LC478, a Novel Di-Substituted Adamantyl Derivative, Enhances the Oral Bioavailability of Docetaxel in Rats



Abstract
:1. Introduction
2. Materials and Methods
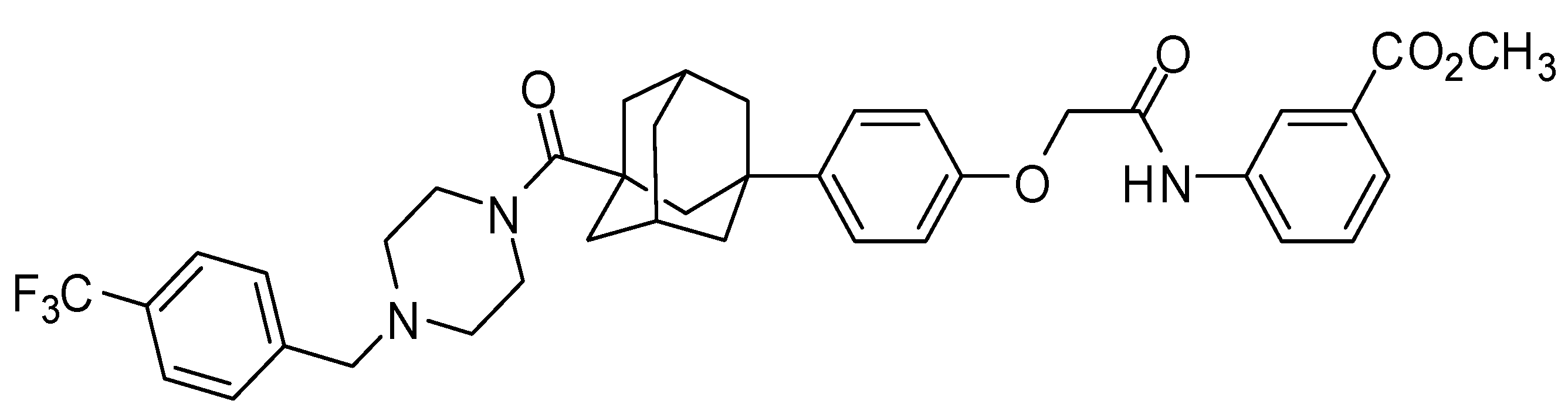
2.1. Chemicals
2.2. Cell Culture and Cell Viability Assay
2.3. Effect of LC478 on P-gp Mediated Efflux of Rhodamine-123, a P-gp Substate, in Caco-2 Cells
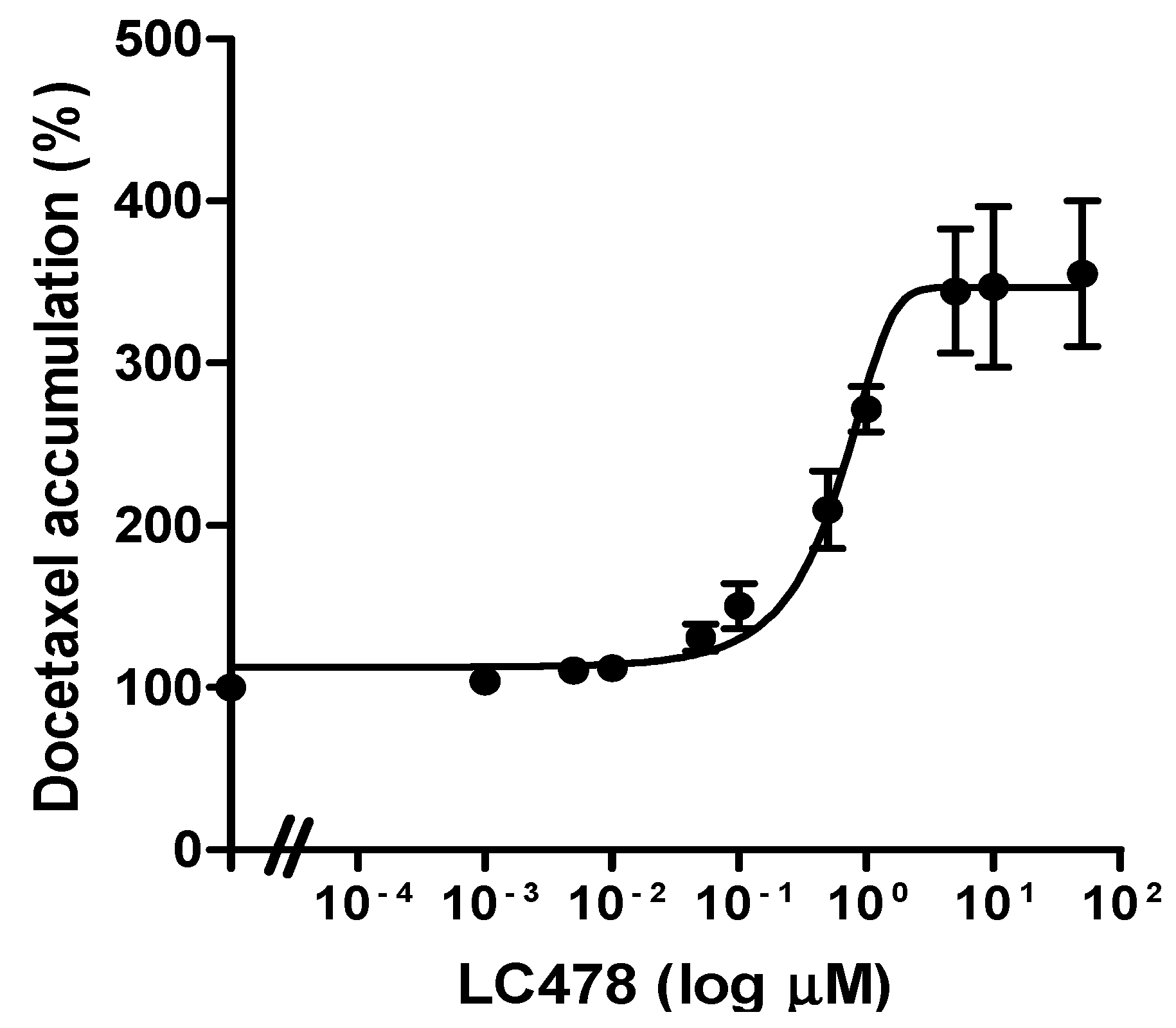
2.4. Effect of LC478 on P-gp Mediated Efflux of Docetaxel in Caco-2 Cells
2.5. Animals
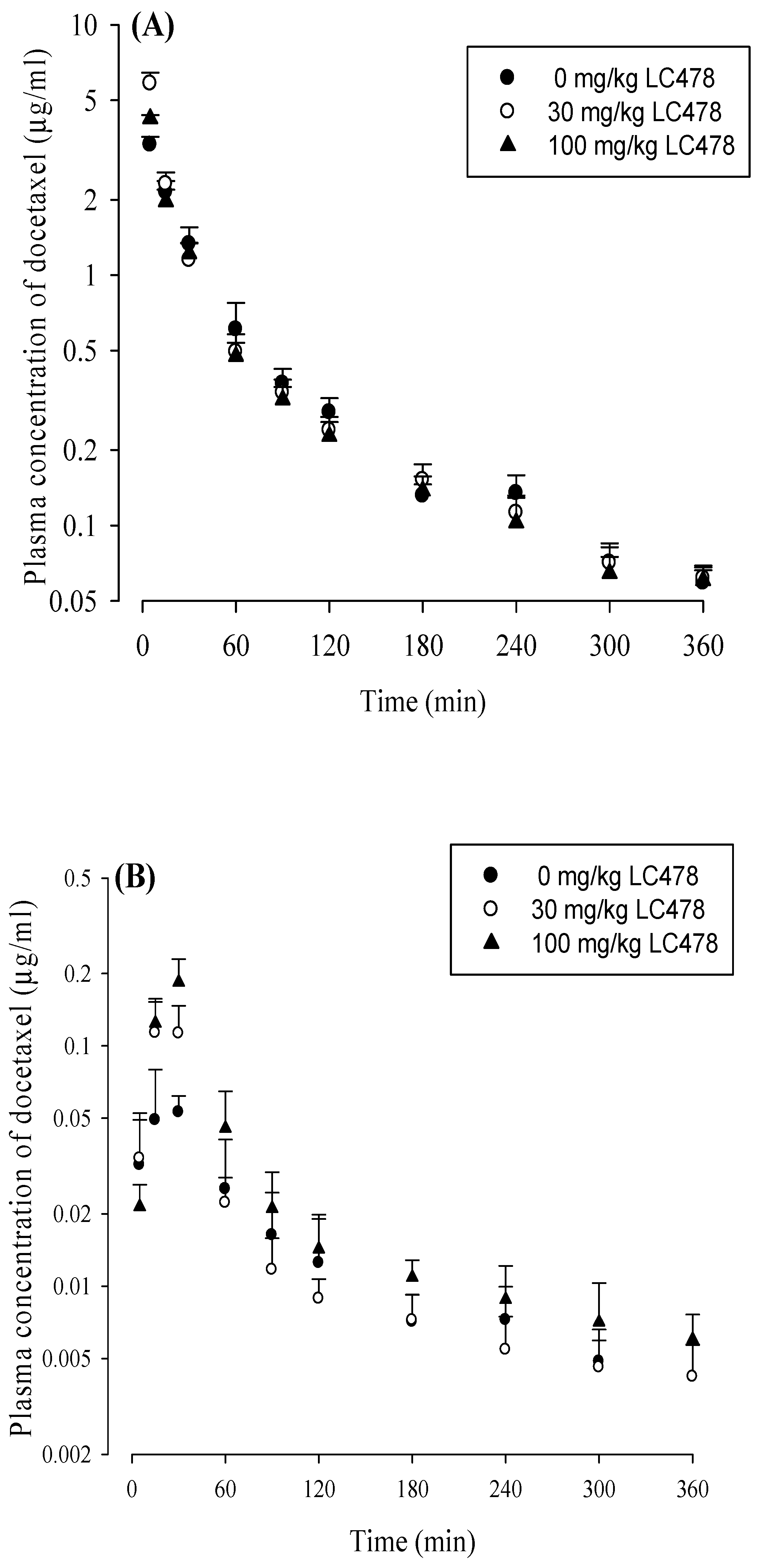
2.6. Pharmacokinetic Studies of Docetaxel with LC478
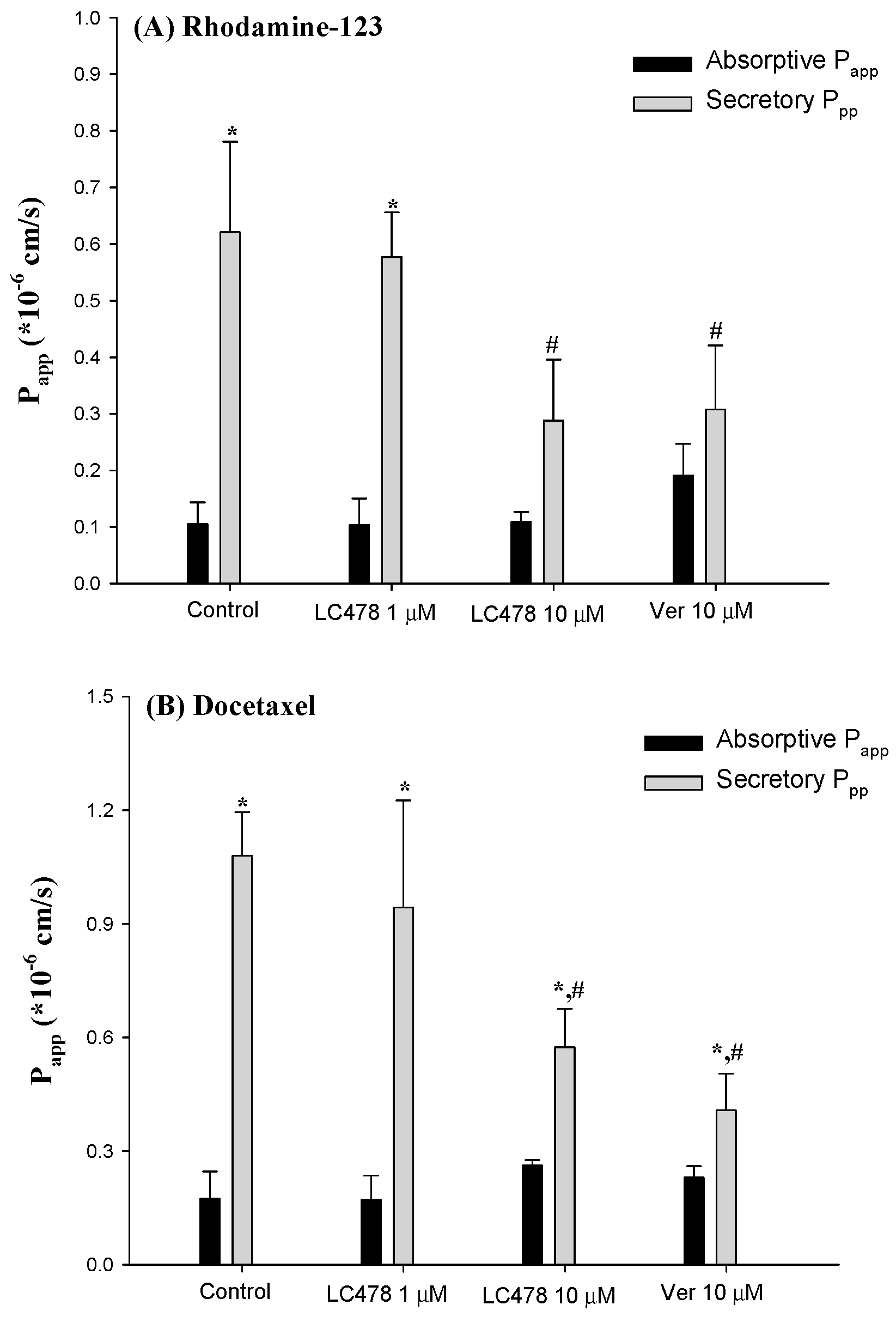
2.7. Effects of LC478 on Bi-Directional Transport of Rhodamine-123 or Docetaxel Across Rat Duodenum Using the Ussing Chamber
2.8. Effect of LC478 for Disappearance of Docetaxel in Rat Hepatic and Intestinal Microsomes
2.9. Effect of LC478 on Rat Plasma Protein Binding of Docetaxel with LC478
2.10. Analytical Methods of Docetaxel and LC478
2.11. Statistical Analysis
3. Results
3.1. Effect of LC478 on Cace-2 Cell Viability
3.2. Effect of LC478 on P-gp Mediated Efflux of Rhodamine-123, a P-gp Substate, in Caco-2 Cells
3.3. Effect of LC478 on P-gp Mediated Efflux of Docetaxel in Caco-2 Cells
3.4. Effect of LC478 on Pharmacokinetics of Docetaxel
3.5. Effect of LC478 on Bi-Directional Transport of Rhodamine-123 or Docetaxel across Small Intestine in Rats
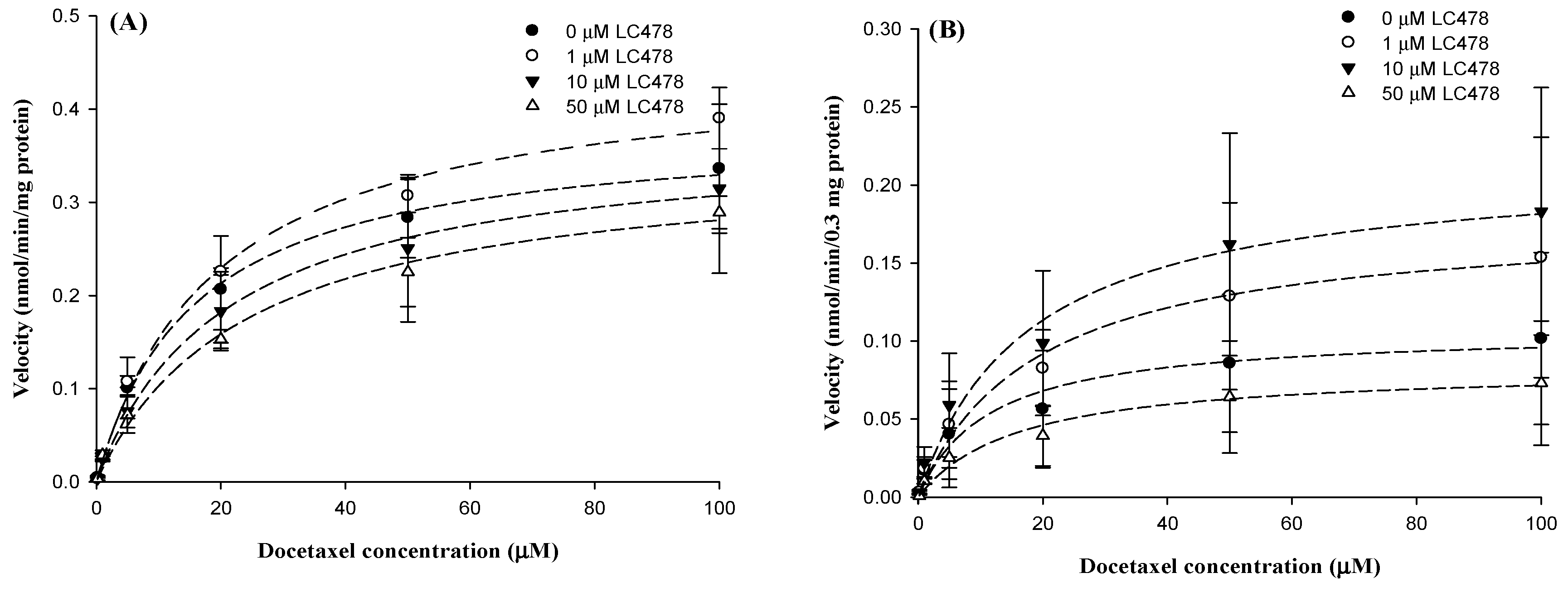
3.6. Effect of LC478 for Docetaxel Metabolism in Liver and Small Intestine
3.7. Effect of LC478 on Rat Plasma Protein Binding of Docetaxel Using Equilibrium Dialysis
4. Discussion
0.365 = Funabs + (0.0101 × 0.0229) 30 mg/kg oral LC478
0.200 = Funabs + (0.0176 × 0.0396) 100 mg/kg oral LC478
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Clarke, S.J.; Rivory, L.P. Clinical pharmacokinetics of docetaxel. Clin. Pharmacokinet. 1999, 36, 99–144. [Google Scholar] [CrossRef] [PubMed]
- van Waterschoot, R.A.; Lagas, J.S.; Wagenaar, E.; van der Kruijssen, C.M.; van Herwaarden, A.E.; Song, J.Y.; Rooswinkel, R.W.; van Tellingen, O.; Rosing, H.; et al. Absence of both cytochrome P450 3A and P-glycoprotein dramatically increases docetaxel oral bioavailability and risk of intestinal toxicity. Cancer Res. 2009, 23, 8996–9002. [Google Scholar] [CrossRef] [PubMed]
- Gelderblom, H.; Verweij, J.; Nooter, K.; Sparreboom, A. Cremophor EL: The drawbacks and advantages of vehicle selection for drug formulation. Eur. J. Cancer 2001, 37, 1590–1598. [Google Scholar] [CrossRef]
- Marks, D.H.; Qureshi, A.; Friedman, A. Evaluation of Prevention Interventions for Taxane-Induced Dermatologic Adverse Events: A Systematic Review. JAMA Dermatol. 2018, 154, 1465–1472. [Google Scholar] [CrossRef]
- Sohail, M.F.; Rehman, M.; Sarwar, H.S.; Naveed, S.; Salman, O.; Bukhari, N.I.; Hussain, I.; Webster, T.J.; Shahnaz, G. Advancements in the oral delivery of Docetaxel: Challenges, current state-of-the-art and future trends. Int. J. Nanomed. 2018, 13, 3145–3161. [Google Scholar] [CrossRef]
- DeMario, M.D.; Ratain, M.J. Oral chemotherapy: Rationale and future directions. J. Clin. Oncol. 1998, 16, 2557–2567. [Google Scholar] [CrossRef] [PubMed]
- Baker, S.D.; Sparreboom, A.; Verweij, J. Clinical pharmacokinetics of docetaxel: Recent developments. Clin. Pharmacokinet. 2006, 45, 235–252. [Google Scholar] [CrossRef]
- Cho, H.J.; Park, J.W.; Yoon, I.S.; Kim, D.D. Surface-modified solid lipid nanoparticles for oral delivery of docetaxel: Enhanced intestinal absorption and lymphatic uptake. Int. J. Nanomed. 2014, 9, 495–504. [Google Scholar]
- Dong, Y.; Feng, S.S. Poly(d,l-lactide-co-glycolide)/montmorillonite nanoparticles for oral delivery of anticancer drugs. Biomaterials 2005, 26, 6068–6076. [Google Scholar] [CrossRef]
- Ben Reguiga, M.; Bonhomme-Faivre, L.; Farinotti, R. Bioavailability and tissular distribution of docetaxel, a P-glycoprotein substrate, are modified by interferon-alpha in rats. J. Pharm. Pharmacol. 2007, 59, 401–408. [Google Scholar] [CrossRef]
- Malingré, M.M.; Richel, D.J.; Beijnen, J.H.; Rosing, H.; Koopman, F.J.; Ten Bokkel Huinink, W.W.; Schot, M.E.; Schellens, J.H. Co-administration of cyclosporine strongly enhances the oral bioavailability of docetaxel. J. Clin. Oncol. 2001, 19, 1160–1166. [Google Scholar] [CrossRef] [PubMed]
- Shirakawa, K.; Takara, K.; Tanigawara, Y.; Aoyama, N.; Kasuga, M.; Komada, F.; Sakaeda, T.; Okumura, K. Interaction of docetaxel (“Taxotere”) with human P-glycoprotein. Jpn. J. Cancer Res. 1999, 90, 1380–1386. [Google Scholar] [CrossRef] [PubMed]
- Sparreboom, A.; van Tellingen, O.; Nooijen, W.J.; Beijnen, J.H. Preclinical pharmacokinetics of paclitaxel and docetaxel. Anticancer Drugs 1998, 9, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Varma, M.V.; Ashokraj, Y.; Dey, C.S.; Panchagnula, R. P-glycoprotein inhibitors and their screening: A perspective from bioavailability enhancement. Pharmacol. Res. 2003, 48, 347–359. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 12, 2615–2623. [Google Scholar] [CrossRef]
- Kenmotsu, H.; Tanigawara, Y. Pharmacokinetics, dynamics and toxicity of docetaxel: Why the Japanese dose differs from the Western dose. Cancer Sci. 2015, 106, 497–504. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Hendrikx, J.J.; Rottenberg, S.; Schellens, J.H.; Beijnen, J.H.; Huitema, A.D. Development of a Tumour Growth Inhibition Model to Elucidate the Effects of Ritonavir on Intratumoural Metabolism and Anti-tumour Effect of Docetaxel in a Mouse Model for Hereditary Breast Cancer. AAPS J. 2016, 18, 362–371. [Google Scholar] [CrossRef]
- Chen, S.; Sutiman, N.; Zhang, C.Z.; Yu, Y.; Lam, S.; Khor, C.C.; Chowbay, B. Pharmacogenetics of irinotecan, doxorubicin and docetaxel transporters in Asian and Caucasian cancer patients: A comparative review. Drug Metab. Rev. 2016, 48, 502–540. [Google Scholar] [CrossRef] [PubMed]
- Chung, F.S.; Santiago, J.S.; Jesus, M.F.; Trinidad, C.V.; See, M.F. Disrupting P-glycoprotein function in clinical settings: What can we learn from the fundamental aspects of this transporter? Am. J. Cancer Res. 2016, 6, 1583–1598. [Google Scholar]
- Saneja, A.; Khare, V.; Alam, N.; Dubey, R.D.; Gupta, P.N. Advances in P-glycoprotein-based approaches for delivering anticancer drugs: Pharmacokinetic perspective and clinical relevance. Expert Opin. Drug Deliv. 2014, 11, 121–138. [Google Scholar] [CrossRef]
- Sun, X.; Li, J.; Guo, C.; Xing, H.; Xu, J.; Wen, Y.; Qiu, Z.; Zhang, Q.; Zheng, Y.; Chen, X.; et al. Pharmacokinetic effects of curcumin on docetaxel mediated by OATP1B1, OATP1B3 and CYP450s. Drug Metab. Pharmacokinet. 2016, 31, 269–275. [Google Scholar] [CrossRef]
- Fojo, A.T.; Ueda, K.; Slamon, D.J.; Poplack, D.G.; Gottesman, M.M.; Pastan, I. Expression of a multidrug-resistance gene in human tumors and tissues. Proc. Natl. Acad. Sci. USA 1987, 84, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Saaby, L.; Brodin, B. A critical view on in vitro analysis of P-glycoprotein (P-gp) transport kinetics. J. Pharm. Sci. 2017, 106, 2257–2264. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Li, Y.; Yu, H.; Zhang, L.; Hou, T. Computational models for predicting substrates or inhibitors of P-glycoprotein. Drug Discov. Today 2012, 17, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Dou, L.; Mai, Y.; Madla, C.M.; Orlu, M.; Basit, A.W. P-glycoprotein expression in the gastrointestinal tract of male and female rats is influenced differently by food. Eur. J. Pharm. Sci. 2018, 123, 569–575. [Google Scholar] [CrossRef] [PubMed]
- Kwak, J.O.; Lee, S.H.; Lee, G.S.; Kim, M.S.; Ahn, Y.G.; Lee, J.H.; Kim, S.W.; Kim, K.H.; Lee, M.G. Selective inhibition of MDR1 (ABCB1) by HM30181 increases oral bioavailability and therapeutic efficacy of paclitaxel. Eur. J. Pharmacol. 2010, 627, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Kuppens, I.E.; Bosch, T.M.; van Maanen, M.J.; Rosing, H.; Fitzpatrick, A.; Beijnen, J.H.; Schellens, J.H. Oral bioavailability of docetaxel in combination with OC144-093 (ONT-093). Cancer Chemother. Pharmacol. 2005, 55, 72–78. [Google Scholar] [CrossRef]
- Oostendorp, R.L.; Huitema, A.; Rosing, H.; Jansen, R.S.; Ter Heine, R.; Keessen, M.; Beijnen, J.H.; Schellens, J.H. Coadministration of ritonavir strongly enhances the apparent oral bioavailability of docetaxel in patients with solid tumors. Clin. Cancer Res. 2009, 15, 4228–4233. [Google Scholar] [CrossRef]
- Engels, F.K.; Sparreboom, A.; Mathot, R.A.; Verweij, J. Potential for improvement of docetaxel-based chemotherapy: A pharmacological review. Br. J. Cancer 2005, 93, 173–177. [Google Scholar] [CrossRef]
- Prueksaritanont, T.; Tatosian, D.A.; Chu, X.; Railkar, R.; Evers, R.; Chavez-Eng, C.; Lutz, R.; Zeng, W.; Yabut, J.; Chan, G.H.; et al. Validation of a microdose probe drug cocktail for clinical drug interaction assessments for drug transporters and CYP3A. Clin. Pharmacol. Ther. 2017, 101, 519–530. [Google Scholar] [CrossRef]
- Soucek, P.; Gut, I. Cytochromes P-450 in rats: Structures, functions, properties and relevant human forms. Xenobiotica 1992, 22, 83–103. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.D.; Kim, D.H.; Sung, J.H.; Yong, C.S.; Choi, H.G. Enhanced oral bioavailability of docetaxel in rats by four consecutive days of pre-treatment with curcumin. Int. J. Pharm. 2010, 399, 116–120. [Google Scholar] [CrossRef] [PubMed]
- Min, K.H.; Xia, Y.; Kim, E.K.; Kaur, N.; Kim, E.S.; Kim, D.K.; Jung, H.Y.; Choi, Y.; Park, M.K.; Min, Y.K.; et al. A novel class of highly potent multidrug resistance reversal agents: Disubstituted adamantyl derivatives. Bioorg. Med. Chem. Lett. 2009, 19, 5376–5379. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Huang, M.; Guan, S.; Bi, H.C.; Pan, Y.; Duan, W.; Chan, S.Y.; Chen, X.; Hong, Y.H.; Bian, J.S.; et al. A mechanistic study of the intestinal absorption of cryptotanshinone, the major active constituent of Salvia miltiorrhiza. J. Pharmacol. Exp. Ther. 2006, 317, 1285–1294. [Google Scholar] [CrossRef] [PubMed]
- Bae, J.K.; Kim, Y.J.; Chae, H.S.; Kim, D.Y.; Choi, H.S.; Chin, Y.W.; Choi, Y.H. Korean red ginseng extract enhances paclitaxel distribution to mammary tumors and its oral bioavailability by P-glycoprotein inhibition. Xenobiotica 2017, 47, 450–459. [Google Scholar] [CrossRef] [PubMed]
- Bentz, J.; O’Connor, M.P.; Bednarczyk, D.; Coleman, J.; Lee, C.; Palm, J.; Pak, Y.A.; Perloff, E.S.; Reyner, E.; Balimane, P.; et al. Variability in P-glycoprotein inhibitory potency (IC₅₀) using various in vitro experimental systems: Implications for universal digoxin drug-drug interaction risk assessment decision criteria. Drug Metab. Dispos. 2013, 41, 1347–1366. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.H.; Yoon, I.; Kim, T.G.; Lee, M.G. Effects of cysteine on the pharmacokinetics of docetaxel in rats with protein-calorie malnutrition. Xenobiotica 2012, 42, 442–455. [Google Scholar] [CrossRef] [PubMed]
- Kishimoto, W.; Ishiguro, N.; Ludwig-Schwellinger, E.; Ebner, T.; Maeda, K.; Sugiyama, Y. Usefulness of A Model-Based Approach for Estimating In Vitro P-Glycoprotein Inhibition Potency in a Transcellular Transport Assay. J. Pharm. Sci. 2016, 105, 891–896. [Google Scholar] [CrossRef] [PubMed]
- Senthilkumari, S.; Velpandian, T.; Biswas, N.R.; Saxena, R.; Ghose, S. Evaluation of the modulation of P-glycoprotein on the intraocular disposition of its substrate in rabbits. Curr. Eye Res 2008, 33, 333–343. [Google Scholar] [CrossRef]
- Jia, J.X.; Wasan, K.M. Effects of monoglycerides on rhodamine 123 accumulation, estradiol 17 beta-D-glucuronide bidirectional transport and MRP2 protein expression within Caco-2 cells. J. Pharm. Pharm. Sci. 2008, 11, 45–62. [Google Scholar] [CrossRef] [PubMed]
- Gibaldi, M.; Perrier, D. Pharmacokinetics, 2nd ed.; Marcel-Dekker: New York, NY, USA, 1982; p. 494. [Google Scholar]
- Benet, L.Z.; Zia-Amirhosseini, P. Basic principles of pharmacokinetics. Toxicol. Pathol. 1995, 23, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Toutain, P.L.; Bousquet-Melou, A. Volumes of distribution. J. Vet. Pharmacol. Ther. 2004, 27, 441–453. [Google Scholar] [CrossRef] [PubMed]
- Saari, T.I.; Laine, K.; Leino, K.; Valtonen, M.; Neuvonen, P.J.; Olkkola, K.T. Effect of voriconazole on the pharmacokinetics and pharmacodynamics of intravenous and oral midazolam. Clin. Pharmacol. Ther. 2005, 79, 362–370. [Google Scholar] [CrossRef] [PubMed]
- Chiou, W.L.; Barve, A. Linear correlation of the fraction of oral dose absorbed of 64 drugs between humans and rats. Pharm. Res. 1998, 15, 1792–1795. [Google Scholar] [CrossRef]
- Werle, M.; Hoffer, M. Glutathione and thiolated chitosan inhibit multidrug resistance P-glycoprotein activity in excised small intestine. J. Control Release 2006, 111, 41–46. [Google Scholar] [CrossRef]
- Yerasi, N.; Vurimindi, H.; Devarakonda, K. Frog intestinal perfusion to evaluate drug permeability: Application to p-gp and cyp3a4 substrates. Front. Pharmacol. 2015, 14, 141. [Google Scholar] [CrossRef]
- Duggleby, R.G. Analysis of enzyme progress curves by nonlinear regression. Methods Enzymol. 1995, 249, 61–90. [Google Scholar]
- Lee, M.G.; Chiou, W.L. Evaluation of potential causes for the incomplete bioavailability of furosemide: Gastric first-pass metabolism. J. Pharmacokinet. Biopharm. 1983, 11, 623–640. [Google Scholar] [CrossRef]
- Stephens, R.H.; O‘Neill, C.A.; Warhurst, A.; Carlson, G.L.; Rowland, M.; Warhurst, G. Kinetic profiling of P-glycoprotein-mediated drug efflux in rat and human intestinal epithelia. J. Pharmacol. Exp. Ther. 2001, 296, 584–591. [Google Scholar]
- Lewis, D.F.V. Chapter 4. P450. Substrate specificity and metabolism. In Cytochromes P450, Structure, Function, and Mechanism, 2nd ed.; Taylor & Fancis: Bristol, PA, USA, 1996; p. 123. [Google Scholar]
- Takahashi, R.; Ma, S.; Yue, Q.; Kim-Kang, H.; Yi, Y.; Lyssikatos, J.P.; Regal, K.; Hunt, K.W.; Kallan, N.C.; Siu, M.; et al. Dose-dependent exposure and metabolism of GNE-892, a β-secretase inhibitor, in monkeys: Contributions by P450, AO, and P-gp. Eur. J. Drug Metab. Pharmacokinet. 2015, 40, 171–185. [Google Scholar] [CrossRef]
- Crommentuyn, K.M.; Schellens, J.H.; van den Berg, J.D.; Beijnen, J.H. In-vitro metabolism of anti-cancer drugs, methods and applications: Paclitaxel, docetaxel, tamoxifen and ifosfamide. Cancer Treat. Rev. 1998, 24, 345–366. [Google Scholar] [CrossRef]
- Wilkinson, G.R.; Shand, D.G. Commentary: A physiological approach to hepatic drug clearance. Clin. Pharmacol. Ther. 1975, 18, 377–390. [Google Scholar] [CrossRef] [PubMed]
- Davies, B.; Morris, T. Physiological parameters in laboratory animals and humans. Pharm. Res. 1993, 10, 1093–1095. [Google Scholar] [CrossRef] [PubMed]
- Jouan, E.; Le Vée, M.; Mayati, A.; Denizot, C.; Parmentier, Y.; Fardel, O. Evaluation of P-Glycoprotein Inhibitory Potential Using a Rhodamine 123 Accumulation Assay. Pharmaceutics 2016, 8, E12. [Google Scholar] [CrossRef] [PubMed]
- Poirier, A.; Cascais, A.C.; Bader, U.; Portmann, R.; Brun, M.E.; Walter, I.; Hillebrecht, A.; Ullah, M.; Funk, C. Calibration of in vitro multidrug resistance protein 1 substrate and inhibition assay as a basis to support the prediction of clinically relevant interactions in vivo. Drug Metab. Dispos. 2014, 42, 1411–1422. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | Papp (× 10−6 cm/s) | Efflux Ratio | |
|---|---|---|---|
| Absorptive (A to B) n = 3 | Secretory (B to A) n = 3 | ||
| Control | 0.231 ± 0.0388 | 1.50 ± 0.113 a | 6.49 |
| 1 μM LC478 | 0.226 ± 0.0429 | 1.03 ± 0.264 a,b | 4.56 |
| 10 μM LC478 | 0.245 ± 0.0469 | 0.555 ± 0.0564 a,b | 2.27 |
| 10 μM Verapamil | 0.270 ± 0.0634 | 0.634 ± 0.207 a,b | 2.35 |
| Parameter | 0 mg/kg | 30 mg/kg | 100 mg/kg |
|---|---|---|---|
| Intravenous Study | n = 6 | n = 6 | n = 6 |
| Body weight (g) | 302 ± 7.45 | 296 ± 4.49 | 301 ± 18.8 |
| AUClast (µg min/mL) | 253 ± 38.1 | 290 ± 46.6 | 268 ± 39.2 |
| AUCinf (µg min/mL) | 265 ± 39.0 | 303 ± 46.5 | 275 ± 40.2 |
| Terminal half-life (min) | 122 ± 36.6 | 144 ± 37.6 | 151 ± 34.5 |
| MRT (min) | 57.9 ± 4.63 | 58.0 ± 11.5 | 67.1 ± 14.6 |
| CL (mL/min/kg) | 78.4 ± 13.1 | 67.9 ± 11.7 | 70.4 ± 9.10 |
| CLR (mL/min/kg) a | 1.13 ± 0.431 | 1.06 ± 0.235 | 0.678 ± 0.253 |
| CLNR (mL/min/kg) | 77.2 ± 12.8 | 66.8 ± 11.4 | 69.7 ± 9.13 |
| Vss (L/kg) | 4.57 ± 1.03 | 4.02 ± 1.36 | 4.91 ± 1.44 |
| Ae0–24 h (% of dose) b | 1.42 ± 0.418 | 1.56 ± 0.129 | 0.979 ± 0.365 |
| GI24 h (% of dose) | 0.881 ± 0.411 | 1.01 ± 0.731 | 1.76 ± 0.958 |
| Frel (%) | - | 114 | 104 |
| Oral Study | n = 5 | n = 6 | n = 6 |
| Body weight (g) | 247 ± 2.74 | 244 ± 6.07 | 253 ± 8.54 |
| AUClast (µg min/mL) b | 4.97 ± 1.44 | 6.38 ± 1.60 | 9.67 ± 2.05 |
| AUCinf (µg min/mL) b | 5.21 ± 1.27 | 6.94 ± 1.74 | 10.9 ± 3.02 |
| Cmax (µg/mL) c | 0.0583 ± 0.0207 | 0.124 ± 0.446 | 0.184 ± 0.459 |
| Tmax (min) | 15 (5−60) | 30 (15−30) | 30 (15−30) |
| CL/F (mL/min/kg) b | 3039 ± 805 | 3167 ± 1021 | 1803 ± 515 |
| Vz/F (L/kg) b | 670 ± 170 | 794 ± 175 | 471 ± 117 |
| CLR (mL/min/kg) b | 3.50 ± 1.10 | 3.43 ± 1.15 | 1.79 ± 0.474 |
| Ae0–24 h (% of dose) | 0.103 ± 0.0510 | 0.104 ± 0.0362 | 0.0870 ± 0.0383 |
| GI24 h (% of dose) b | 41.7 ± 13.3 | 36.5 ± 12.6 | 20.0 ± 7.44 |
| Fabs (%) | 58.3 | 63.5 | 80.1 |
| F (%) | 1.97 | 2.29 | 3.96 |
| Frel (%) | - | 133 | 209 |
| Compounds | Papp (× 10−6 cm/s) | Efflux Ratio | |
|---|---|---|---|
| Absorptive (M to S) | Secretory (S to M) | ||
| Rhodamine-123 | n = 3 | n = 3 | |
| Control | 0.105 ± 0.0387 | 0.629 ± 0.160 a | 5.99 |
| 1 µM LC478 | 0.103 ± 0.0472 | 0.577 ± 0.0791 a | 5.60 |
| 10 µM LC478 | 0.188 ± 0.0177 | 0.288 ± 0.108 b | 1.53 |
| 10 µM verapamil | 0.191 ± 0.0562 | 0.308 ± 0.113 b | 1.61 |
| Docetaxel | n = 3 | n = 3 | |
| Control | 0.174 ± 0.0721 | 1.08 ± 0.115 a | 6.21 |
| 1 µM LC478 | 0.171 ± 0.0639 | 0.943 ± 0.283 a | 5.51 |
| 10 µM LC478 | 0.262 ± 0.0139 b | 0.574 ± 0.101 a,b | 2.19 |
| 10 µM verapamil | 0.230 ± 0.0303 b | 0.408 ± 0.0959 a,b | 1.77 |
| Parameters | Concentrations of LC478 (µM) | |||
|---|---|---|---|---|
| 0 | 1 | 5 | 10 | |
| Hepatic microcomes | n = 3 | n = 3 | n = 3 | n = 3 |
| Km (µM) | 20.2 ± 2.78 | 20.6 ± 2.09 | 21.4 ± 2.65 | 23.1 ± 1.74 |
| Vmax (nmol/min/mg protein) | 0.464 ± 0.107 | 0.461 ± 0.0474 | 0.438 ± 0.0578 | 0.427 ± 0.0411 |
| CLint (µL/min/mg protein) | 0.0229 ± 0.00204 | 0.0226 ± 0.00454 | 0.0206 ± 0.00280 | 0.0187 ± 0.00314 |
| Intestinal microsomes | n = 3 | n = 3 | n = 3 | n = 3 |
| Km (µM) | 18.1 ± 1.20 | 18.3 ± 7.07 | 20.2 ± 8.94 | 17.4 ± 1.01 |
| Vmax (nmol/min/0.3 mg protein) | 0.109 ± 0.0502 | 0.133 ± 0.0343 | 0.133 ± 0.0737 | 0.105 ± 0.0355 |
| CLint (µL/min/0.3 mg protein) | 0.00603 ± 0.00291 | 0.00767 ± 0.00158 | 0.00757 ± 0.00559 | 0.00688 ± 0.00349 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, S.Y.; Lu, Q.; Lee, K.; Choi, Y.H. LC478, a Novel Di-Substituted Adamantyl Derivative, Enhances the Oral Bioavailability of Docetaxel in Rats. Pharmaceutics 2019, 11, 135. https://doi.org/10.3390/pharmaceutics11030135
Han SY, Lu Q, Lee K, Choi YH. LC478, a Novel Di-Substituted Adamantyl Derivative, Enhances the Oral Bioavailability of Docetaxel in Rats. Pharmaceutics. 2019; 11(3):135. https://doi.org/10.3390/pharmaceutics11030135
Chicago/Turabian StyleHan, Seung Yon, Qili Lu, Kyeong Lee, and Young Hee Choi. 2019. "LC478, a Novel Di-Substituted Adamantyl Derivative, Enhances the Oral Bioavailability of Docetaxel in Rats" Pharmaceutics 11, no. 3: 135. https://doi.org/10.3390/pharmaceutics11030135