n.a. | n.a. | - | 0.22 ± 0.11 | 0.11 ± 0.05 | (3) | 0.74 ± 0.09 | 0.23 ± 0.08 | (6) |
| Drug | Papp,pig Ex Vivo (10−6 cm/s) | QDEP (%) QPERM (%) (n) a | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Duodenum (n) | Jejunum (n) | Ileum (n) | Colon (n) | Duodenum | Jejunum | Ileum | Colon | |||||
| Antipyrine | 13.69 ± 5.24 (5) | 8.06 ± 7.91 (9) | 27.26 ± 5.47 (6) | 28.56 ± 3.97 (6) | 1.10 ± 0.47 | 1.57 ± 0.58 (5) | 0.72 ± 0.20 | 2.18 ± 0.53 (5) | 1.97 ± 0.34 | 1.39 ± 0.56 (6) | 0.87 ± 0.16 | 3.79 ± 0.80 (5) |
| Cimetidine | 1.12 ± 0.30 (4) | 15.01 ± 0.07 (4) | 1.66 ± 0.48 (4) | 6.91 ± 2.74 (6) | 0.67 ± 0.06 | 0.91 ± 0.27 (4) | 3.28 ± 0.56 | 1.66 ± 0.64 (6) | 0.07 ± 0.01 | 0.26± 0.08 (3) | 0.05 ± 0.01 | 0.81 ± 0.25 (6) |
| Metoprolol | 0.39 ± 0.16 (5) | 10.64 ± 2.92 (3) | 6.87 ± 1.23 (6) | 17.55 ± 5.41 (6) | 2.35 ± 0.25 | 0.14 ± 0.08 (5) | 0.27 ± 0.04 | 0.18 ± 0.05 (4) | 1.89 ± 0.67 | 0.83 ± 0.09 (5) | 0.02 ± 0.00 | 1.65 ± 0.87 (6) |
| Drug | Papp,pig Ex Vivo (10−6 cm/s) | QDEP (%) QPERM (%) (n) a | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Jejunum | Ileum | Jejunum | Jejunum | Ileum | Ileum | |||||||
| (−VER) (n) | (+VER) (n) | (−VER) (n) | (+VER) (n) | (−VER) | (+VER) | (−VER) | (+VER) | |||||
| Cimetidine | 15.01 ± 0.07 (4) | 10.07 ± 4.80 (6) | 1.66 ± 0.48 (4) | 4.28 ± 0.70 (4) | 2.98 ± 0.29 | 1.38 ± 0.03 (4) | 4.32 ± 0.97 | 1.49 ± 0.67(6) | 0.07 ± 0.01 | 0.26 ± 0.08 (3) | 0.08 ± 0.02 | 0.71 ± 0.14 (4) |
| Ranitidine | 5.42 ± 0.61 (6) | 4.92 ± 0.30 (5) | 1.04 ± 0.83 (5) | 4.78 ± 0.31 (6) | 1.32 ± 0.31 | 0.67 ± 0.12 (6) | 5.47 ± 0.75 | 0.23 ± 0.15 (5) | 1.10 ± 0.59 | 0.55 ± 0.35 (4) | 7.91 ± 0.74 | 0.25 ± 0.03 (6) |
| Drug | Papp,pig Ex Vivo (10−6 cm/s) | QDEP (%) QPERM (%) (n) a | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| (−CLOTR) | (n) | (+CLOTR) | (n) | (−CLOTR) | (+CLOTR) | |||||
| Midazolam | 0.183 ± 0.138 | (4) | 0.460 ± 0.070 | (4) | 8.06 ± 1.33 | 0.13 ± 0.09 | (6) | 7.49 ± 2.50 | 0.18 ± 0.08 | (5) |
| Tamoxifen | 0.124 ± 0.046 | (3) | 1.381 ± 1.080 | (3) | 1.82 ± 1.39 | 0.07 ± 0.07 | (3) | 1.82 ± 0.58 | 0.06 ± 0.06 | (4) |
| Verapamil | 0.008 ± 0.003 | (4) | 0.211 ± 0.071 | (3) | 1.88 ± 0.12 | 0.00 ± 0.00 | (3) | 3.00 ± 0.63 | 0.18 ± 0.07 | (6) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arnold, Y.E.; Thorens, J.; Bernard, S.; Kalia, Y.N. Drug Transport across Porcine Intestine Using an Ussing Chamber System: Regional Differences and the Effect of P-Glycoprotein and CYP3A4 Activity on Drug Absorption. Pharmaceutics 2019, 11, 139. https://doi.org/10.3390/pharmaceutics11030139
Arnold YE, Thorens J, Bernard S, Kalia YN. Drug Transport across Porcine Intestine Using an Ussing Chamber System: Regional Differences and the Effect of P-Glycoprotein and CYP3A4 Activity on Drug Absorption. Pharmaceutics. 2019; 11(3):139. https://doi.org/10.3390/pharmaceutics11030139
Chicago/Turabian StyleArnold, Yvonne E., Julien Thorens, Stéphane Bernard, and Yogeshvar N. Kalia. 2019. "Drug Transport across Porcine Intestine Using an Ussing Chamber System: Regional Differences and the Effect of P-Glycoprotein and CYP3A4 Activity on Drug Absorption" Pharmaceutics 11, no. 3: 139. https://doi.org/10.3390/pharmaceutics11030139