Pharmacokinetics and Pharmacodynamics Modeling and Simulation Systems to Support the Development and Regulation of Liposomal Drugs


Abstract
:1. Introduction
2. Liposomal Drugs Approved for Use in Humans
2.1. Liposomal Drugs for Cancer Treatment
2.2. Liposomal Drugs for Other Indications
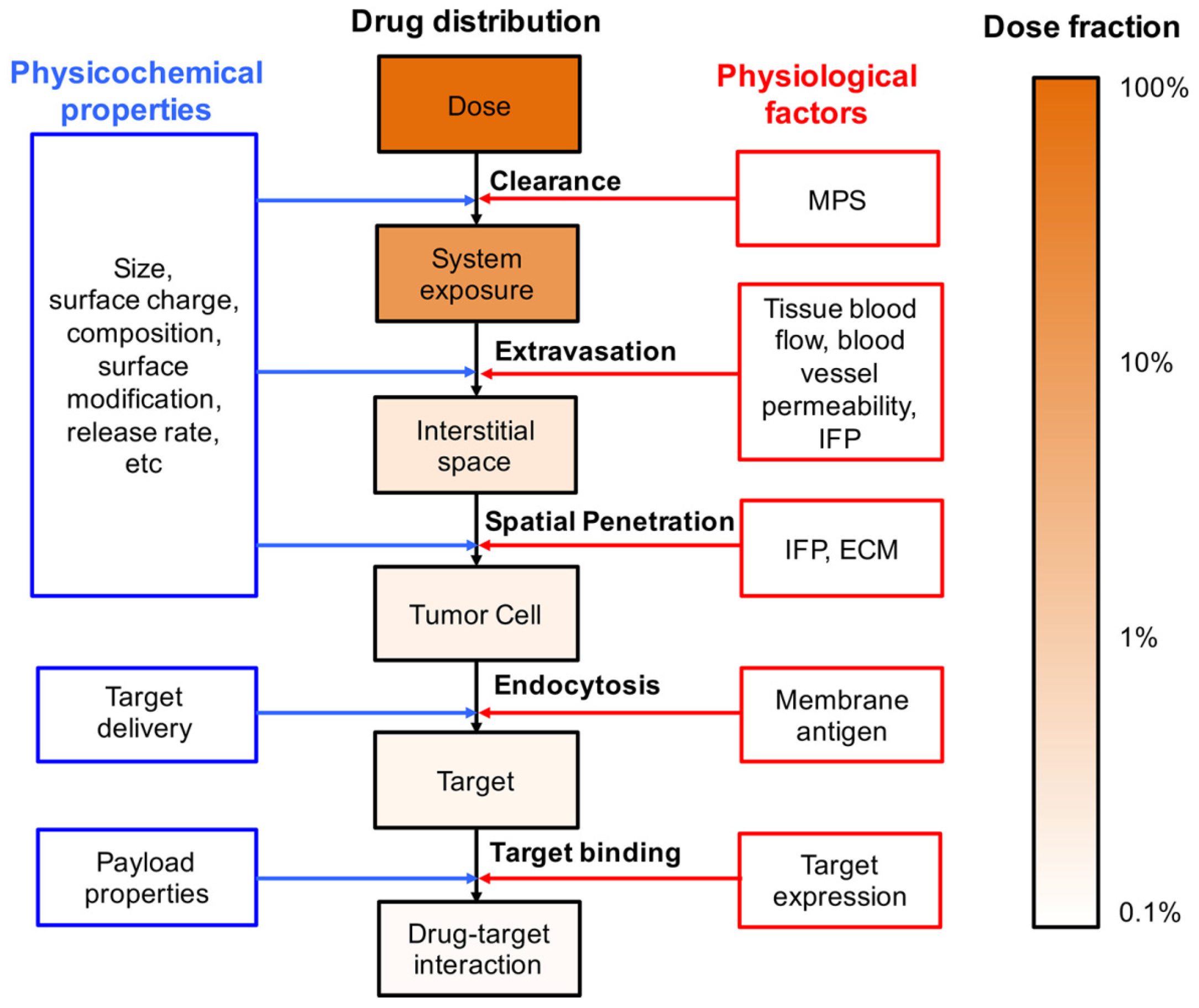
3. Physiological Barriers to the Targeted Delivery of Liposomes
3.1. Systemic Clearance
3.2. Extravasation
3.3. Spatial Penetration in the Tumor Tissues
3.4. Intracellular Uptake
4. Challenges to Liposomal Drug Development and a Regulatory Review
4.1. The Identification of Critical Quantity Attributes
4.2. The Gap in Translation from Animal Models to the Clinic
4.3. The Quantitative Pharmacokinetic–Pharmacodynamic Relationship
5. The Application of Pharmacokinetic–Pharmacodynamic Modeling and Simulation Systems in Liposomal Drug Delivery
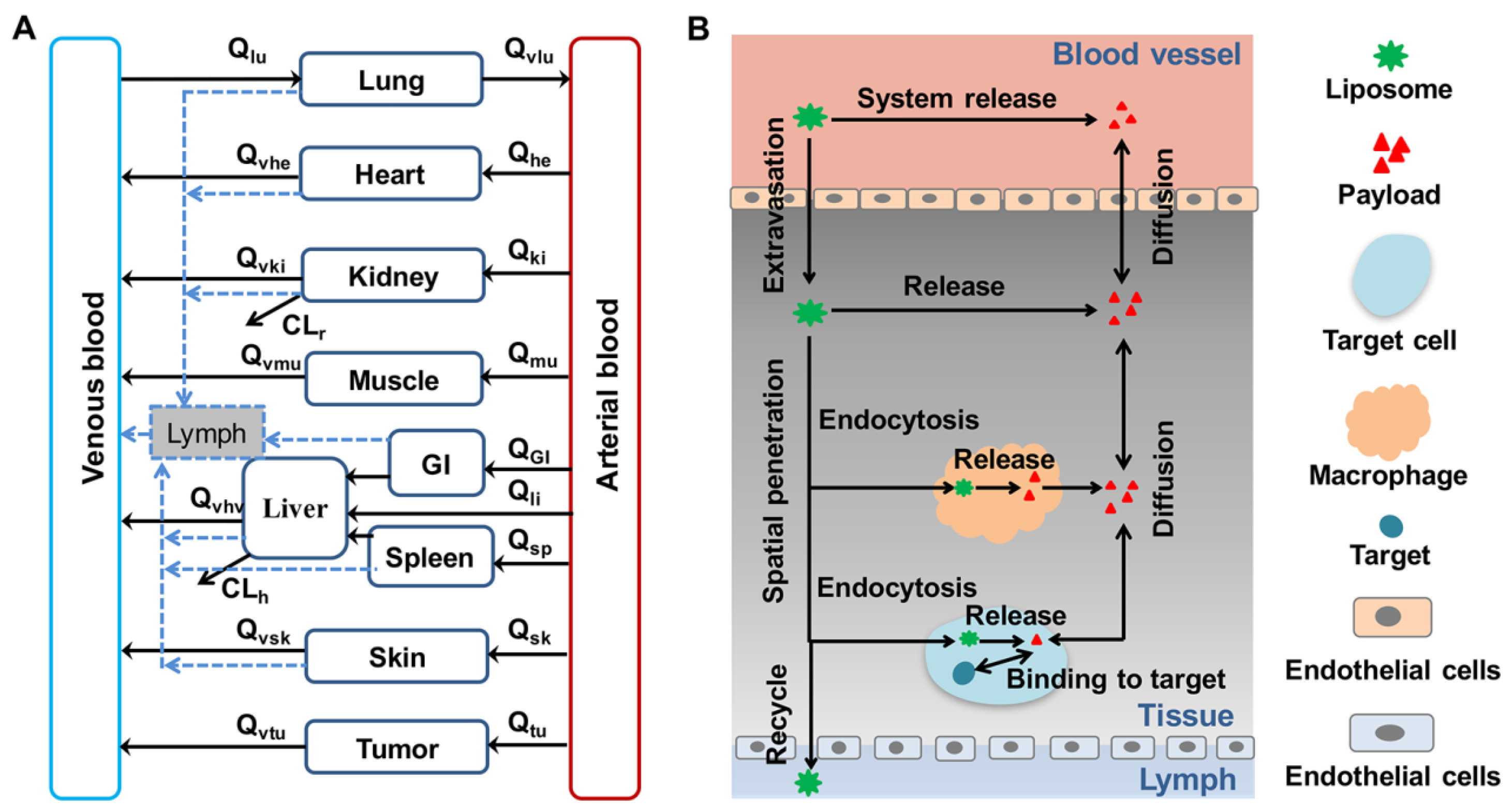
5.1. Physiologically-Based Pharmacokinetic Modeling and Simulation
5.1.1. Simplified PBPK Model
5.1.2. Whole-Body PBPK Model
5.2. PK–PD Modeling System with Spatiotemporal Characterization
5.3. Combination of In Vitro Study and the PK–PD Modeling System
6. Perspective and Future Direction
Funding
Acknowledgments
Conflicts of Interest
References
- Bangham, A.D.; Horne, R.W. Negatie staining of phospholipids and their structural modification by surface-active agents as observed in the electron microscope. J. Mol. Biol. 1964, 8, 660–668. [Google Scholar] [CrossRef]
- Bangham, A.D.; Standish, M.M.; Watkins, J.C. Diffusion of univalent ions across the lamellae of swollen phospholipids. J. Mol. Biol. 1965, 13, 238–252. [Google Scholar] [CrossRef]
- Gregoriadis, G.; Ryman, B.E. Liposomes as carriers of enzymes or drugs: A new approach to the treatment of storage diseases. Biochem. J. 1971, 124, 58p. [Google Scholar] [CrossRef] [PubMed]
- Gregoriadis, G. Drug entrapment in liposomes. FEBS Lett. 1973, 36, 292–296. [Google Scholar] [CrossRef] [Green Version]
- Gregoriadis, G. The carrier potential of liposomes in biology and medicine (second of two parts). N. Engl. J. Med. 1976, 295, 765–770. [Google Scholar] [CrossRef] [PubMed]
- Gregoriadis, G. The carrier potential of liposomes in biology and medicine (first of two parts). N. Engl. J. Med. 1976, 295, 704–710. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Tsukagoshi, S.; Sakurai, Y. Enhancement of the cancer chemotherapeutic effect of cytosine arabinoside entrapped in liposomes on mouse leukemia L-1210. Gann 1975, 66, 719–720. [Google Scholar] [PubMed]
- Alving, C.R.; Steck, E.A.; Chapman, W.L., Jr.; Waits, V.B.; Hendricks, L.D.; Swartz, G.M., Jr.; Hanson, W.L. Therapy of leishmaniasis: Superior efficacies of liposome-encapsulated drugs. Proc. Natl. Acad. Sci. USA 1978, 75, 2959–2963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez-Berestein, G.; Fainstein, V.; Hopfer, R.; Mehta, K.; Sullivan, M.P.; Keating, M.; Rosenblum, M.G.; Mehta, R.; Luna, M.; Hersh, E.M.; et al. Liposomal amphotericin B for the treatment of systemic fungal infections in patients with cancer: a preliminary study. J. Infect. Dis. 1985, 151, 704–710. [Google Scholar] [CrossRef] [PubMed]
- Gabizon, A.; Goren, D.; Fuks, Z.; Barenholz, Y.; Dagan, A.; Meshorer, A. Enhancement of adriamycin delivery to liver metastatic cells with increased tumoricidal effect using liposomes as drug carriers. Cancer Res. 1983, 43, 4730–4735. [Google Scholar] [PubMed]
- Maeda, H. Toward a full understanding of the EPR effect in primary and metastatic tumors as well as issues related to its heterogeneity. Adv. Drug Deliv. Rev. 2015, 91, 3–6. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, Y.; Maeda, H. A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986, 46, 6387–6392. [Google Scholar] [PubMed]
- Barenholz, Y. Doxil(R)--the first FDA-approved nano-drug: Lessons learned. J. Control. Release 2012, 160, 117–134. [Google Scholar] [CrossRef] [PubMed]
- Bulbake, U.; Doppalapudi, S.; Kommineni, N.; Khan, W. Liposomal Formulations in Clinical Use: An Updated Review. Pharmaceutics 2017, 9, 12. [Google Scholar] [CrossRef] [PubMed]
- Lucas, A.T.; Madden, A.J.; Zamboni, W.C. Formulation and physiologic factors affecting the pharmacology of carrier-mediated anticancer agents. Expert Opin. Drug Metab. Toxicol. 2015, 11, 1419–1433. [Google Scholar] [CrossRef] [PubMed]
- Garralda, E.; Dienstmann, R.; Tabernero, J. Pharmacokinetic/Pharmacodynamic Modeling for Drug Development in Oncology. Am. Soc. Clin. Oncol. Educ. Book 2017, 37, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Zou, P.; Tyner, K.; Lee, S. Physiologically Based Pharmacokinetic (PBPK) Modeling of Pharmaceutical Nanoparticles. AAPS J. 2017, 19, 26–42. [Google Scholar] [CrossRef] [PubMed]
- Yuan, D.; He, H.; Wu, Y.; Fan, J.; Cao, Y. Physiologically Based Pharmacokinetic Modeling of Nanoparticles. J. Pharm. Sci. 2019, 108, 58–72. [Google Scholar] [CrossRef] [PubMed]
- Weiss, R.B. The anthracyclines: will we ever find a better doxorubicin? Semin. Oncol. 1992, 19, 670–686. [Google Scholar] [PubMed]
- Chatterjee, K.; Zhang, J.; Honbo, N.; Karliner, J.S. Doxorubicin cardiomyopathy. Cardiology 2010, 115, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Octavia, Y.; Tocchetti, C.G.; Gabrielson, K.L.; Janssens, S.; Crijns, H.J.; Moens, A.L. Doxorubicin-induced cardiomyopathy: From molecular mechanisms to therapeutic strategies. J. Mol. Cell. Cardiol. 2012, 52, 1213–1225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, T.M.; Martin, F.J. Advantages of liposomal delivery systems for anthracyclines. Semin. Oncol. 2004, 31, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Gabizon, A.; Catane, R.; Uziely, B.; Kaufman, B.; Safra, T.; Cohen, R.; Martin, F.; Huang, A.; Barenholz, Y. Prolonged circulation time and enhanced accumulation in malignant exudates of doxorubicin encapsulated in polyethylene-glycol coated liposomes. Cancer Res. 1994, 54, 987–992. [Google Scholar] [PubMed]
- Mross, K.; Niemann, B.; Massing, U.; Drevs, J.; Unger, C.; Bhamra, R.; Swenson, C.E. Pharmacokinetics of liposomal doxorubicin (TLC-D99; Myocet) in patients with solid tumors: an open-label, single-dose study. Cancer Chemother. Pharmacol. 2004, 54, 514–524. [Google Scholar] [CrossRef] [PubMed]
- Batist, G. Cardiac safety of liposomal anthracyclines. Cardiovasc. Toxicol. 2007, 7, 72–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batist, G.; Ramakrishnan, G.; Rao, C.S.; Chandrasekharan, A.; Gutheil, J.; Guthrie, T.; Shah, P.; Khojasteh, A.; Nair, M.K.; Hoelzer, K.; et al. Reduced cardiotoxicity and preserved antitumor efficacy of liposome-encapsulated doxorubicin and cyclophosphamide compared with conventional doxorubicin and cyclophosphamide in a randomized, multicenter trial of metastatic breast cancer. J. Clin. Oncol. 2001, 19, 1444–1454. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, M.E.; Wigler, N.; Inbar, M.; Rosso, R.; Grischke, E.; Santoro, A.; Catane, R.; Kieback, D.G.; Tomczak, P.; Ackland, S.P.; et al. Reduced cardiotoxicity and comparable efficacy in a phase III trial of pegylated liposomal doxorubicin HCl (CAELYX/Doxil) versus conventional doxorubicin for first-line treatment of metastatic breast cancer. Ann. Oncol. 2004, 15, 440–449. [Google Scholar]
- Bhowmik, S.; Bhowmick, S.; Maiti, K.; Chakra, A.; Shahi, P.; Jain, D.; Rajamannar, T. Two multicenter Phase I randomized trials to compare the bioequivalence and safety of a generic doxorubicin hydrochloride liposome injection with Doxil((R)) or Caelyx((R)) in advanced ovarian cancer. Cancer Chemother. Pharmacol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Mamidi, R.N.; Weng, S.; Stellar, S.; Wang, C.; Yu, N.; Huang, T.; Tonelli, A.P.; Kelley, M.F.; Angiuoli, A.; Fung, M.C. Pharmacokinetics, efficacy and toxicity of different pegylated liposomal doxorubicin formulations in preclinical models: is a conventional bioequivalence approach sufficient to ensure therapeutic equivalence of pegylated liposomal doxorubicin products? Cancer Chemother. Pharmacol. 2010, 66, 1173–1184. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.A.; Mathew, L.; Burney, M.; Nyshadham, P.; Coleman, R.L. Equivalency challenge: Evaluation of Lipodox(R) as the generic equivalent for Doxil(R) in a human ovarian cancer orthotropic mouse model. Gynecol. Oncol. 2016, 141, 357–363. [Google Scholar] [CrossRef] [PubMed]
- Burade, V.; Bhowmick, S.; Maiti, K.; Zalawadia, R.; Ruan, H.; Thennati, R. Lipodox(R) (generic doxorubicin hydrochloride liposome injection): In vivo efficacy and bioequivalence versus Caelyx(R) (doxorubicin hydrochloride liposome injection) in human mammary carcinoma (MX-1) xenograft and syngeneic fibrosarcoma (WEHI 164) mouse models. BMC Cancer 2017, 17, 405. [Google Scholar] [CrossRef]
- Burade, V.; Bhowmick, S.; Maiti, K.; Zalawadia, R.; Jain, D.; Rajamannar, T. Comparative plasma and tissue distribution of Sun Pharma’s generic doxorubicin HCl liposome injection versus Caelyx((R)) (doxorubicin HCl liposome injection) in syngeneic fibrosarcoma-bearing BALB/c mice and Sprague-Dawley rats. Cancer Chemother. Pharmacol. 2017, 79, 899–913. [Google Scholar] [CrossRef] [PubMed]
- Petre, C.E.; Dittmer, D.P. Liposomal daunorubicin as treatment for Kaposi’s sarcoma. Int. J. Nanomed. 2007, 2, 277–288. [Google Scholar]
- Forssen, E.A.; Coulter, D.M.; Proffitt, R.T. Selective in vivo localization of daunorubicin small unilamellar vesicles in solid tumors. Cancer Res. 1992, 52, 3255–3261. [Google Scholar] [PubMed]
- Gill, P.S.; Espina, B.M.; Muggia, F.; Cabriales, S.; Tulpule, A.; Esplin, J.A.; Liebman, H.A.; Forssen, E.; Ross, M.E.; Levine, A.M. Phase I/II clinical and pharmacokinetic evaluation of liposomal daunorubicin. J. Clin. Oncol. 1995, 13, 996–1003. [Google Scholar] [CrossRef] [PubMed]
- Fumagalli, L.; Zucchetti, M.; Parisi, I.; Vigano, M.G.; Zecca, B.; Careddu, A.; D’Incalci, M.; Lazzarin, A. The pharmacokinetics of liposomal encapsulated daunorubicin are not modified by HAART in patients with HIV-associated Kaposi’s sarcoma. Cancer Chemother. Pharmacol. 2000, 45, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.C.; Fathi, A.T.; Brunner, A.M. Reformulating acute myeloid leukemia: Liposomal cytarabine and daunorubicin (CPX-351) as an emerging therapy for secondary AML. Onco. Targets Ther. 2018, 11, 3425–3434. [Google Scholar] [CrossRef] [PubMed]
- Tardi, P.; Johnstone, S.; Harasym, N.; Xie, S.; Harasym, T.; Zisman, N.; Harvie, P.; Bermudes, D.; Mayer, L. In vivo maintenance of synergistic cytarabine:daunorubicin ratios greatly enhances therapeutic efficacy. Leuk. Res. 2009, 33, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Krauss, A.C.; Gao, X.; Li, L.; Manning, M.L.; Patel, P.; Fu, W.; Janoria, K.G.; Gieser, G.; Bateman, D.A.; Przepiorka, D.; et al. FDA Approval Summary: (Daunorubicin and Cytarabine) Liposome for Injection for the Treatment of Adults with High-Risk Acute Myeloid Leukemia. Clin. Cancer Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Lim, W.S.; Tardi, P.G.; Dos Santos, N.; Xie, X.; Fan, M.; Liboiron, B.D.; Huang, X.; Harasym, T.O.; Bermudes, D.; Mayer, L.D. Leukemia-selective uptake and cytotoxicity of CPX-351, a synergistic fixed-ratio cytarabine:daunorubicin formulation, in bone marrow xenografts. Leuk. Res. 2010, 34, 1214–1223. [Google Scholar] [CrossRef] [PubMed]
- Lancet, J.E.; Ritchie, E.K.; Uy, G.L.; Medeiros, B.C.; Newell, L.F.; Lin, T.L.; Hogge, D.; Stuart, R.K.; Strickland, S.A.; Solomon, S.R.; et al. Efficacy and Safety of CPX-351 Versus 7+3 in Older Adults with Secondary Acute Myeloid Leukemia: Combined Subgroup Analysis of Phase 2 and Phase 3 Studies. Blood 2017, 130, 4. [Google Scholar]
- Sethi, V.S.; Jackson, D.V., Jr.; White, D.R.; Richards, F., 2nd; Stuart, J.J.; Muss, H.B.; Cooper, M.R.; Spurr, C.L. Pharmacokinetics of vincristine sulfate in adult cancer patients. Cancer Res. 1981, 41, 3551–3555. [Google Scholar] [PubMed]
- Hamada, A.; Kawaguchi, T.; Nakano, M. Clinical pharmacokinetics of cytarabine formulations. Clin. Pharmacokinet. 2002, 41, 705–718. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Khatibi, S.; Howell, S.B.; McCully, C.; Balis, F.M.; Poplack, D.G. Prolongation of drug exposure in cerebrospinal fluid by encapsulation into DepoFoam. Cancer Res. 1993, 53, 1596–1598. [Google Scholar] [PubMed]
- Phuphanich, S.; Maria, B.; Braeckman, R.; Chamberlain, M. A pharmacokinetic study of intra-CSF administered encapsulated cytarabine (DepoCyt) for the treatment of neoplastic meningitis in patients with leukemia, lymphoma, or solid tumors as part of a phase III study. J. Neurooncol. 2007, 81, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Chatelut, E.; Kim, J.C.; Howell, S.B.; Cates, C.; Kormanik, P.A.; Chamberlain, M.C. Extended CSF cytarabine exposure following intrathecal administration of DTC 101. J. Clin. Oncol. 1993, 11, 2186–2193. [Google Scholar] [CrossRef] [PubMed]
- Glantz, M.J.; Jaeckle, K.A.; Chamberlain, M.C.; Phuphanich, S.; Recht, L.; Swinnen, L.J.; Maria, B.; LaFollette, S.; Schumann, G.B.; Cole, B.F.; et al. A randomized controlled trial comparing intrathecal sustained-release cytarabine (DepoCyt) to intrathecal methotrexate in patients with neoplastic meningitis from solid tumors. Clin. Cancer Res. 1999, 5, 3394–3402. [Google Scholar] [PubMed]
- Glantz, M.J.; LaFollette, S.; Jaeckle, K.A.; Shapiro, W.; Swinnen, L.; Rozental, J.R.; Phuphanich, S.; Rogers, L.R.; Gutheil, J.C.; Batchelor, T.; et al. Randomized trial of a slow-release versus a standard formulation of cytarabine for the intrathecal treatment of lymphomatous meningitis. J. Clin. Oncol. 1999, 17, 3110–3116. [Google Scholar] [CrossRef] [PubMed]
- FDA approves liposomal vincristine (Marqibo) for rare leukemia. Oncology (Williston Park) 2012, 26, 841.
- Rodriguez, M.A.; Pytlik, R.; Kozak, T.; Chhanabhai, M.; Gascoyne, R.; Lu, B.; Deitcher, S.R.; Winter, J.N. Vincristine sulfate liposomes injection (Marqibo) in heavily pretreated patients with refractory aggressive non-Hodgkin lymphoma: report of the pivotal phase 2 study. Cancer 2009, 115, 3475–3482. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, S.M.; Aulitzky, W.; Ben Yehuda, D.; Lister, J.; Schiller, G.J.; Selter, K.; Smith, S.E.; Stock, W.; Silverman, J.A.; Kantarjian, H. Phase II study of marqibo in adult patients with refractory or relapsed philadelphia chromosome negative (Ph-) acute lymphoblastic leukemia (ALL). J. Clin. Oncol. 2010, 28, 1. [Google Scholar] [CrossRef]
- Frampton, J.E. Mifamurtide: A review of its use in the treatment of osteosarcoma. Paediatr. Drugs. 2010, 12, 141–153. [Google Scholar] [CrossRef] [PubMed]
- Nardin, A.; Lefebvre, M.L.; Labroquere, K.; Faure, O.; Abastado, J.P. Liposomal muramyl tripeptide phosphatidylethanolamine: Targeting and activating macrophages for adjuvant treatment of osteosarcoma. Curr. Cancer Drug Targets 2006, 6, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Fogler, W.E.; Wade, R.; Brundish, D.E.; Fidler, I.J. Distribution and fate of free and liposome-encapsulated [3H]nor-muramyl dipeptide and [3H]muramyl tripeptide phosphatidylethanolamine in mice. J. Immunol. 1985, 135, 1372–1377. [Google Scholar] [PubMed]
- Fogler, W.E.; Fidler, I.J. Comparative interaction of free and liposome-encapsulated nor-muramyl dipeptide or muramyl tripeptide phosphatidylethanolamine (3H-labelled) with human blood monocytes. Int. J. Immunopharmacol. 1987, 9, 141–150. [Google Scholar] [CrossRef]
- Fidler, I.J.; Brown, N.O.; Hart, I.R. Species variability for toxicity of free and liposome-encapsulated muramyl peptides administered intravenously. J. Biol. Response Mod. 1985, 4, 298–309. [Google Scholar] [PubMed]
- Chang, T.C.; Shiah, H.S.; Yang, C.H.; Yeh, K.H.; Cheng, A.L.; Shen, B.N.; Wang, Y.W.; Yeh, C.G.; Chiang, N.J.; Chang, J.Y.; et al. Phase I study of nanoliposomal irinotecan (PEP02) in advanced solid tumor patients. Cancer Chemother. Pharmacol. 2015, 75, 579–586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang-Gillam, A.; Li, C.P.; Bodoky, G.; Dean, A.; Shan, Y.S.; Jameson, G.; Macarulla, T.; Lee, K.H.; Cunningham, D.; Blanc, J.F.; et al. Nanoliposomal irinotecan with fluorouracil and folinic acid in metastatic pancreatic cancer after previous gemcitabine-based therapy (NAPOLI-1): a global, randomised, open-label, phase 3 trial. Lancet 2016, 387, 545–557. [Google Scholar] [CrossRef]
- Laniado-Laborin, R.; Cabrales-Vargas, M.N. Amphotericin B: Side effects and toxicity. Rev. Iberoam. Micol. 2009, 26, 223–227. [Google Scholar] [CrossRef] [PubMed]
- Stone, N.R.; Bicanic, T.; Salim, R.; Hope, W. Liposomal Amphotericin B (AmBisome((R))): A Review of the Pharmacokinetics, Pharmacodynamics, Clinical Experience and Future Directions. Drugs 2016, 76, 485–500. [Google Scholar] [CrossRef] [PubMed]
- Bekersky, I.; Fielding, R.M.; Dressler, D.E.; Lee, J.W.; Buell, D.N.; Walsh, T.J. Pharmacokinetics, excretion, and mass balance of liposomal amphotericin B (AmBisome) and amphotericin B deoxycholate in humans. Antimicrob. Agents Chemother. 2002, 46, 828–833. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Leifer, F.; Rose, S.; Chun, D.Y.; Thaisz, J.; Herr, T.; Nashed, M.; Joseph, J.; Perkins, W.R.; DiPetrillo, K. Amikacin Liposome Inhalation Suspension (ALIS) Penetrates Non-tuberculous Mycobacterial Biofilms and Enhances Amikacin Uptake Into Macrophages. Front. Microbiol. 2018, 9, 915. [Google Scholar] [CrossRef] [PubMed]
- Bovier, P.A. Epaxal: A virosomal vaccine to prevent hepatitis A infection. Expert Rev. Vaccines 2008, 7, 1141–1150. [Google Scholar] [CrossRef] [PubMed]
- Gluck, R.; Mischler, R.; Finkel, B.; Que, J.U.; Scarpa, B.; Cryz, S.J., Jr. Immunogenicity of new virosome influenza vaccine in elderly people. Lancet 1994, 344, 160–163. [Google Scholar] [CrossRef]
- Alam, M.; Hartrick, C.T. Extended-release epidural morphine (DepoDur): An old drug with a new profile. Pain Pract. 2005, 5, 349–353. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.; Murdande, S.; Gruber, A.; Kim, S. Sustained-release morphine for epidural analgesia in rats. Anesthesiology 1996, 85, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Davidson, E.M.; Barenholz, Y.; Cohen, R.; Haroutiunian, S.; Kagan, L.; Ginosar, Y. High-dose bupivacaine remotely loaded into multivesicular liposomes demonstrates slow drug release without systemic toxic plasma concentrations after subcutaneous administration in humans. Anesth. Analg. 2010, 110, 1018–1023. [Google Scholar] [CrossRef] [PubMed]
- Blumenkranz, M.S.; Bressler, N.M.; Bressler, S.B.; Donati, G.; Fish, G.E.; Haynes, L.A.; Lewis, H.; Miller, J.W.; Mones, J.M.; Potter, M.J.; et al. Verteporfin therapy for subfoveal choroidal neovascularization in age-related macular degeneration: three-year results of an open-label extension of 2 randomized clinical trials--TAP Report no. 5. Arch. Ophthalmol. 2002, 120, 1307–1314. [Google Scholar] [PubMed]
- Richter, A.M.; Waterfield, E.; Jain, A.K.; Canaan, A.J.; Allison, B.A.; Levy, J.G. Liposomal delivery of a photosensitizer, benzoporphyrin derivative monoacid ring A (BPD), to tumor tissue in a mouse tumor model. Photochem. Photobiol. 1993, 57, 1000–1006. [Google Scholar] [CrossRef] [PubMed]
- Laginha, K.M.; Verwoert, S.; Charrois, G.J.; Allen, T.M. Determination of doxorubicin levels in whole tumor and tumor nuclei in murine breast cancer tumors. Clin. Cancer Res. 2005, 11, 6944–6949. [Google Scholar] [CrossRef] [PubMed]
- Gabizon, A.; Tzemach, D.; Mak, L.; Bronstein, M.; Horowitz, A.T. Dose dependency of pharmacokinetics and therapeutic efficacy of pegylated liposomal doxorubicin (DOXIL) in murine models. J. Drug Target. 2002, 10, 539–548. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Scherphof, G.L.; Kamps, J.A. Liposome opsonization. J. Liposome Res. 2005, 15, 109–139. [Google Scholar] [CrossRef] [PubMed]
- Ishida, T.; Harashima, H.; Kiwada, H. Liposome clearance. Biosci. Rep. 2002, 22, 197–224. [Google Scholar] [CrossRef] [PubMed]
- Yingchoncharoen, P.; Kalinowski, D.S.; Richardson, D.R. Lipid-Based Drug Delivery Systems in Cancer Therapy: What Is Available and What Is Yet to Come. Pharmacol. Rev. 2016, 68, 701–787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Immordino, M.L.; Dosio, F.; Cattel, L. Stealth liposomes: Review of the basic science, rationale, and clinical applications, existing and potential. Int. J. Nanomed. 2006, 1, 297–315. [Google Scholar]
- Jain, R.K. Normalizing tumor microenvironment to treat cancer: bench to bedside to biomarkers. J. Clin. Oncol. 2013, 31, 2205–2218. [Google Scholar] [CrossRef] [PubMed]
- Stapleton, S.; Allen, C.; Pintilie, M.; Jaffray, D.A. Tumor perfusion imaging predicts the intra-tumoral accumulation of liposomes. J. Control. Release 2013, 172, 351–357. [Google Scholar] [CrossRef] [PubMed]
- Stapleton, S.; Milosevic, M.; Tannock, I.F.; Allen, C.; Jaffray, D.A. The intra-tumoral relationship between microcirculation, interstitial fluid pressure and liposome accumulation. J. Control. Release 2015, 211, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Folkman, J. Tumor angiogenesis: Therapeutic implications. N. Engl. J. Med. 1971, 285, 1182–1186. [Google Scholar] [CrossRef] [PubMed]
- Sorensen, A.G.; Emblem, K.E.; Polaskova, P.; Jennings, D.; Kim, H.; Ancukiewicz, M.; Wang, M.; Wen, P.Y.; Ivy, P.; Batchelor, T.T.; et al. Increased survival of glioblastoma patients who respond to antiangiogenic therapy with elevated blood perfusion. Cancer Res. 2012, 72, 402–407. [Google Scholar] [CrossRef] [PubMed]
- Sorensen, A.G.; Batchelor, T.T.; Zhang, W.T.; Chen, P.J.; Yeo, P.; Wang, M.; Jennings, D.; Wen, P.Y.; Lahdenranta, J.; Ancukiewicz, M.; et al. A “vascular normalization index” as potential mechanistic biomarker to predict survival after a single dose of cediranib in recurrent glioblastoma patients. Cancer Res. 2009, 69, 5296–5300. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.K. Normalization of tumor vasculature: An emerging concept in antiangiogenic therapy. Science 2005, 307, 58–62. [Google Scholar] [CrossRef] [PubMed]
- Hashizume, H.; Baluk, P.; Morikawa, S.; McLean, J.W.; Thurston, G.; Roberge, S.; Jain, R.K.; McDonald, D.M. Openings between defective endothelial cells explain tumor vessel leakiness. Am. J. Pathol. 2000, 156, 1363–1380. [Google Scholar] [CrossRef]
- Chauhan, V.P.; Stylianopoulos, T.; Boucher, Y.; Jain, R.K. Delivery of molecular and nanoscale medicine to tumors: transport barriers and strategies. Annu. Rev. Chem. Biomol. Eng. 2011, 2, 281–298. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.M.; Faix, P.H.; Schnitzer, J.E. Overcoming key biological barriers to cancer drug delivery and efficacy. J. Control. Release 2017, 267, 15–30. [Google Scholar] [CrossRef] [PubMed]
- Yuan, F.; Dellian, M.; Fukumura, D.; Leunig, M.; Berk, D.A.; Torchilin, V.P.; Jain, R.K. Vascular permeability in a human tumor xenograft: Molecular size dependence and cutoff size. Cancer Res. 1995, 55, 3752–3756. [Google Scholar] [PubMed]
- Ojha, T.; Pathak, V.; Shi, Y.; Hennink, W.E.; Moonen, C.T.W.; Storm, G.; Kiessling, F.; Lammers, T. Pharmacological and physical vessel modulation strategies to improve EPR-mediated drug targeting to tumors. Adv. Drug Deliv. Rev. 2017, 119, 44–60. [Google Scholar] [CrossRef] [PubMed]
- Milosevic, M.F.; Fyles, A.W.; Wong, R.; Pintilie, M.; Kavanagh, M.C.; Levin, W.; Manchul, L.A.; Keane, T.J.; Hill, R.P. Interstitial fluid pressure in cervical carcinoma: Within tumor heterogeneity, and relation to oxygen tension. Cancer 1998, 82, 2418–2426. [Google Scholar] [CrossRef]
- Heldin, C.H.; Rubin, K.; Pietras, K.; Ostman, A. High interstitial fluid pressure—An obstacle in cancer therapy. Nat. Rev. Cancer. 2004, 4, 806–813. [Google Scholar] [CrossRef] [PubMed]
- Netti, P.A.; Berk, D.A.; Swartz, M.A.; Grodzinsky, A.J.; Jain, R.K. Role of extracellular matrix assembly in interstitial transport in solid tumors. Cancer Res. 2000, 60, 2497–2503. [Google Scholar] [PubMed]
- Davies Cde, L.; Berk, D.A.; Pluen, A.; Jain, R.K. Comparison of IgG diffusion and extracellular matrix composition in rhabdomyosarcomas grown in mice versus in vitro as spheroids reveals the role of host stromal cells. Br. J. Cancer 2002, 86, 1639–1644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, F.; Leunig, M.; Huang, S.K.; Berk, D.A.; Papahadjopoulos, D.; Jain, R.K. Microvascular permeability and interstitial penetration of sterically stabilized (stealth) liposomes in a human tumor xenograft. Cancer Res. 1994, 54, 3352–3356. [Google Scholar] [PubMed]
- Tong, R.T.; Boucher, Y.; Kozin, S.V.; Winkler, F.; Hicklin, D.J.; Jain, R.K. Vascular normalization by vascular endothelial growth factor receptor 2 blockade induces a pressure gradient across the vasculature and improves drug penetration in tumors. Cancer Res. 2004, 64, 3731–3736. [Google Scholar] [CrossRef] [PubMed]
- McKee, T.D.; Grandi, P.; Mok, W.; Alexandrakis, G.; Insin, N.; Zimmer, J.P.; Bawendi, M.G.; Boucher, Y.; Breakefield, X.O.; Jain, R.K. Degradation of fibrillar collagen in a human melanoma xenograft improves the efficacy of an oncolytic herpes simplex virus vector. Cancer Res. 2006, 66, 2509–2513. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, V.P.; Stylianopoulos, T.; Martin, J.D.; Popovic, Z.; Chen, O.; Kamoun, W.S.; Bawendi, M.G.; Fukumura, D.; Jain, R.K. Normalization of tumour blood vessels improves the delivery of nanomedicines in a size-dependent manner. Nat. Nanotechnol. 2012, 7, 383–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duncan, R.; Richardson, S.C. Endocytosis and intracellular trafficking as gateways for nanomedicine delivery: opportunities and challenges. Mol. Pharm. 2012, 9, 2380–2402. [Google Scholar] [CrossRef] [PubMed]
- Kirpotin, D.B.; Drummond, D.C.; Shao, Y.; Shalaby, M.R.; Hong, K.; Nielsen, U.B.; Marks, J.D.; Benz, C.C.; Park, J.W. Antibody targeting of long-circulating lipidic nanoparticles does not increase tumor localization but does increase internalization in animal models. Cancer Res. 2006, 66, 6732–6740. [Google Scholar] [CrossRef] [PubMed]
- Baru, M.; Nahum, O.; Jaaro, H.; Sha’anani, J.; Nur, I. Lysosome-disrupting peptide increases the efficiency of in-vivo gene transfer by liposome-encapsulated DNA. J. Drug Target. 1998, 6, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Lu, Z.; Wientjes, M.G.; Au, J.L. Delivery of siRNA therapeutics: Barriers and carriers. AAPS J. 2010, 12, 492–503. [Google Scholar] [CrossRef] [PubMed]
- Drummond, D.C.; Noble, C.O.; Hayes, M.E.; Park, J.W.; Kirpotin, D.B. Pharmacokinetics and in vivo drug release rates in liposomal nanocarrier development. J. Pharm. Sci. 2008, 97, 4696–4740. [Google Scholar] [CrossRef] [PubMed]
- Ernsting, M.J.; Murakami, M.; Roy, A.; Li, S.D. Factors controlling the pharmacokinetics, biodistribution and intratumoral penetration of nanoparticles. J. Control. Release 2013, 172, 782–794. [Google Scholar] [CrossRef] [PubMed]
- Sarin, H.; Kanevsky, A.S.; Wu, H.; Sousa, A.A.; Wilson, C.M.; Aronova, M.A.; Griffiths, G.L.; Leapman, R.D.; Vo, H.Q. Physiologic upper limit of pore size in the blood-tumor barrier of malignant solid tumors. J. Transl. Med. 2009, 7, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maeda, H.; Nakamura, H.; Fang, J. The EPR effect for macromolecular drug delivery to solid tumors: Improvement of tumor uptake, lowering of systemic toxicity, and distinct tumor imaging in vivo. Adv. Drug Deliv. Rev. 2013, 65, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Zamboni, W.C.; Torchilin, V.; Patri, A.K.; Hrkach, J.; Stern, S.; Lee, R.; Nel, A.; Panaro, N.J.; Grodzinski, P. Best practices in cancer nanotechnology: Perspective from NCI nanotechnology alliance. Clin. Cancer Res. 2012, 18, 3229–3241. [Google Scholar] [CrossRef] [PubMed]
- Lammers, T.; Kiessling, F.; Hennink, W.E.; Storm, G. Drug targeting to tumors: Principles, pitfalls and (pre-) clinical progress. J. Control. Release 2012, 161, 175–187. [Google Scholar] [CrossRef] [PubMed]
- Caron, W.P.; Clewell, H.; Dedrick, R.; Ramanathan, R.K.; Davis, W.L.; Yu, N.; Tonda, M.; Schellens, J.H.; Beijnen, J.H.; Zamboni, W.C. Allometric scaling of pegylated liposomal anticancer drugs. J. Pharmacokinet. Pharmacodyn. 2011, 38, 653–669. [Google Scholar] [CrossRef] [PubMed]
- Ait-Oudhia, S.; Mager, D.E.; Straubinger, R.M. Application of pharmacokinetic and pharmacodynamic analysis to the development of liposomal formulations for oncology. Pharmaceutics. 2014, 6, 137–174. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Ramanathan, R.K.; Zamboni, B.A.; Strychor, S.; Ramalingam, S.; Edwards, R.P.; Friedland, D.M.; Stoller, R.G.; Belani, C.P.; Maruca, L.J.; et al. Population pharmacokinetics of pegylated liposomal CKD-602 (S-CKD602) in patients with advanced malignancies. J. Clin. Pharmacol. 2012, 52, 180–194. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Infante, J.R.; Keedy, V.L.; Jones, S.F.; Chan, E.; Bendell, J.C.; Lee, W.; Zamboni, B.A.; Ikeda, S.; Kodaira, H.; et al. Population pharmacokinetics of PEGylated liposomal CPT-11 (IHL-305) in patients with advanced solid tumors. Eur. J. Clin. Pharmacol. 2013, 69, 2073–2081. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Infante, J.R.; Keedy, V.L.; Jones, S.F.; Chan, E.; Bendell, J.C.; Lee, W.; Kirschbrown, W.P.; Zamboni, B.A.; Ikeda, S.; et al. Factors affecting the pharmacokinetics and pharmacodynamics of PEGylated liposomal irinotecan (IHL-305) in patients with advanced solid tumors. Int. J. Nanomed. 2015, 10, 1201–1209. [Google Scholar] [CrossRef] [PubMed]
- Zhao, P.; Rowland, M.; Huang, S.M. Best practice in the use of physiologically based pharmacokinetic modeling and simulation to address clinical pharmacology regulatory questions. Clin. Pharmacol. Ther. 2012, 92, 17–20. [Google Scholar] [CrossRef] [PubMed]
- Zhao, P.; Zhang, L.; Grillo, J.A.; Liu, Q.; Bullock, J.M.; Moon, Y.J.; Song, P.; Brar, S.S.; Madabushi, R.; Wu, T.C.; et al. Applications of physiologically based pharmacokinetic (PBPK) modeling and simulation during regulatory review. Clin. Pharmacol. Ther. 2011, 89, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Rowland, M.; Peck, C.; Tucker, G. Physiologically-based pharmacokinetics in drug development and regulatory science. Annu. Rev. Pharmacol. Toxicol. 2011, 51, 45–73. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.M.; Abernethy, D.R.; Wang, Y.; Zhao, P.; Zineh, I. The utility of modeling and simulation in drug development and regulatory review. J. Pharm. Sci. 2013, 102, 2912–2923. [Google Scholar] [CrossRef] [PubMed]
- Jones, H.M.; Chen, Y.; Gibson, C.; Heimbach, T.; Parrott, N.; Peters, S.A.; Snoeys, J.; Upreti, V.V.; Zheng, M.; Hall, S.D. Physiologically based pharmacokinetic modeling in drug discovery and development: a pharmaceutical industry perspective. Clin. Pharmacol. Ther. 2015, 97, 247–262. [Google Scholar] [CrossRef] [PubMed]
- Sinha, V.; Zhao, P.; Huang, S.M.; Zineh, I. Physiologically based pharmacokinetic modeling: From regulatory science to regulatory policy. Clin. Pharmacol. Ther. 2014, 95, 478–480. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Al-Jamal, K.T.; Kostarelos, K.; Reineke, J. Physiologically based pharmacokinetic modeling of nanoparticles. ACS Nano 2010, 4, 6303–6317. [Google Scholar] [CrossRef] [PubMed]
- Harashima, H.; Tsuchihashi, M.; Iida, S.; Doi, H.; Kiwada, H. Pharmacokinetic/pharmacodynamic modeling of antitumor agents encapsulated into liposomes. Adv. Drug Deliv. Rev. 1999, 40, 39–61. [Google Scholar] [CrossRef]
- Harashima, H.; Iida, S.; Urakami, Y.; Tsuchihashi, M.; Kiwada, H. Optimization of antitumor effect of liposomally encapsulated doxorubicin based on simulations by pharmacokinetic/pharmacodynamic modeling. J. Control. Release 1999, 61, 93–106. [Google Scholar] [CrossRef]
- Hendriks, B.S.; Reynolds, J.G.; Klinz, S.G.; Geretti, E.; Lee, H.; Leonard, S.C.; Gaddy, D.F.; Espelin, C.W.; Nielsen, U.B.; Wickham, T.J. Multiscale kinetic modeling of liposomal Doxorubicin delivery quantifies the role of tumor and drug-specific parameters in local delivery to tumors. CPT Pharmacomet. Syst. Pharmacol. 2012, 1, e15. [Google Scholar] [CrossRef] [PubMed]
- Apgar, J.F.; Tang, J.P.; Singh, P.; Balasubramanian, N.; Burke, J.; Hodges, M.R.; Lasaro, M.A.; Lin, L.; Miliard, B.L.; Moore, K.; et al. Quantitative Systems Pharmacology Model of hUGT1A1-modRNA Encoding for the UGT1A1 Enzyme to Treat Crigler-Najjar Syndrome Type 1. CPT Pharmacomet. Syst. Pharmacol. 2018, 7, 404–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kagan, L.; Gershkovich, P.; Wasan, K.M.; Mager, D.E. Dual physiologically based pharmacokinetic model of liposomal and nonliposomal amphotericin B disposition. Pharm. Res. 2014, 31, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Khawar, I.A.; Kim, J.H.; Kuh, H.J. Improving drug delivery to solid tumors: Priming the tumor microenvironment. J. Control. Release 2015, 201, 78–89. [Google Scholar] [CrossRef] [PubMed]
- Stapleton, S.; Milosevic, M.; Allen, C.; Zheng, J.; Dunne, M.; Yeung, I.; Jaffray, D.A. A mathematical model of the enhanced permeability and retention effect for liposome transport in solid tumors. PLoS ONE 2013, 8, e81157. [Google Scholar] [CrossRef] [PubMed]
- Frieboes, H.B.; Wu, M.; Lowengrub, J.; Decuzzi, P.; Cristini, V. A computational model for predicting nanoparticle accumulation in tumor vasculature. PLoS ONE 2013, 8, e56876. [Google Scholar] [CrossRef] [PubMed]
- Troendle, E.P.; Khan, A.; Searson, P.C.; Ulmschneider, M.B. Predicting drug delivery efficiency into tumor tissues through molecular simulation of transport in complex vascular networks. J. Control. Release 2018, 292, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Macklin, P.; McDougall, S.; Anderson, A.R.; Chaplain, M.A.; Cristini, V.; Lowengrub, J. Multiscale modelling and nonlinear simulation of vascular tumour growth. J. Math. Biol. 2009, 58, 765–798. [Google Scholar] [CrossRef] [PubMed]
- Curtis, L.T.; Wu, M.; Lowengrub, J.; Decuzzi, P.; Frieboes, H.B. Computational Modeling of Tumor Response to Drug Release from Vasculature-Bound Nanoparticles. PLoS ONE 2015, 10, e0144888. [Google Scholar] [CrossRef] [PubMed]
- Stylianopoulos, T.; Soteriou, K.; Fukumura, D.; Jain, R.K. Cationic nanoparticles have superior transvascular flux into solid tumors: Insights from a mathematical model. Ann. Biomed. Eng. 2013, 41, 68–77. [Google Scholar] [CrossRef] [PubMed]
- Stylianopoulos, T.; Economides, E.A.; Baish, J.W.; Fukumura, D.; Jain, R.K. Towards Optimal Design of Cancer Nanomedicines: Multi-stage Nanoparticles for the Treatment of Solid Tumors. Ann. Biomed. Eng. 2015, 43, 2291–2300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stylianopoulos, T.; Jain, R.K. Combining two strategies to improve perfusion and drug delivery in solid tumors. Proc. Natl. Acad. Sci. USA 2013, 110, 18632–18637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirtane, A.R.; Siegel, R.A.; Panyam, J. A pharmacokinetic model for quantifying the effect of vascular permeability on the choice of drug carrier: a framework for personalized nanomedicine. J. Pharm. Sci. 2015, 104, 1174–1186. [Google Scholar] [CrossRef] [PubMed]
- FDA. Guidance for Industry: Extended Release Oral Dosage Forms: Development, Evaluation and Application of In Vitro/In Vivo Correlations. Available online: https://www.fda.gov/downloads/drugs/guidances/ucm070239.pdf (accessed on 15 January 2019).
- Shabbits, J.A.; Chiu, G.N.; Mayer, L.D. Development of an in vitro drug release assay that accurately predicts in vivo drug retention for liposome-based delivery systems. J. Control. Release 2002, 84, 161–170. [Google Scholar] [CrossRef]
- Xu, X.; Khan, M.A.; Burgess, D.J. A two-stage reverse dialysis in vitro dissolution testing method for passive targeted liposomes. Int. J. Pharm. 2012, 426, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Crielaard, B.J.; Yousefi, A.; Schillemans, J.P.; Vermehren, C.; Buyens, K.; Braeckmans, K.; Lammers, T.; Storm, G. An in vitro assay based on surface plasmon resonance to predict the in vivo circulation kinetics of liposomes. J. Control. Release 2011, 156, 307–314. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Wang, J.; Wu, Y.; Fan, J.; Zhao, L.; Cao, Y. Physiologically based pharmacokinetic platform to quantify the influence of nano–bio interactions on the biodistribution of liposomal drug (In preparation).



{kind=link}
{kind=link}
| Product | Active Drug | Therapeutic Area | Year of Approval | PK–PD Profiles |
|---|---|---|---|---|
| DaunoXome® | Daunorubicin | Cancer | 1996 | Prolonged retention, increased target distribution, equivalent efficacy, and reduced toxicity. |
| DepoCyt® | Cytarabine | Cancer | 1999 | Prolonged tumor exposure to cytotoxic concentration, increased response rate, and reduced toxicity. |
| Doxil® | Doxorubicin | Cancer | 1995 | Prolonged retention, increased target distribution, equivalent efficacy, and reduced toxicity. |
| Lipodox® | Doxorubicin | Cancer | 2012 | Prolonged retention, increased target distribution, equivalent efficacy, and reduced toxicity. |
| Myocet® | Doxorubicin | Cancer | 2000 | Prolonged retention, increased target distribution, equivalent efficacy, and reduced toxicity. |
| Marqibo® | Vincristine | Cancer | 2012 | Prolonged retention, increased target distribution, superior efficacy, and reduced toxicity. |
| Vyxeos® | Daunorubicin-cytarabine (1:5) | Cancer | 2017 | Increased targeted exposure of daunorubicin and cytarabine in a fixed ratio, superior efficacy, and comparable toxicity. |
| OnivydeTM | Irinotecan | Cancer | 2015 | Prolonged retention and increased exposure of the bioactive metabolite of irinotecan (SN-38), superior efficacy, and reduced toxicity. |
| Mepact® | Mifamurtide | Cancer | 2009 | Prolonged and increased retention in target, superior efficacy, and reduced toxicity. |
| Ambisome® | Amphotericin B | Infection | 1997 | Releases the drug only when the liposome binds to the fungus and significant reduction in toxicity. |
| Arikayce® | Amikacin | Infection | 2018 | Increased target distribution, superior efficacy, and comparable toxicity. |
| Inflexal® V | Flu vaccine | Vaccine | 1997 | Superior immune response. |
| Epaxal® | Hepatitis A vaccine (synthetic lipids, influenza proteins, hepatitis A antigen) | Vaccine | 1994 | Higher tolerability and reduced toxicity. |
| DepoDurTM | Morphine | Analgesics | 2004 | Prolonged retention, reduced peak concentration, superior efficacy, and reduced toxicity. |
| Exparel® | Bupivacaine | Analgesics | 2011 | Prolonged retention, reduced peak concentration, superior efficacy, and reduced toxicity. |
| Visudyne® | Verteporfin | Photodynamic therapy | 2000 | Equivalent clearance, slightly increased tissue distribution, and reduced toxicity. |
| Model Approach | Mathematical Model | Mathematical Model of the Rest of the Body | Drugs | Notes | Ref |
|---|---|---|---|---|---|
| Simplified physiologically-based pharmacokinetic (PBPK) model Simplified PBPK model Simplified PBPK model | Tumor was divided into capillary, interstitial and tumor cell sub-compartments. | 1-compartment PK model for liposome and 2-compartment PK model for drug. | Doxorubicin | Liposomal retention in tumors and the local release rate were identified to play pivotal roles in antitumor efficacy. | [118,119] |
| Tumor was divided into capillary, interstitial, tumor cell and nucleus sub-compartments. | 1-compartment PK model for liposome and2-compartment PK model for drug. | Doxorubicin | The detailed drug transport into and out of the cell, drug-target association and dissociation, and liposome uptake and release in tumor cells were described. | [120] | |
| Liver hepatocyte compartment with endosomal and cytoplastic compartments. | Plasma compartment. | hUGT1A1-modRNA | Endocytosis, release and transcription processes were described. After translated to humans, this model was used to estimate the first-in-human dose. | [121] | |
| Whole-body PBPK model | / | Plasma and tissue (liver, spleen, kidneys, gut, lungs, heart and others) compartments. | Amphotericin B | The first whole-body PBPK model described the disposition of both liposome and drug simultaneously. | [122] |
| Model with spatiotemporal characterization Model with spatiotemporal characterization | The combination of tumor growth, angiogenesis, oxygen transport, nanoparticle transport and antitumor effect models. | / | / | The physiological properties of the tumor were considered. The model described the interactions between tumor progression and liposome disposition. | [125,127,128] |
| Tumor vascular network and nanoparticle transport model. | / | / | Tumor blood vessel properties were simulated in this model. The interactions between the liposome and blood vessel were simulated to optimize the particle properties. | [129,130,131] | |
| Model with in vitro—in vivo correlation (IVIVC) | Using in vitro studies to determine the model parameters and replace in vivo study in liposome optimization study. | Plasma and tissues compartments. | Doxorubicin | Liposome property–disposition relationships were established to facilitate liposome optimization. | [137] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, H.; Yuan, D.; Wu, Y.; Cao, Y. Pharmacokinetics and Pharmacodynamics Modeling and Simulation Systems to Support the Development and Regulation of Liposomal Drugs. Pharmaceutics 2019, 11, 110. https://doi.org/10.3390/pharmaceutics11030110
He H, Yuan D, Wu Y, Cao Y. Pharmacokinetics and Pharmacodynamics Modeling and Simulation Systems to Support the Development and Regulation of Liposomal Drugs. Pharmaceutics. 2019; 11(3):110. https://doi.org/10.3390/pharmaceutics11030110
Chicago/Turabian StyleHe, Hua, Dongfen Yuan, Yun Wu, and Yanguang Cao. 2019. "Pharmacokinetics and Pharmacodynamics Modeling and Simulation Systems to Support the Development and Regulation of Liposomal Drugs" Pharmaceutics 11, no. 3: 110. https://doi.org/10.3390/pharmaceutics11030110