Intestinal Permeation Enhancers for Oral Delivery of Macromolecules: A Comparison between Salcaprozate Sodium (SNAC) and Sodium Caprate (C10)

 , , , and
, , , and 
Abstract
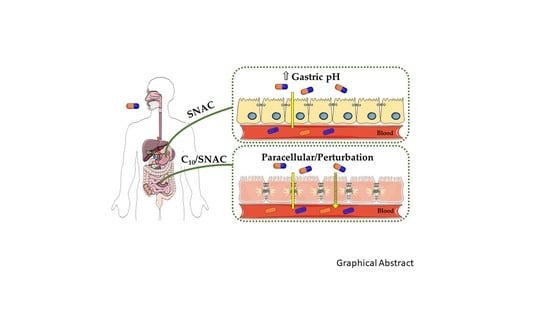
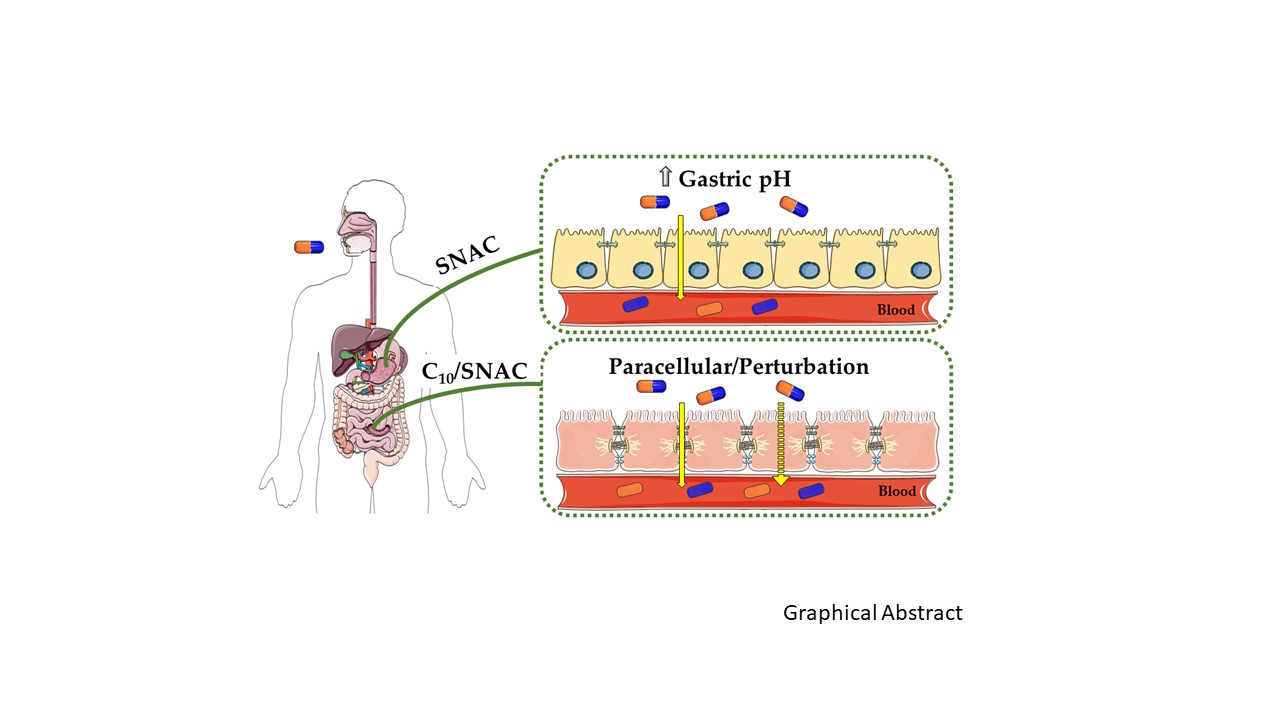
:
1. Introduction
2. Challenges for Oral Delivery of Macromolecules
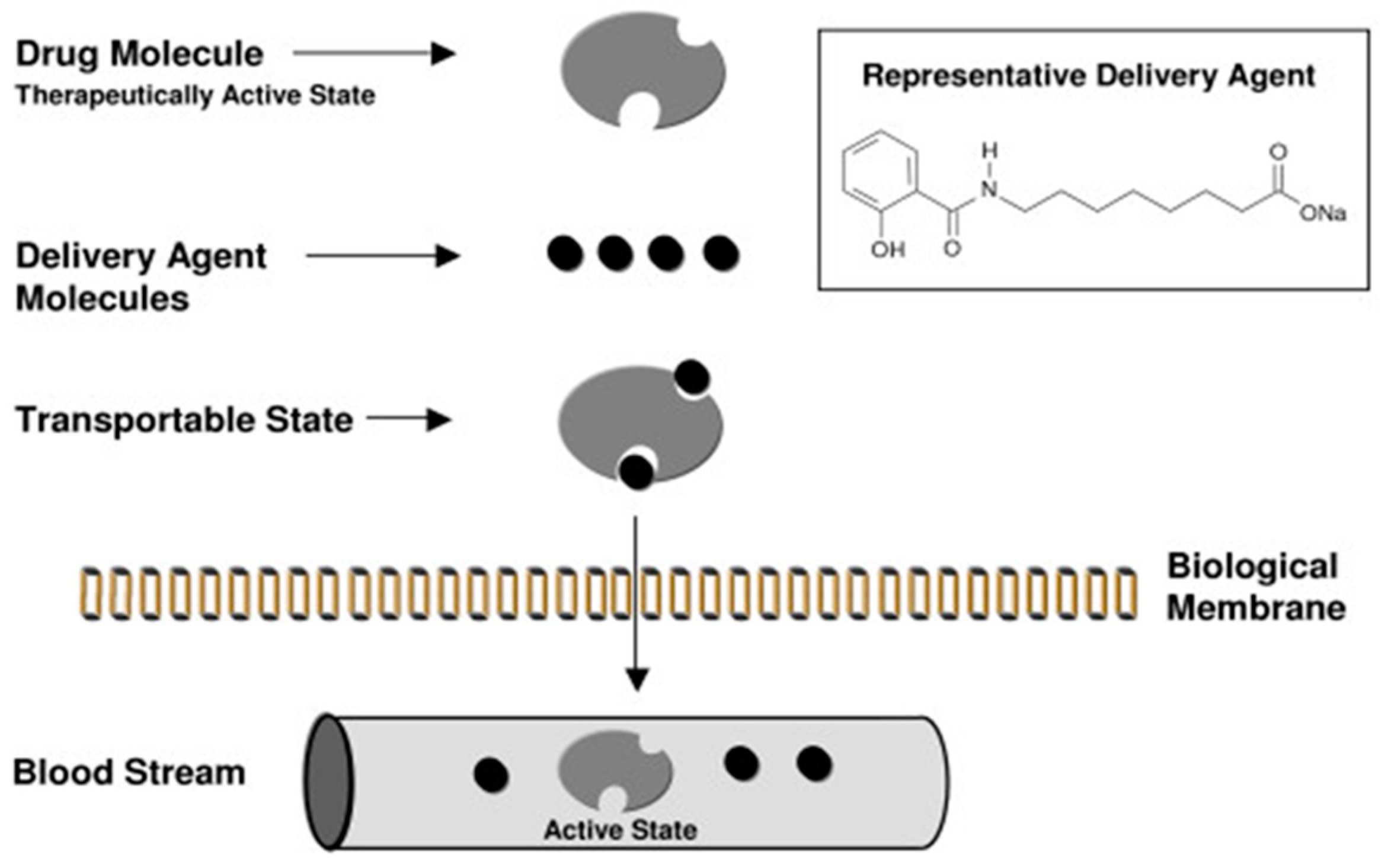
3. Intestinal Permeation Enhancers
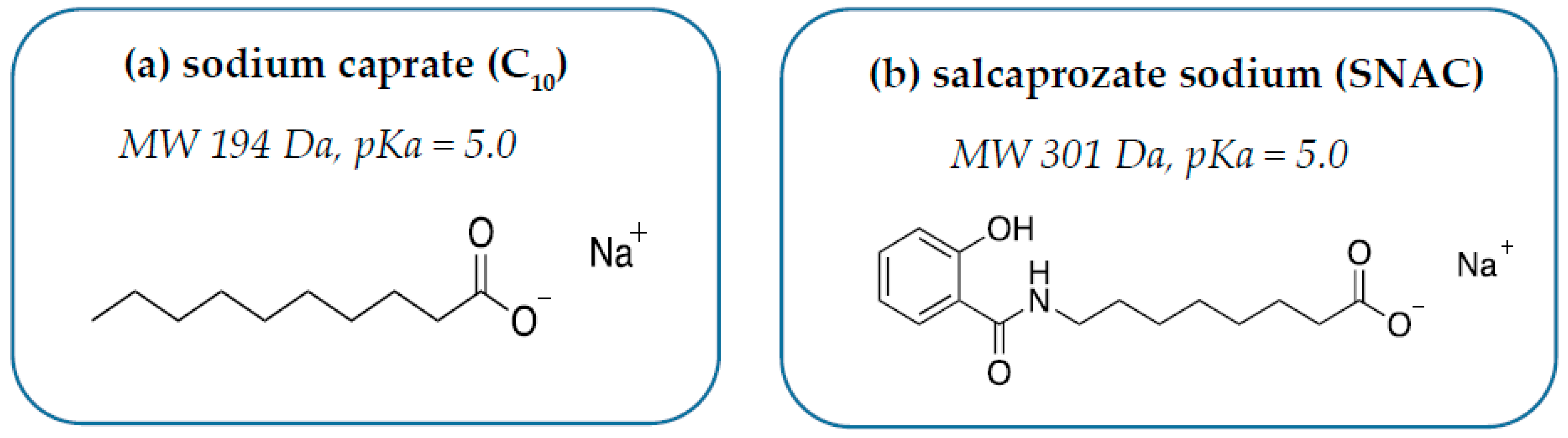
4. Introducing C10
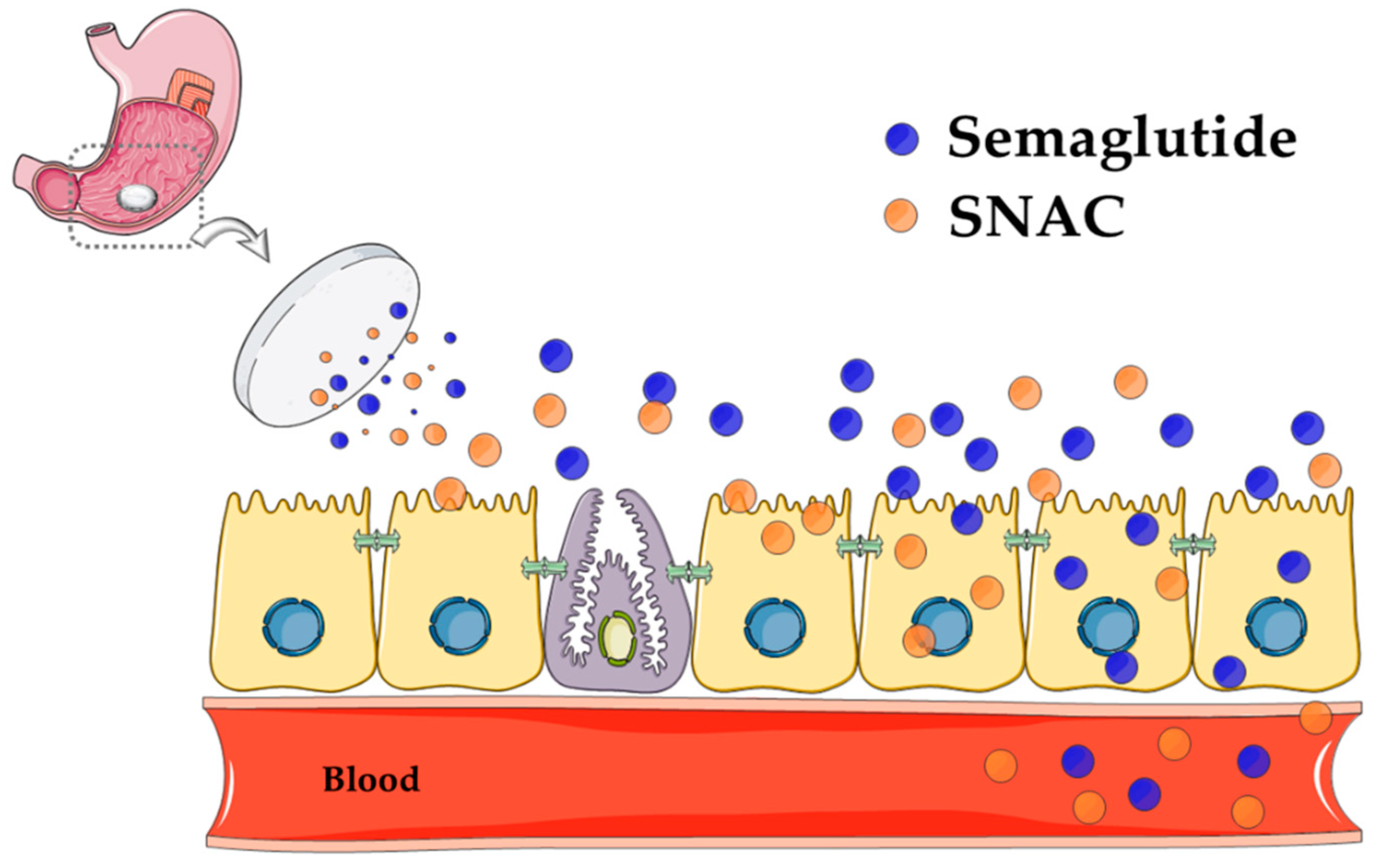
5. Introducing SNAC
6. How Do SNAC and C10 Alter GI Permeability?
6.1. Challenges in Determining Mechanism of Action
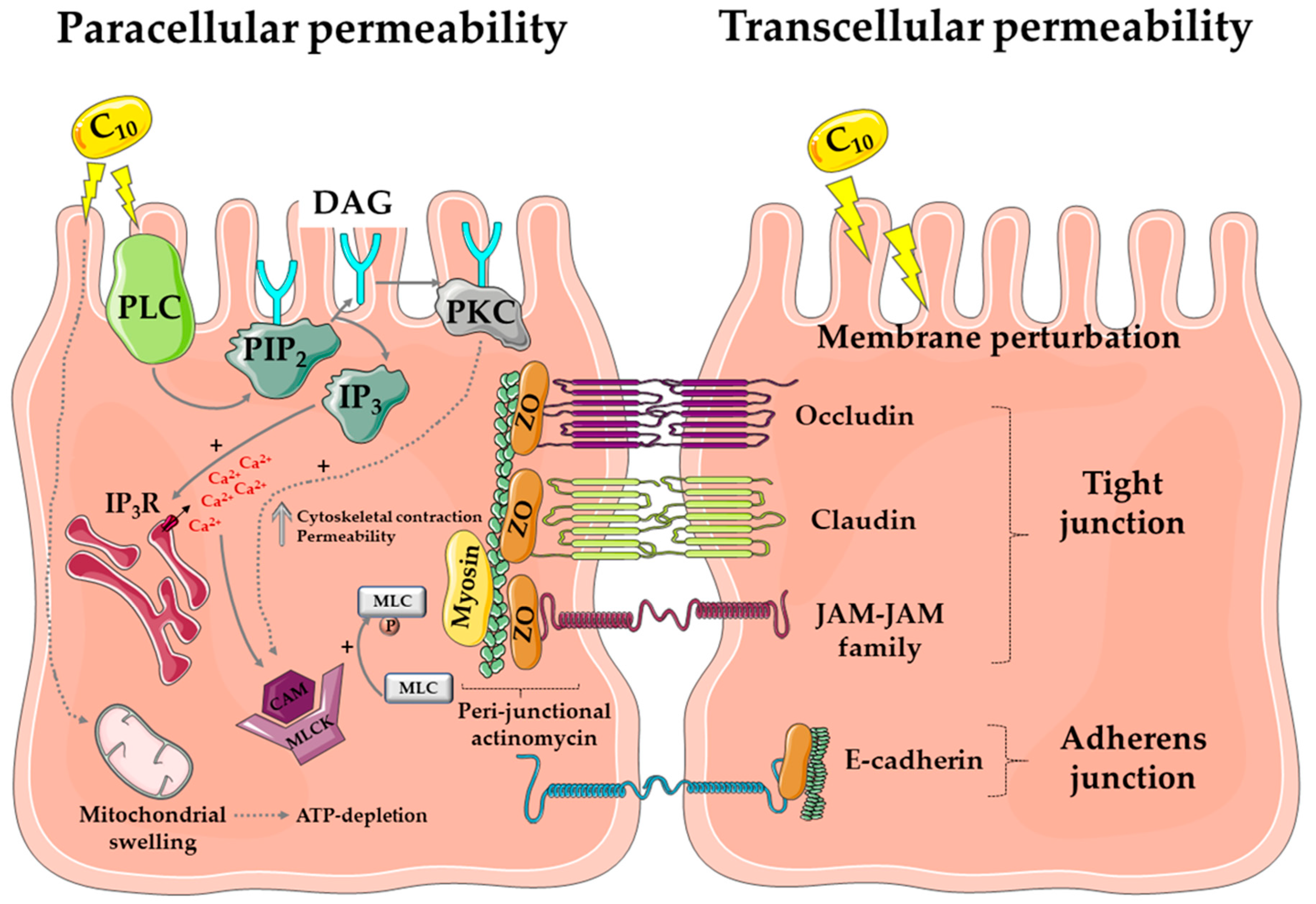
6.2. C10 Mode of Action
6.3. SNAC Mode of Action
7. C10 and SNAC: Pharmacokinetics and Efficacy in Clinical Trials
7.1. C10
7.2. SNAC
8. Safety of SNAC and C10 in Preclinical and Clinical Studies
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Fosgerau, K.; Hoffmann, T. Peptide therapeutics: Current status and future directions. Drug Discov. Today 2015, 20, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Usmani, S.S.; Bedi, G.; Samuel, J.S.; Singh, S.; Kalra, S.; Kumar, P.; Ahuja, A.A.; Sharma, M.; Gautam, A.; Raghava, G.P.S. THPdb: Database of FDA-approved peptide and protein therapeutics. PLoS ONE 2017, 12, e0181748. [Google Scholar] [CrossRef] [PubMed]
- Lakkireddy, H.R.; Urmann, M.; Besenius, M.; Werner, U.; Haack, T.; Brun, P.; Alié, J.; Illel, B.; Hortala, L.; Vogel, R.; et al. Oral delivery of diabetes peptides—Comparing standard formulations incorporating functional excipients and nanotechnologies in the translational context. Adv. Drug Deliv. Rev. 2016, 106, 196–222. [Google Scholar] [CrossRef] [PubMed]
- Cowan Report. Therapeutic Categories Outlook: Comprehensive Study; Cowan & Co.: New York, NY, USA, 2014. [Google Scholar]
- Lewis, A.L.; Richard, J. Challenges in the delivery of peptide drugs: An industry perspective. Ther. Deliv. 2015, 6, 149–163. [Google Scholar] [CrossRef]
- Aguirre, T.A.; Teijeiro-Osorio, D.; Rosa, M.; Coulter, I.S.; Alonso, M.J.; Brayden, D.J. Current status of selected oral peptide technologies in advanced preclinical development and in clinical trials. Adv. Drug Deliv. Rev. 2016, 106, 223–241. [Google Scholar] [CrossRef] [PubMed]
- Benet, L.Z. The drug transporter-metabolism alliance: Uncovering and defining the interplay. Mol. Pharm. 2009, 6, 631–643. [Google Scholar] [CrossRef] [PubMed]
- Eligen® Technology: Summary and Value Proposition. Available online: https://www.emisphere.com/wp-content/uploads/2017/02/Eligen-Technology-Presentation_2.15-Update.pdf (accessed on 12 February 2019).
- Baluom, M.; Friedman, M.; Assaf, P.; Haj-Yehia, A.I.; Rubinstein, A. Synchronized release of sulpiride and sodium decanoate from HPMC matrices: A rational approach to enhance sulpiride absorption in the rat intestine. Pharm. Res. 2000, 17, 1071–1076. [Google Scholar] [CrossRef] [PubMed]
- Maher, S.; Mrsny, R.J.; Brayden, D.J. Intestinal permeation enhancers for oral peptide delivery. Adv. Drug Deliv. Rev. 2016, 106, 277–319. [Google Scholar] [CrossRef] [PubMed]
- Card, J.W.; Magnuson, B.A. A review of the efficacy and safety of nanoparticle-based oral insulin delivery systems. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 301, G956–G967. [Google Scholar] [CrossRef]
- Banerjee, A.; Mitragotri, S. Intestinal patch systems for oral drug delivery. Curr. Opin. Pharmacol. 2017, 36, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Whitehead, K.; Karr, N.; Mitragotri, S. Safe and effective permeation enhancers for oral drug delivery. Pharm. Res. 2008, 25, 1782–1788. [Google Scholar] [CrossRef] [PubMed]
- Maher, S.; Ryan, B.; Duffy, A.; Brayden, D.J. Formulation strategies to improve oral peptide delivery. Pharm. Pat. Anal. 2014, 3, 313–336. [Google Scholar] [CrossRef] [PubMed]
- Aungst, B.J. Absorption enhancers: Applications and advances. AAPS J. 2012, 14, 10–18. [Google Scholar] [CrossRef] [PubMed]
- EFSA Panel on Food Additives and Nutrient Sources added to Food (ANS); Younes, M.; Aggett, P.; Aguilar, F.; Crebelli, R.; Dusemund, B.; Filipič, M.; Frutos, M.J.; Galtier, P.; Gott, D.; et al. Re-evaluation of sodium, potassium and calcium salts of fatty acids (E 470a) and magnesium salts of fatty acids (E 470b) as food additives. EFSA J. 2018. [Google Scholar] [CrossRef]
- Lindmark, T.; Söderholm, J.D.; Olaison, G.; Alván, G.; Ocklind, G.; Artursson, P. Mechanism of absorption enhancement in humans after rectal administration of ampicillin in suppositories containing sodium caprate. Pharm. Res. 1997, 14, 930–955. [Google Scholar] [CrossRef] [PubMed]
- Walsh, E.; Adamczyk, B.; Chalasani, K.B.; Maher, M.; O’Toole, E.B.; Fox, J.; Leonard, T.W.; Brayden, D.J. Oral delivery of macromolecules: Rationale underpinning Gastrointestinal Permeation Enhancement Technology (GIPET®). Ther. Deliv. 2011, 2, 1595–1610. [Google Scholar] [CrossRef]
- Leone-Bay, A.; Santiago, N.; Achan, D.; Chaudhary, K.; DeMorin, F.; Falzarano, L.; Haas, S.; Kalbag, S.; Kaplan, D.; Leipold, H.; et al. N-acylated alpha amino acids as novel oral delivery agents for proteins. J. Med. Chem. 1995, 38, 4263–4269. [Google Scholar] [CrossRef]
- Castelli, M.C.; Wong, D.F.; Friedman, K.; Riley, M.G. Pharmacokinetics of oral cyanocobalamin formulated with sodium N-[8-(2-hydroxybenzoyl)amino]caprylate (SNAC): An open-label, randomized, single-dose, parallel-group study in healthy male subjects. Clin. Ther. 2011, 33, 934–945. [Google Scholar] [CrossRef]
- Smith, L.; Mosley, J.; Ford, M.; Courtney, J. Cyanocobalamin/Salcaprozate Sodium: A novel way to treat vitamin B12 deficiency and anemia. J. Hematol. Oncol. Pharm. 2016, 6, 42–45. [Google Scholar]
- Leone-Bay, A.; Paton, D.R.; Variano, B.; Leipold, H.; Rivera, T.; Miura-Fraboni, J.; Baughman, R.A.; Santiago, N. Acylated non-alpha-amino acids as novel agents for the oral delivery of heparin sodium, USP. J. Control. Release 1998, 50, 41–49. [Google Scholar] [CrossRef]
- Goldberg, M. Gomez-Orellana I. Challenges for the oral delivery of macromolecules. Nat. Rev. Drug Discov. 2003, 2, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Leone-Bay, A.; Leipold, H.; Sarubbi, D.; Variano, B.; Rivera, T.; Baughman, R.A. Oral delivery of sodium cromolyn: Preliminary studies in vivo and in vitro. Pharm. Res. 1996, 13, 222–226. [Google Scholar] [CrossRef] [PubMed]
- Anonymous, Sodium Decanoate, PubChem ID 16211937. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/16211937 (accessed on 12 February 2019).
- Anonymous, Sodium Caprozate PubChem ID 22669833. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/23669833 (accessed on 12 February 2019).
- Brayden, D.J.; Gleeson, J.; Walsh, E. A head-to-head multi-parametric high content analysis of a series of medium chain fatty acid intestinal permeation enhancers in Caco-2 cells. Eur. J. Pharm. Biopharm. 2014, 88, 830–839. [Google Scholar] [CrossRef] [PubMed]
- Khafagy, E.S.; Morishita, M.; Takayama, K. The role of intermolecular interactions with penetratin and its analogue on the enhancement of absorption of nasal therapeutic peptides. Int. J. Pharm. 2010, 388, 209–212. [Google Scholar] [CrossRef]
- Twarog, C.; Hillaireau, H.; Taverna, M.; Noiray, M.; Illel, B.; Vogel, R.; Brayden, D.J.; Fattal, E. Oral peptide delivery: Understanding interactions between a peptide and permeation enhancers. In Proceedings of the 11th World Meeting on Pharmaceutics, Biopharmaceutics and Pharmaceutical Technology, Granada, Spain, 19–22 March 2018. [Google Scholar]
- Lindmark, T.; Nikkila, T.; Artursson, P. Mechanisms of absorption enhancement by medium chain fatty acids in intestinal epithelial Caco-2 cell monolayers. J. Pharmacol. Exp. Ther. 1995, 275, 958–964. [Google Scholar]
- Sawada, T.; Ogawa, T.; Tomita, M.; Hayashi, M.; Awazu, S. Role of paracellular pathway in nonelectrolyte permeation across rat colon epithelium enhanced by sodium caprate and sodium caprylate. Pharm. Res. 1991, 8, 1365–1371. [Google Scholar] [CrossRef]
- Maher, S.; Kennelly, R.; Bzik, V.A.; Baird, A.W.; Wang, X.; Winter, D.; Brayden, D.J. Evaluation of intestinal absorption enhancement and local mucosal toxicity of two promoters. I. Studies in isolated rat and human colonic mucosae. Eur. J. Pharm. Sci. 2009, 38, 291–300. [Google Scholar] [CrossRef]
- Tomita, M.; Hayashi, M.; Awazu, S. Absorption-enhancing mechanism of EDTA, caprate, and decanoylcarnitine in Caco-2 cells. J. Pharm. Sci. 1996, 85, 608–611. [Google Scholar] [CrossRef]
- Lindmark, T.; Kimura, Y.; Artursson, P. Absorption enhancement through intracellular regulation of tight junction permeability by medium chain fatty acids in Caco-2 cells. J. Pharmacol. Exp. Ther. 1998, 284, 362–369. [Google Scholar]
- Feighery, L.M.; Cochrane, S.W.; Quinn, T.; Baird, A.W.; O’Toole, D.; Owens, S.E.; O’Donoghue, D.; Mrsny, R.J.; Brayden, D.J. Myosin light chain kinase inhibition: Correction of increased intestinal epithelial permeability in vitro. Pharm. Res. 2008, 2, 1377–1386. [Google Scholar] [CrossRef]
- Shimazaki, T.; Tomita, M.; Sadahiro, S.; Hayashi, M.; Awazu, S. Absorption-enhancing effects of sodium caprate and palmitoyl carnitine in rat and human colons. Dig. Dis. Sci. 1998, 43, 641–645. [Google Scholar] [CrossRef] [PubMed]
- Krug, S.M.; Amasheh, M.; Dittmann, I.; Christoffel, I.; Fromm, M.; Amasheh, S. Sodium caprate as an enhancer of macromolecule permeation across tricellular tight junctions of intestinal cells. Biomaterials 2013, 34, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Blikslager, A.T.; Moeser, A.J.; Gookin, J.L.; Jones, S.L.; Odle, J. Restoration of barrier function in injured intestinal mucosa. Physiol. Rev. 2007, 87, 545–564. [Google Scholar] [CrossRef]
- Sugibayashi, K.; Onuki, Y.; Takayama, K. Displacement of tight junction proteins from detergent-resistant membrane domains by treatment with sodium caprate. Eur. J. Pharm. Sci. 2009, 36, 46–253. [Google Scholar] [CrossRef]
- Maher, S.; Heade, J.; McCartney, F.; Waters, S.; Bleiel, S.B.; Brayden, D.J. Effects of surfactant-based permeation enhancers on mannitol permeability, histology, and electrogenic ion transport responses in excised rat colonic mucosae. Int. J. Pharm. 2018, 539, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Brayden, D.J.; Maher, S.; Bahar, B.; Walsh, E. Sodium caprate-induced increases in intestinal permeability and epithelial damage are prevented by misoprostol. Eur. J. Pharm. Biopharm. 2015, 94, 194–206. [Google Scholar] [CrossRef]
- Leonard, T.W.; Lynch, J.; McKenna, M.J.; Brayden, D.J. Promoting absorption of drugs in humans using medium-chain fatty acid-based solid dosage forms: GIPET. Exp. Opin. Drug Deliv. 2006, 3, 685–692. [Google Scholar] [CrossRef]
- Gradauer, K.; Nishiumi, A.; Unrinin, K.; Higashino, H.; Kataoka, M.; Pedersen, B.L.; Buckley, S.T.; Yamashita, S. Interaction with mixed micelles in the intestine attenuates the permeation enhancing potential of alkyl-maltosides. Mol. Pharm. 2015, 12, 2245–2253. [Google Scholar] [CrossRef]
- Brayden, D.; Creed, E.; O’Connell, A.; Leipold, H.; Agarwal, R.; Leone-Bay, A. Heparin absorption across the intestine: Effects of sodium N-[8-(2-hydroxybenzoyl)amino]caprylate in rat in situ intestinal instillations and in Caco-2 monolayers. Pharm. Res. 1997, 14, 1772–1779. [Google Scholar] [CrossRef]
- Malkov, D.; Angelo, R.; Wang, H.Z.; Flanders, E.; Tang, H.; Gomez-Orellana, I. Oral delivery of insulin with the eligen technology: Mechanistic studies. Curr. Drug Deliv. 2005, 2, 191–197. [Google Scholar] [CrossRef]
- Hess, S.; Rotshild, V.; Hoffman, A. Investigation of the enhancing mechanism of sodium N-[8-(2-hydroxybenzoyl)amino]caprylate effect on the intestinal permeability of polar molecules utilizing a voltage clamp method. Eur. J. Pharm. Sci. 2005, 25, 307–312. [Google Scholar] [CrossRef] [PubMed]
- Malkov, D.; Wang, H.Z.; Dinh, S.; Gomez-Orellana, I. Pathway of oral absorption of heparin with sodium N-[8-(2-hydroxybenzoyl)amino] caprylate. Pharm. Res. 2002, 19, 1180–1184. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Rath, P.; Angelo, R.; Stringfellow, T.; Flanders, E.; Dinh, S.; Gomez-Orellana, I.; Robinson, J.R. Oral absorption enhancement of cromolyn sodium through noncovalent complexation. Pharm. Res. 2004, 21, 2196–2206. [Google Scholar] [CrossRef] [PubMed]
- Arbit, E.; Goldberg, M.; Gomez-Orellana, I.; Majuru, S. Oral heparin: Status review. Thromb. J. 2006, 4, 6. [Google Scholar] [CrossRef] [PubMed]
- Alani, A.W.; Robinson, J.R. Mechanistic understanding of oral drug absorption enhancement of cromolyn sodium by an amino acid derivative. Pharm. Res. 2008, 25, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Rehmani, S.; Dixon, J.E. Oral delivery of anti-diabetes therapeutics using cell penetrating and transcytosing peptide strategies. Peptides 2018, 100, 24–35. [Google Scholar] [CrossRef] [PubMed]
- Buckley, S.T.; Bækdal, T.A.; Vegge, A.; Maarbjerg, S.J.; Pyke, C.; Ahnfelt-Rønne, J.; Madsen, K.G.; Schéele, S.G.; Alanentalo, T.; Kirk, R.K.; et al. Transcellular stomach absorption of a derivatized glucagon-like peptide-1 receptor agonist. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef]
- Amory, J.K.; Leonard, T.W.; Page, S.T.; O’Toole, E.; McKenna, M.J.; Bremner, W.J. Oral administration of the GnRH antagonist acyline, in a GIPET-enhanced tablet form, acutely suppresses serum testosterone in normal men: Single-dose pharmacokinetics and pharmacodynamics. Cancer Chemother. Pharmacol. 2009, 64, 641–645. [Google Scholar] [CrossRef]
- Leonard, T.W. Composition and Drug Delviery of Bisphosphonates. U.S. Patent 0,215,743 A1, 26 August 2010. [Google Scholar]
- Halberg, I.B.; Lyby, K.; Wassermann, K.; Heise, T.; Zijlstra, E.; Plum-Morchel, L. Efficacy and safety of oral basal insulin versus subcutaneous insulin in type 2 diabetes: A randomised, double-blind Phase II trial. Lancet Diabetes Endocrinol. 2019. [Google Scholar] [CrossRef]
- Khedkar, A.; Lebovitz, H.; Fleming, A.; Cherrington, A.; Jose, V.; Athalye, S.N.; Vishweswaramurthy, A. Impact of insulin tregopil and its permeation enhancer on pharmacokinetics of metformin in healthy volunteers: Randomized, open-label, placebo-controlled, crossover study. Clin. Transl. Sci. 2018. [Google Scholar] [CrossRef]
- Raoof, A.A.; Chiu, P.; Ramtoola, Z.; Cumming, I.K.; Teng, C.; Weinbach, S.P.; Hardee, G.E.; Levin, A.A.; Geary, R.S. Oral bioavailability and multiple dose tolerability of an antisense oligonucleotide tablet formulated with sodium caprate. J. Pharm. Sci. 2004, 93, 1431–1439. [Google Scholar] [CrossRef] [PubMed]
- Tillman, L.G.; Geary, R.S.; Hardee, G.E. Oral delivery of antisense oligonucleotides in man. J. Pharm. Sci. 2008, 97, 225–236. [Google Scholar] [CrossRef] [PubMed]
- Available online: http://ir.ionispharma.com/news-releases/news-release-details/ionis-pharmaceuticals-licenses-first-oral-antisense-drug-acting (accessed on 12 February 2019).
- Lennernäs, H.; Gjellan, K.; Hallgren, R.; Graffner, C. The influence of caprate on rectal absorption of phenoxymethylpenicillin: Experience from an in-vivo perfusion in humans. J. Pharm. Pharmacol. 2002, 54, 499–508. [Google Scholar] [CrossRef] [PubMed]
- Baughman, R.A.; Kapoor, S.C.; Agarwal, R.K.; Kisicki, J.; Catella-Lawson, F.; FitzGerald, G.A. Oral delivery of anticoagulant doses of heparin. A randomized, double-blind, controlled study in humans. Circulation 1998, 98, 1610–1615. [Google Scholar] [CrossRef] [PubMed]
- Berkowitz, S.D.; Marder, V.J.; Kosutic, G.; Baughman, R.A. Oral heparin administration with a novel drug delivery agent (SNAC) in healthy volunteers and patients undergoing elective total hip arthroplasty. J. Thromb. Haemost. 2003, 1, 1914–1919. [Google Scholar] [CrossRef] [PubMed]
- Mousa, S.A.; Zhang, F.; Aljada, A.; Chaturvedi, S.; Takieddin, M.; Zhang, H.; Chi, L.; Castelli, M.C.; Friedman, K.; Goldberg, M.M.; et al. Pharmacokinetics and pharmacodynamics of oral heparin solid dosage form in healthy human subjects. J. Clin. Pharmacol. 2007, 47, 1508–1520. [Google Scholar] [CrossRef] [PubMed]
- Buclin, T.; Cosma Rochat, M.; Burckhardt, P.; Azria, M.; Attinger, M. Bioavailability and biological efficacy of a new oral formulation of salmon calcitonin in healthy volunteers. J. Bone Miner. Res. 2002, 17, 1478–1485. [Google Scholar] [CrossRef] [PubMed]
- Kapitza, C.; Zijlstra, E.; Heinemann, L.; Castelli, M.C.; Riley, G.; Heise, T. Oral insulin: A comparison with subcutaneous regular human insulin in patients with type 2 diabetes. Diabetes Care 2010, 33, 1288–1290. [Google Scholar] [CrossRef] [PubMed]
- Karsdal, M.A.; Byrjalsen, I.; Alexandersen, P.; Bihlet, A.; Andersen, J.R.; Riis, B.J.; Bay-Jensen, A.C.; Christiansen, C.; CSMC021C2301/2 investigators. Treatment of symptomatic knee osteoarthritis with oral salmon calcitonin: Results from two phase 3 trials. Osteoarthr. Cartil. 2015, 23, 532–543. [Google Scholar] [CrossRef]
- Henriksen, K.; Byrjalsen, I.; Andersen, J.R.; Bihlet, A.R.; Russo, L.A.; Alexandersen, P.; Valter, I.; Qvist, P.; Lau, E.; Riis, B.J.; et al. SMC021 investigators., A randomized, double-blind, multicenter, placebo-controlled study to evaluate the efficacy and safety of oral salmon calcitonin in the treatment of osteoporosis in postmenopausal women taking calcium and vitamin D. Bone 2016, 91, 122–129. [Google Scholar] [CrossRef]
- Karsdal, M.A.; Henriksen, K.; Bay-Jensen, A.C.; Molloy, B.; Arnold, M.; John, M.R.; Byrjalsen, I.; Azria, M.; Riis, B.J.; Qvist, P.; et al. Lessons learned from the development of oral calcitonin: The first tablet formulation of a protein in phase III clinical trials. J. Clin. Pharmacol. 2011, 51, 460–471. [Google Scholar] [CrossRef] [PubMed]
- Davies, M.; Pieber, T.R.; Hartoft-Nielsen, M.L.; Hansen, O.K.H.; Jabbour, S.; Rosenstock, J. Effect of oral semaglutide compared with placebo and subcutaneous semaglutide on glycemic control in patients with Type 2 diabetes: A Randomized Clinical Trial. JAMA 2017, 318, 1460–1470. [Google Scholar] [CrossRef] [PubMed]
- Bjerregaard, S.; Nielsen, F.S.; Sauerberg, P. Solid Compositions Comprising a GLP-1 Agonist and a Salt of n-(8-(2-Hydroxybenzoyl)Amino)Caprylic Acid. U.S. Patent WO2012080471A1, 8 March 2016. [Google Scholar]
- Available online: https://globenewswire.com/news-release/2018/02/22/1379640/0/en/Novo-Nordisk-successfully-completes-the-first-phase-3a-trial-PIONEER-1-with-oral-semaglutide.html (accessed on 28 November 2018).
- Granhall, C.; Søndergaard, F.L.; Thomsen, M.; Anderson, T.W. Pharmacokinetics, safety and tolerability of oral semaglutide in subjects with renal impairment. Clin. Pharm. 2018, 57, 1571–1580. [Google Scholar] [CrossRef] [PubMed]
- Baekdal, T.A.; Thomsen, M.; Kupčová, V.; Hansen, C.W.; Anderson, T.W. Pharmacokinetics, safety, and tolerability of oral semaglutide in subjects with hepatic impairment. J. Clin. Pharmacol. 2018, 58, 1314–1323. [Google Scholar] [CrossRef] [PubMed]
- Bækdal, T.A.; Breitschaft, A.; Navarria, A.; Hansen, C.W. A randomized study investigating the effect of omeprazole on the pharmacokinetics of oral semaglutide. Expert Opin. Drug Metab. Toxicol. 2018, 14, 869–877. [Google Scholar] [CrossRef] [PubMed]
- Bain, S.C.; Mosenzon, O.; Arechavaleta, R.; Bogdański, P.; Comlekci, A.; Consoli, A.; Deerochanawong, C.; Dungan, K.; Faingold, M.C.; Farkouh, M.E.; et al. Cardiovascular safety of oral semaglutide in patients with type 2 diabetes: Rationale, design and patient baseline characteristics for the PIONEER 6 trial. Diabetes Obes. Metab. 2018. [Google Scholar] [CrossRef]
- Maher, S.; Leonard, T.W.; Jacobsen, J.; Brayden, D.J. Safety and efficacy of sodium caprate in promoting oral drug absorption: From in vitro to the clinic. Adv. Drug Deliv. Rev. 2009, 61, 1427–1449. [Google Scholar] [CrossRef]
- Raoof, A.A.; Ramtoola, Z.; McKenna, B.; Yu, R.Z.; Hardee, G.; Geary, R.S. Effect of sodium caprate on the intestinal absorption of two modified antisense oligonucleotides in pigs. Eur. J. Pharm. Sci. 2002, 17, 131–138. [Google Scholar] [CrossRef]
- Riley, M.G.; Castelli, M.C.; Paehler, E.A. Subchronic oral toxicity of salcaprozate sodium (SNAC) in Sprague-Dawley and Wistar rats. Int. J. Toxicol. 2009, 28, 278–293. [Google Scholar] [CrossRef]
- Riley, M.G.; York, R.G. Peri-and postnatal developmental toxicity of salcaprozate sodium (SNAC) in Sprague-Dawley rats. Int. J. Toxicol. 2009, 28, 266–277. [Google Scholar] [CrossRef]
- McCartney FGleeson, J.; Brayden, D.J. Safety concerns over the use of intestinal permeation enhancers: A mini-review. Tissue Barriers 2016, 4, e1176822. [Google Scholar] [CrossRef] [PubMed]
- Van der Flier, L.G.; Clevers, H. Stem cells, self-renewal, and differentiation in the intestinal epithelium. Annu. Rev. Physiol. 2009, 71, 241–260. [Google Scholar] [CrossRef]
- Laine, L.; Takeuchi, K.; Tarnawski, A. Gastric mucosal defense and cytoprotection: Bench to bedside. Gastroenterology 2008, 135, 41–60. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Maher, S.; Brayden, D.J. Restoration of rat colonic epithelium after in situ intestinal instillation of the absorption promoter, sodium caprate. Ther. Deliv. 2010, 1, 75–82. [Google Scholar] [CrossRef]
- Gookin, J.L.; Galanko, J.A.; Blikslager, A.T.; Argenzio, R.A. PG-mediated closure of paracellular pathway and not restitution is the primary determinant of barrier recovery in acutely injured porcine ileum. Am. J. Physiol. 2003, 285, G967–G979. [Google Scholar] [CrossRef] [PubMed]
- Narkar, Y.; Burnette, R.; Bleher, R.; Albrecht, R.; Kandela, A.; Robinson, J.R. Evaluation of mucosal damage and recovery in the gastrointestinal tract of rats by a penetration enhancer. Pharm. Res. 2008, 25, 25–38. [Google Scholar] [CrossRef] [PubMed]
- König, J.; Wells, J.; Cani, P.D.; García-Ródenas, C.L.; MacDonald, T.; Mercenier, A.; Whyte, J.; Troost, F.; Brummer, R.J. Human intestinal barrier function in health and disease. Clin. Transl. Gastroenterol. 2016, 7, e196. [Google Scholar] [CrossRef]
- Cani, P.D. Human gut microbiome: Hopes, threats and promises. Gut 2018, 67, 1716–1725. [Google Scholar] [CrossRef]
- Chassaing, B.; Koren, O.; Goodrich, J.K.; Poole, A.C.; Srinivasan, S.; Ley, R.E.; Gewirtz, A.T. Dietary emulsifiers impact the mouse gut microbiota promoting colitis and metabolic syndrome. Nature 2015, 519, 92–96. [Google Scholar] [CrossRef]
- Choonara, B.F.; Choonara, Y.E.; Kumar, P.; Bijukumar, D.; du Toit, L.C.; Pillay, V. A review of advanced oral drug delivery technologies facilitating the protection and absorption of protein and peptide molecules. Biotechnol. Adv. 2014, 32, 1269–1282. [Google Scholar] [CrossRef]
- Tscheik, C.; Blasig, I.E.; Winkler, L. Trends in drug delivery through tissue barriers containing tight junctions. Tissue Barriers 2013, 1, e24565. [Google Scholar] [CrossRef] [PubMed]
- Bala, S.; Marcos, M.; Gattu, A.; Catalano, D.; Szabo, G. Acute binge drinking increases serum endotoxin and bacterial DNA levels in healthy individuals. PLoS ONE 2014, 9, e96864. [Google Scholar] [CrossRef] [PubMed]
- Cox, A.B.; Rawlinson, L.; Baird, A.W.; Bzik, V.; Brayden, D.J. In vitro interactions between the oral absorption promoter, sodium caprate (C10) and S. typhimurium in rat intestinal ileal mucosae. Pharm. Res. 2008, 25, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Petschow, B.W.; Batema, R.P.; Ford, L.L. Susceptibility of Helicobacter pylori to bactericidal properties of medium-chain monoglycerides and free fatty acids. Antimicrob. Agents Chemother. 1996, 40, 302–306. [Google Scholar] [CrossRef]
- Van Immerseel, F.; De Buck, J.; Boyen, F.; Bohez, L.; Pasmans, F.; Volf, J.; Sevcik, M.; Rychlik, I.; Haesebrouck, F.; Ducatelle, R. Medium-chain fatty acids decrease colonization and invasion through hilA suppression shortly after infection of chickens with Salmonella enterica serovar Enteritidis. Appl. Environ. Microbiol. 2004, 70, 3582–3587. [Google Scholar] [CrossRef] [PubMed]
- Chadeganipour, M.; Haims, A. Antifungal activities of pelargonic and capric acid on Microsporum gypseum. Mycoses 2001, 44, 109–112. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.B.; Alimova, Y.; Myers, T.M.; Ebersole, J.L. Short and medium-chain fatty acids exhibit antimicrobial activity for oral microorganisms. Arch. Oral Biol. 2011, 56, 650–654. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Description | Treatment | Outcome | Reference |
|---|---|---|---|
| Ampicillin with C10 in healthy subjects (n = 12). | Rectal suppository containing 250 mg of ampicillin and 25 mg of C10. | Cmax increased 2.6-fold compared to ampicillin alone and BA increased 1.8-fold. Some local tissue damage not ascribed to C10. | [17] |
| Phenoxymethylpenicillin, antipyrine with C10 in healthy subjects (n = 6). | Rectal perfusion containing 2 g of phenoxymethylpenicillin, 8 mg of antipyrine, and 0.7 g of C10. Two treatments (T), T1: pH 6 and T2: pH 7.4. Each subject received control (no C10) and treatment. | C10 was ineffective at increasing permeability across rectal epithelium. | [60] |
| GIPET™: oral acyline in healthy subjects (n = 8). | 3 oral tablet doses of acyline: 10, 20, and 40 mg. Subjects received all doses, 1 week apart, under fasting conditions. | Significant reduction in LH, FSH, and testosterone. No serious treatment related adverse effects. | [53] |
| GIPET™: oral zoledronic acid in prostate cancer patients with bone metastasis (n = 30). | Once-weekly enteric-coated Orazol™ tablets containing 20 mg of zoledronic acid versus weekly Zometa® (4 mg) i.v. infusion over 49 days. | Equivalent urine output biomarkers; claim of 5% bioavailability (BA) in patent. | [54] |
| Antisense oligonucleotide with C10 (ISIS 104838) in healthy subjects (n = 15). | Enteric-coated tablets, four formulations, and one after a high-fat meal. Subjects received all treatments. | 9.5% bioavailability compared to s.c. No study-related adverse effects. | [58] |
| Basal insulin in C10 formulation versus insulin glargine in Type 2 diabetics (s.c.) (n = 25). | Daily tablets of a long-acting insulin (I338) over 8 weeks. | 1.5–2.0% bioavailability compared to s.c. Comparable reductions in plasma glucose. | [55] |
| Insulin tregopil (IN-105) in C10 tablets in healthy subjects. | Single treatments of insulin along with metoformin over 4 periods of 2 days. | No effects on the pharmacokinetics (PK) of metformin; good safety. | [56] |
| Description | Treatment | Outcome | Reference |
|---|---|---|---|
| Vitamin B12 with SNAC in tablets in healthy subjects (n = 20). Medical food clinical study. | (A) Two tablets, each with 5 mg of vitamin B12 with 100 mg of SNAC (B) One tablet: 5 mg of vitamin B12 with 100 mg of SNAC (C) One commercial tablet: 5 mg of vitamin B12 (D) 1 mg of vitamin B12 via i.v. injection. | Treatment (B) achieved 3% higher absolute BA compared to the commercial oral formulation. No adverse effects. | [20] |
| Heparin with SNAC in hip replacement patients, (n = 123). Phase II. | Two studies: one dose every 8 h (max 16 doses), and two doses every 8 h (max 12 doses). | Achieved anti-factor Xa activity comparable to s.c. heparin. No change in major bleeding events compared to s.c. | [62] |
| Insulin with 4-CNAB in untreated T2D (n = 10). Phase II. | 300 mg of insulin with 400 mg of 4-CNAB, or 15 IU of insulin s.c. Performed under fasting conditions. | Cmax was higher and was reached faster compared to s.c. Shorter duration and high subject variability. No adverse effects. | [65] |
| sCT with 5-CNAC in osteoarthritic patients over 24 months (n = 1176 and n = 1030) Phase III. | 0.8 mg of sCT in tablets twice daily for 24 months. | No significant effect compared to placebo. | [66] |
| sCT with 5-CNAC in postmenopausal women with osteoporosis (n = 4665). Phase III. | 0.8 mg or placebo in tablets daily, together with vitamin D and calcium for 36 months. | No beneficial effect on fractures was observed. No change in quality of life. | [6] |
| Description | Parameters | Comment | Reference |
|---|---|---|---|
| Phase II dose-ranging 26-week study in patients (n = 632) (NCT01923181). | 0.7–1.9% reduction in glycated hemoglobin (HbA1c); some weight reduction; mild gastro-intestinal (GI) side effects common. | The key trial which supported moving to Phase III. | [69] |
| PIONEER-1 Phase IIIa 26-week study in patients (n = 703) (NCT02906930). | Mean 1.5% reduction in HbA1c confirmed with 14-mg dose; 4.1-kg weight reduction; mild–moderate nausea in 16% versus 6% in placebo. | 14 mg established as semaglutide dose with 300 mg of SNAC in all studies. | [71] |
| PIONEER 5 Phase IIIa in renal-impaired patients (n = 71) (NCT02014259). | 5 mg of semaglutide for 5 days; 10 mg for 5 days, assessed up to 21 days after; no change in PK overall. | Area under curve (AUC) and half-life (t½) similar to regular T2D patients, no need to change dose regime. | [72] |
| Trial in hepatic-impaired patients (n = 56) (NCT02016911). | Design as for PIONEER-5. | AUC, Cmax, and t½ unchanged, no need to change in dose regime. | [73] |
| Trial in healthy subjects 1 taking omeprazole (n = 54) (NCT02249871). | 5 mg for 5 days, followed by 10 mg for 5 days) ± 40 mg omeprazole. | AUC and stomach pH slightly higher in semaglutide/omeprazole group, but no need to change dose regime. | [74] |
| PIONEER-6 Phase IIIa assessed cardiovascular (CV) risk in T2D patients (n = 3183) (NCT02692716). | Primary end-points: reduction in major CV events over median 16-month period. | Cardiovascular (CV) outcomes not different from placebo, but suggestion of a mortality benefit of oral tablet. | [75] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Twarog, C.; Fattah, S.; Heade, J.; Maher, S.; Fattal, E.; Brayden, D.J. Intestinal Permeation Enhancers for Oral Delivery of Macromolecules: A Comparison between Salcaprozate Sodium (SNAC) and Sodium Caprate (C10). Pharmaceutics 2019, 11, 78. https://doi.org/10.3390/pharmaceutics11020078
Twarog C, Fattah S, Heade J, Maher S, Fattal E, Brayden DJ. Intestinal Permeation Enhancers for Oral Delivery of Macromolecules: A Comparison between Salcaprozate Sodium (SNAC) and Sodium Caprate (C10). Pharmaceutics. 2019; 11(2):78. https://doi.org/10.3390/pharmaceutics11020078
Chicago/Turabian StyleTwarog, Caroline, Sarinj Fattah, Joanne Heade, Sam Maher, Elias Fattal, and David J. Brayden. 2019. "Intestinal Permeation Enhancers for Oral Delivery of Macromolecules: A Comparison between Salcaprozate Sodium (SNAC) and Sodium Caprate (C10)" Pharmaceutics 11, no. 2: 78. https://doi.org/10.3390/pharmaceutics11020078