Effect of Carrier Lipophilicity and Preparation Method on the Properties of Andrographolide–Solid Dispersion

Abstract
:
1. Introduction
2. Materials and Methods
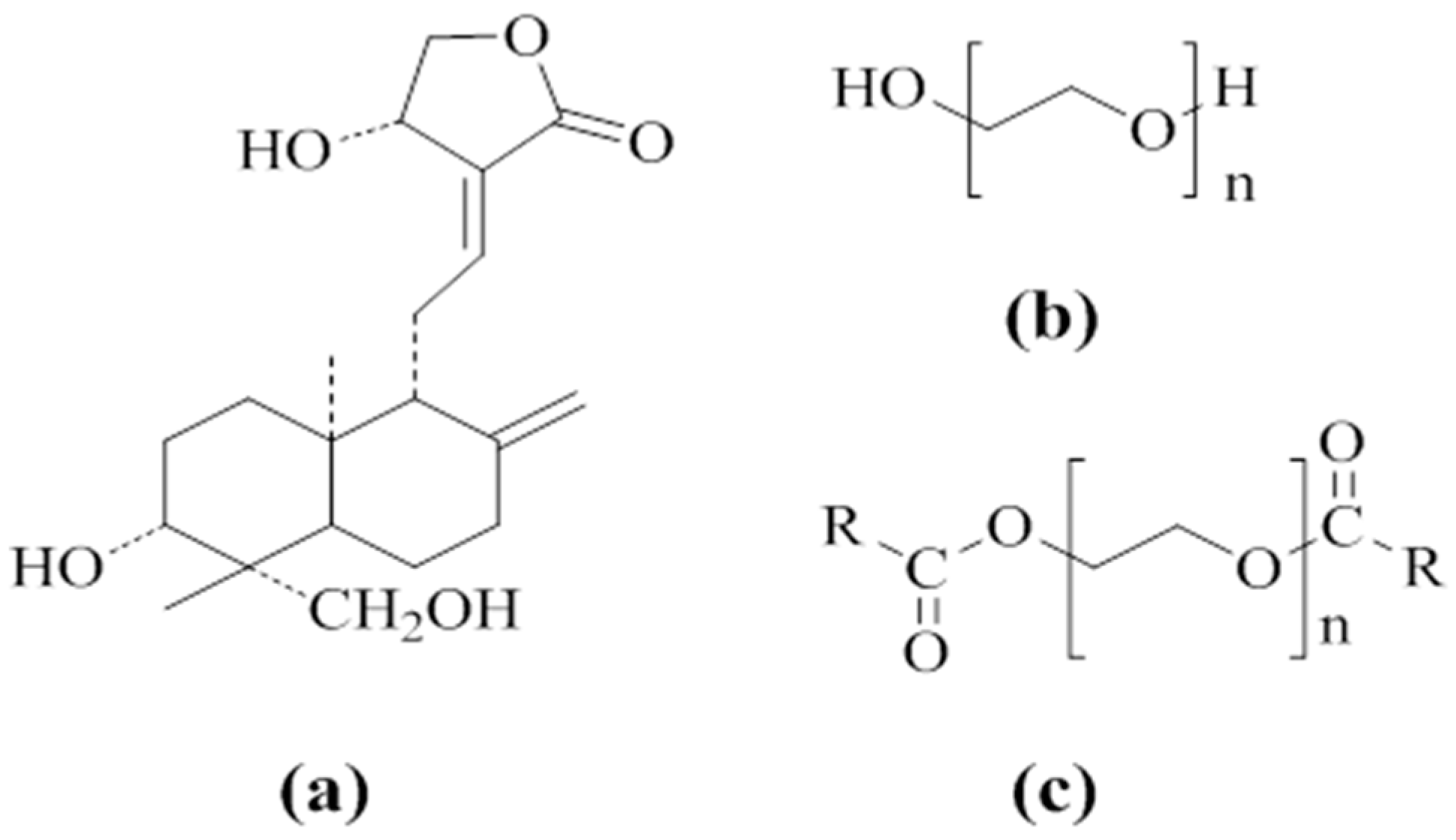
2.1. Materials
2.2. Preparation of Physical Mixture (PM)
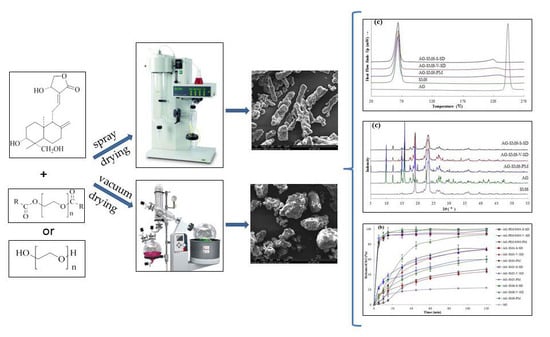
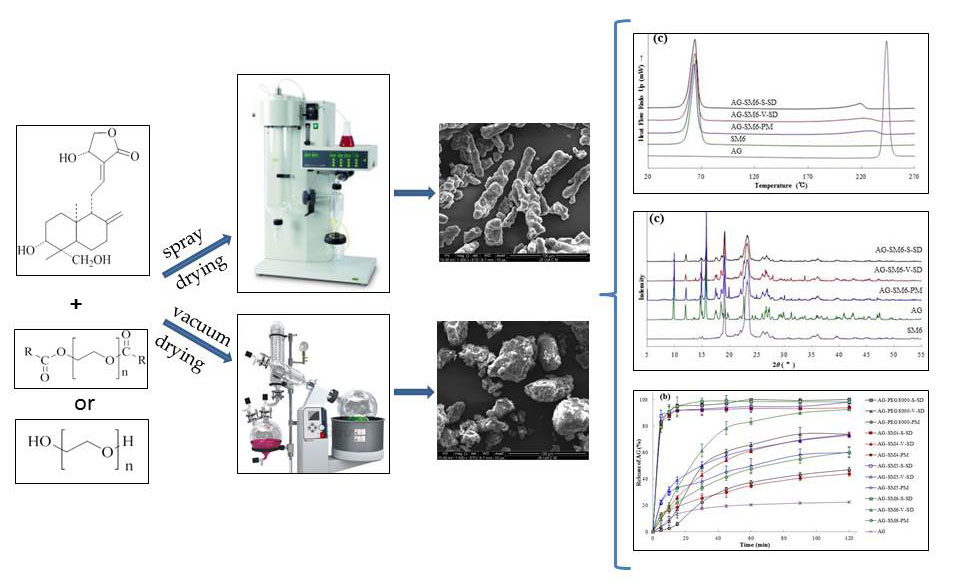
2.3. Preparation of AG–SD by Spray-Drying Method (S)
2.4. Preparation of AG–SD by Vacuum-Drying Method (V)
2.5. Fourier Transform Infrared Spectroscopy (FT-IR)
2.6. Thermogravimetric Analysis
2.7. Differential Scanning Calorimetry
2.8. Powder X-ray Diffraction
2.9. Scanning Electron Microscopy (SEM)
2.10. Specific Surface Area and Pore Volume
2.11. Particle Size
2.12. High Performance Liquid Chromatography (HPLC) Analysis
2.13. Dissolution Testing
3. Results
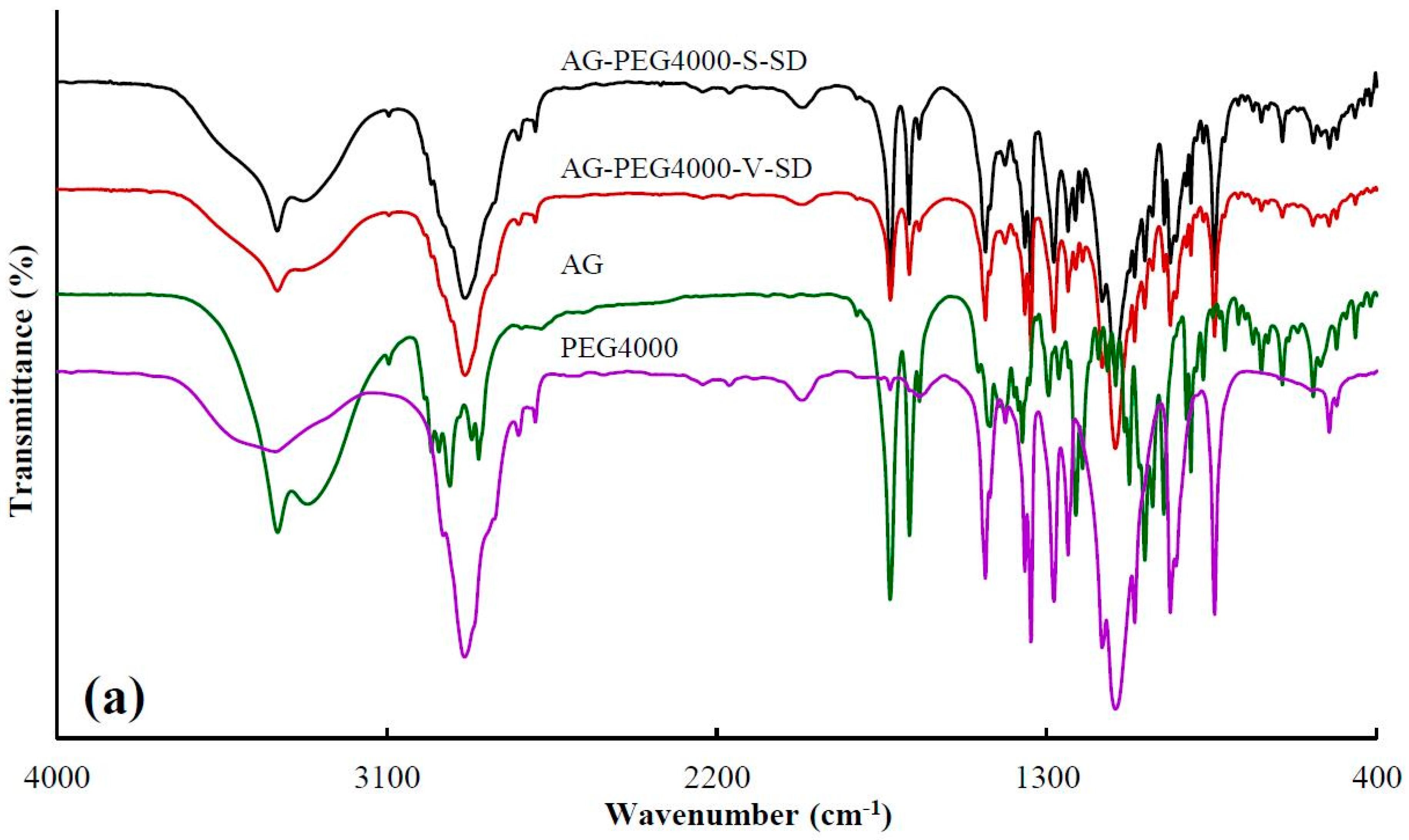
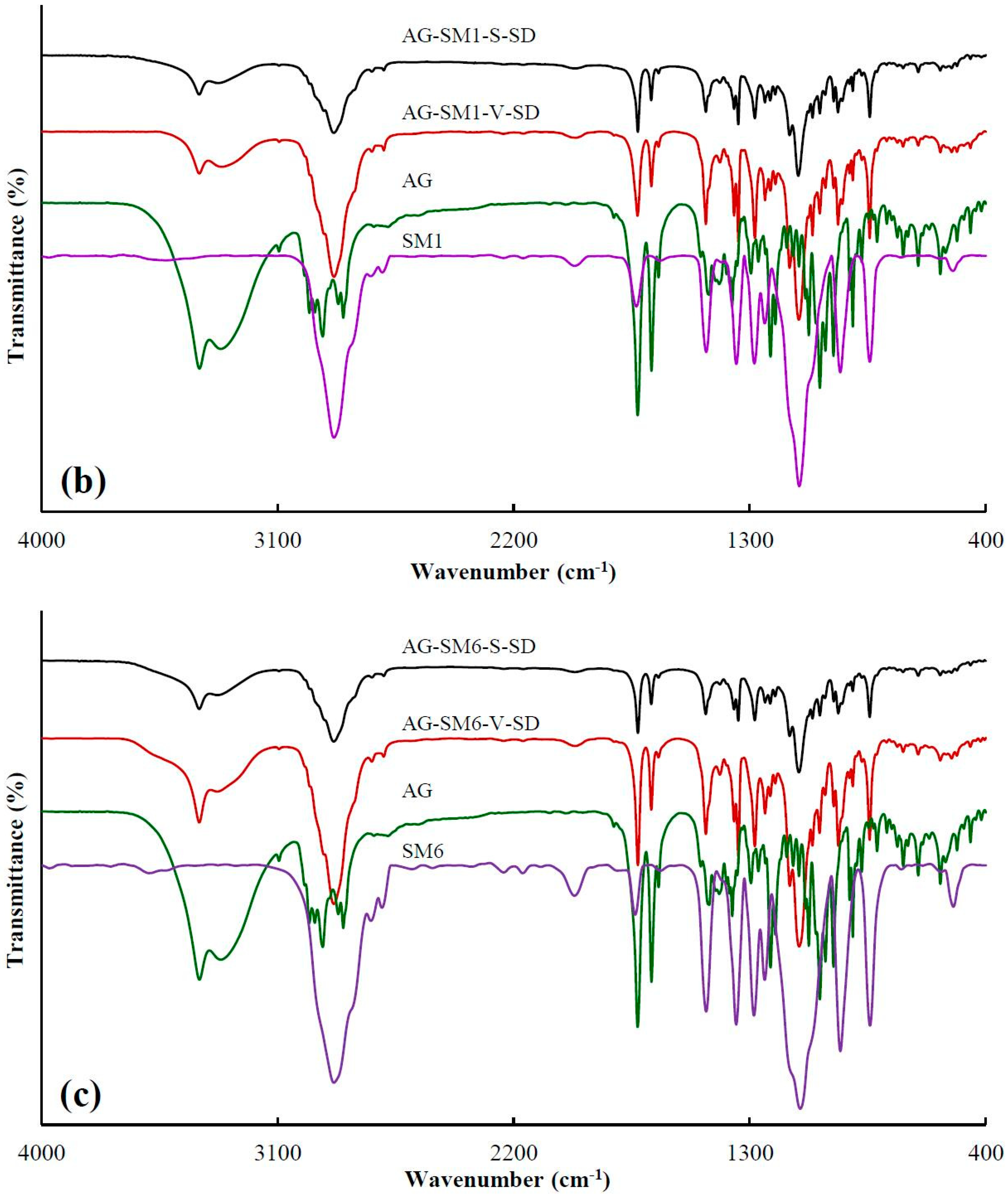
3.1. Investigation of Drug–Carrier Interactions by FT-IR
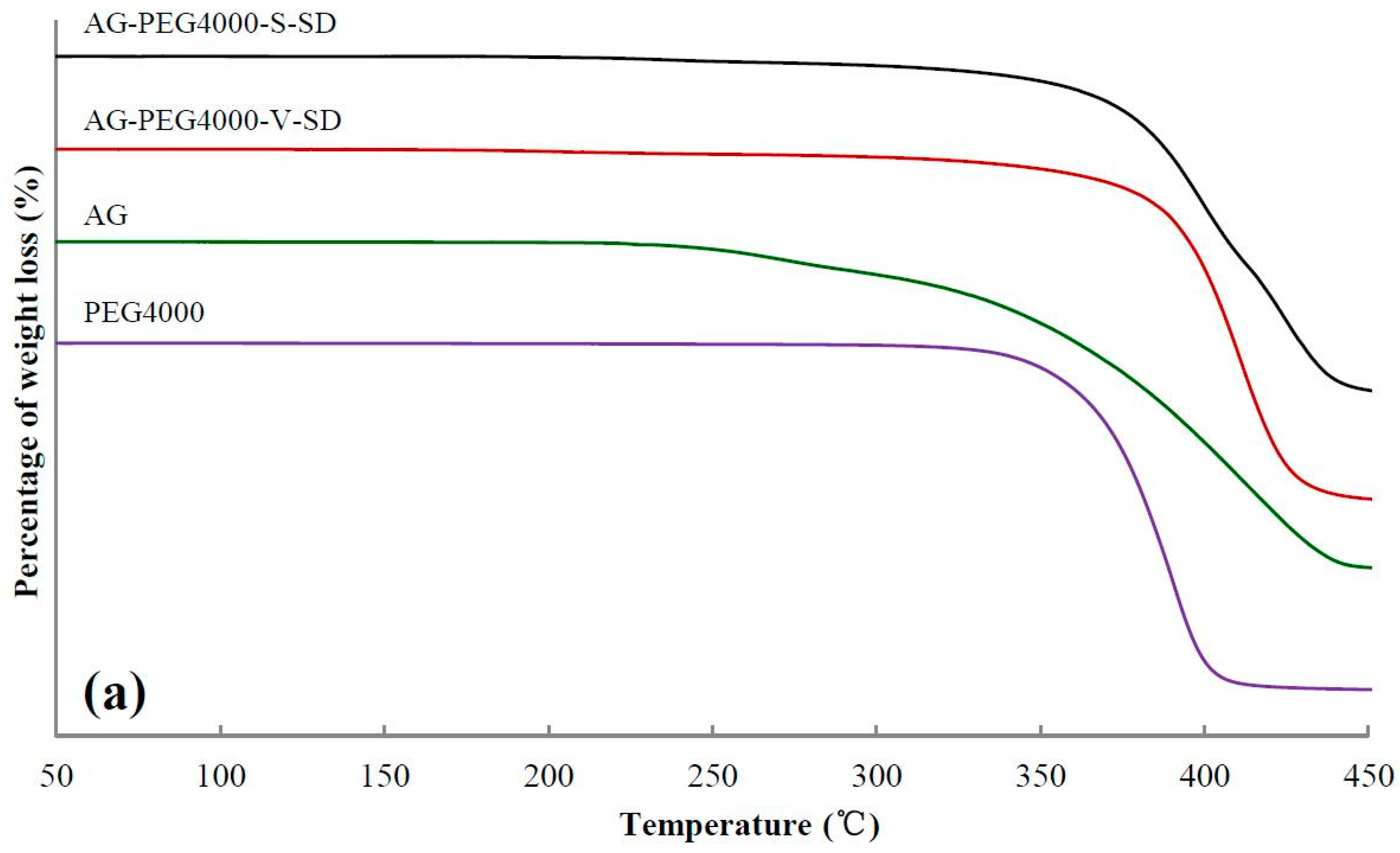
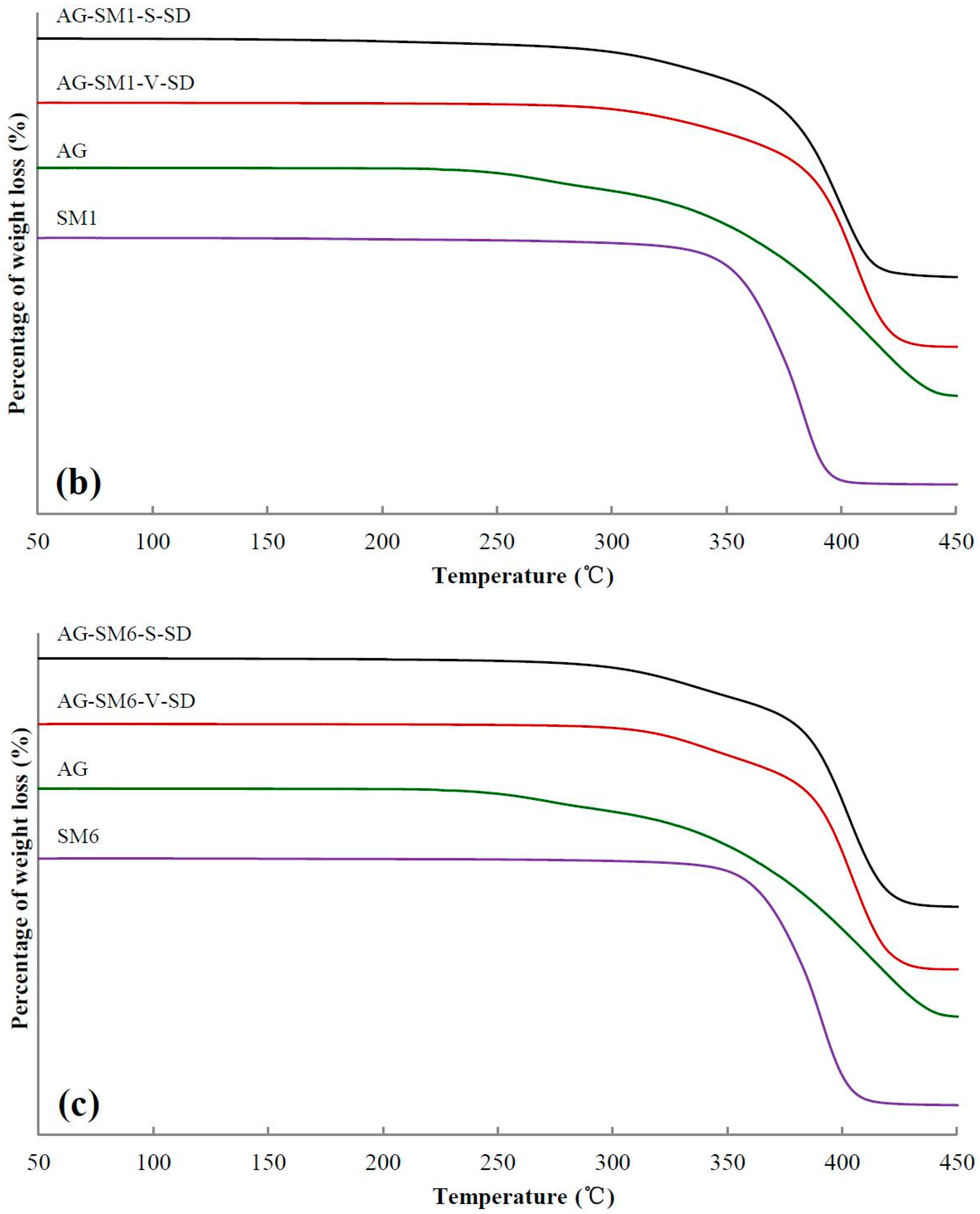
3.2. Investigation of Thermal Stability by TG
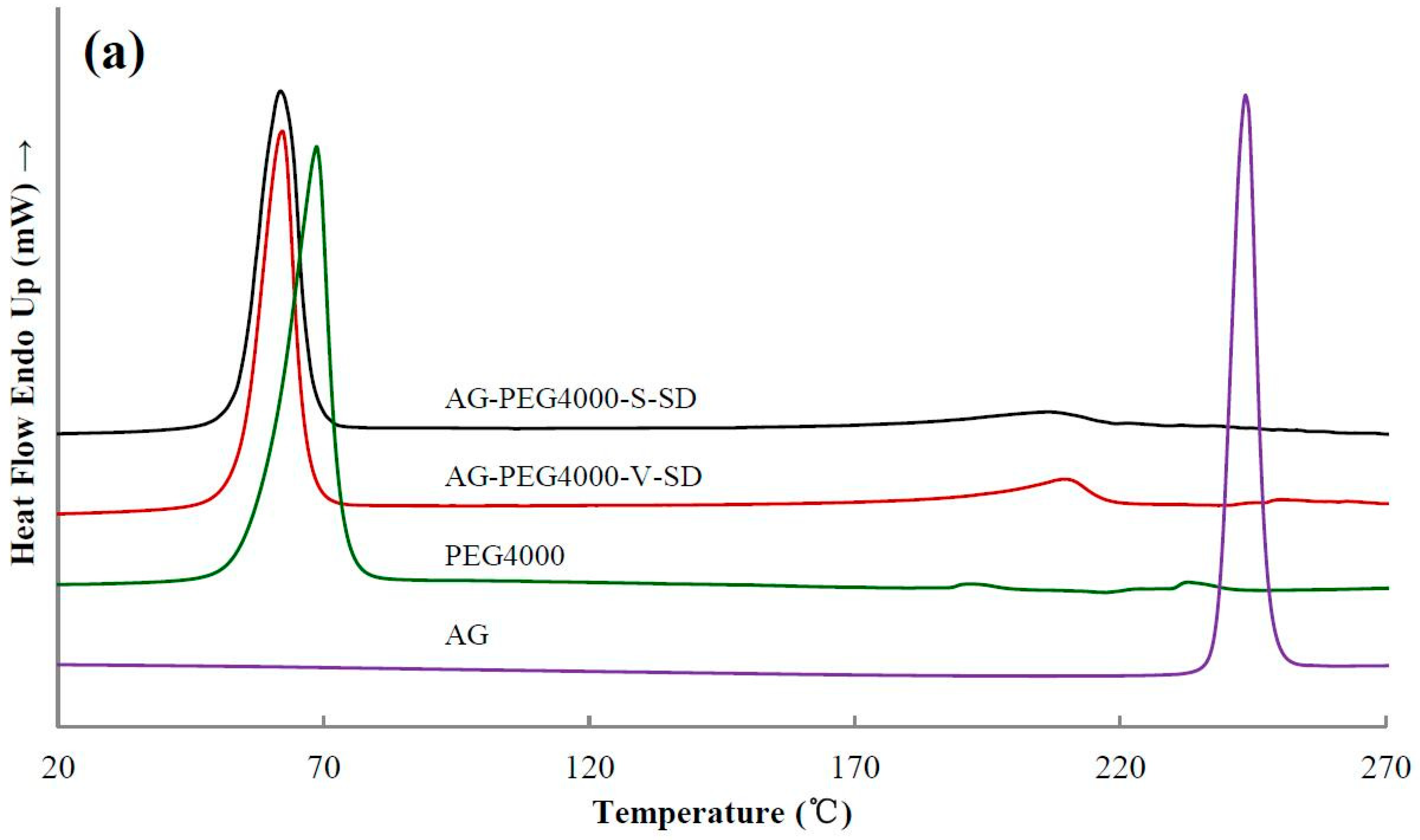
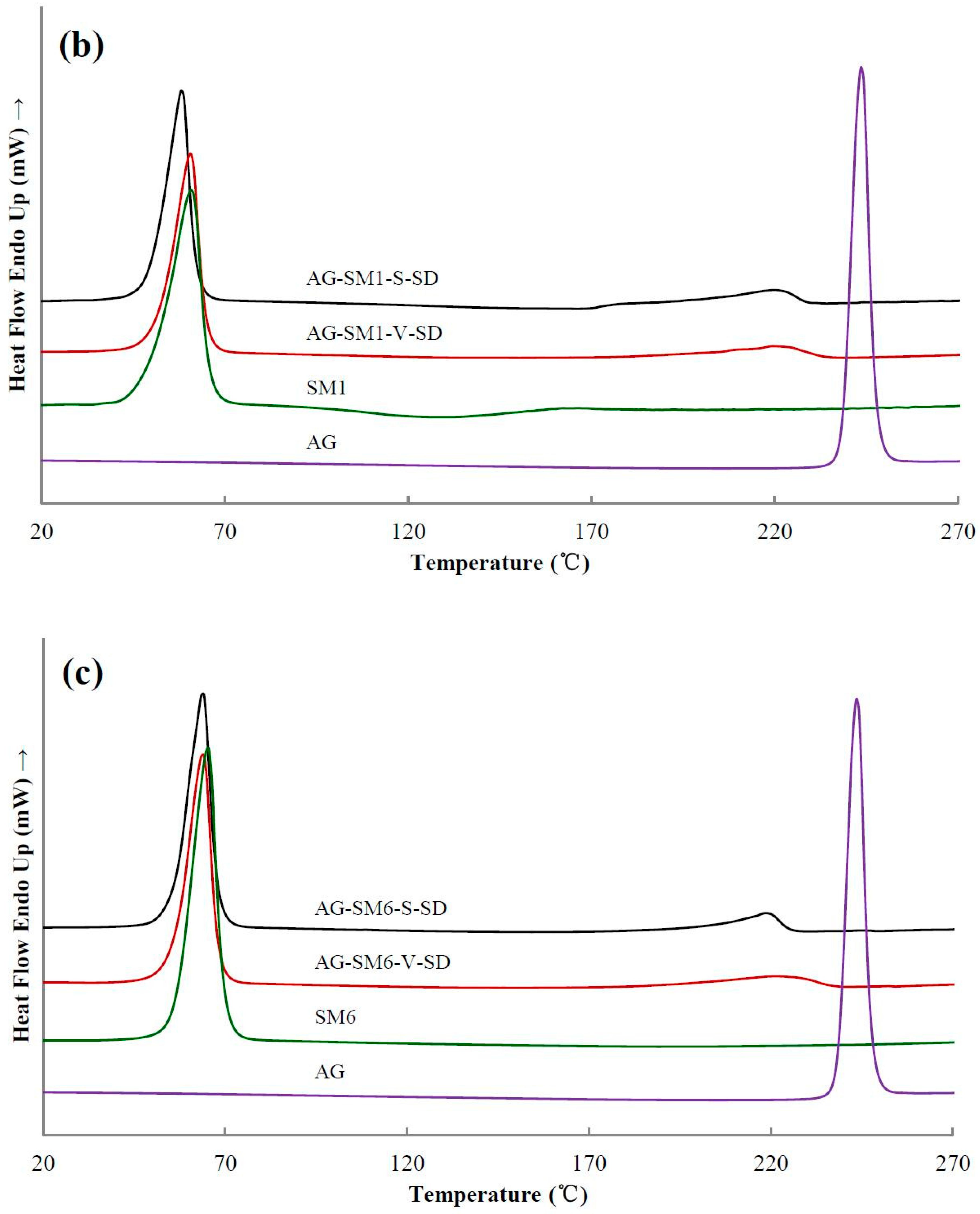
3.3. Solid State Characterization by DSC
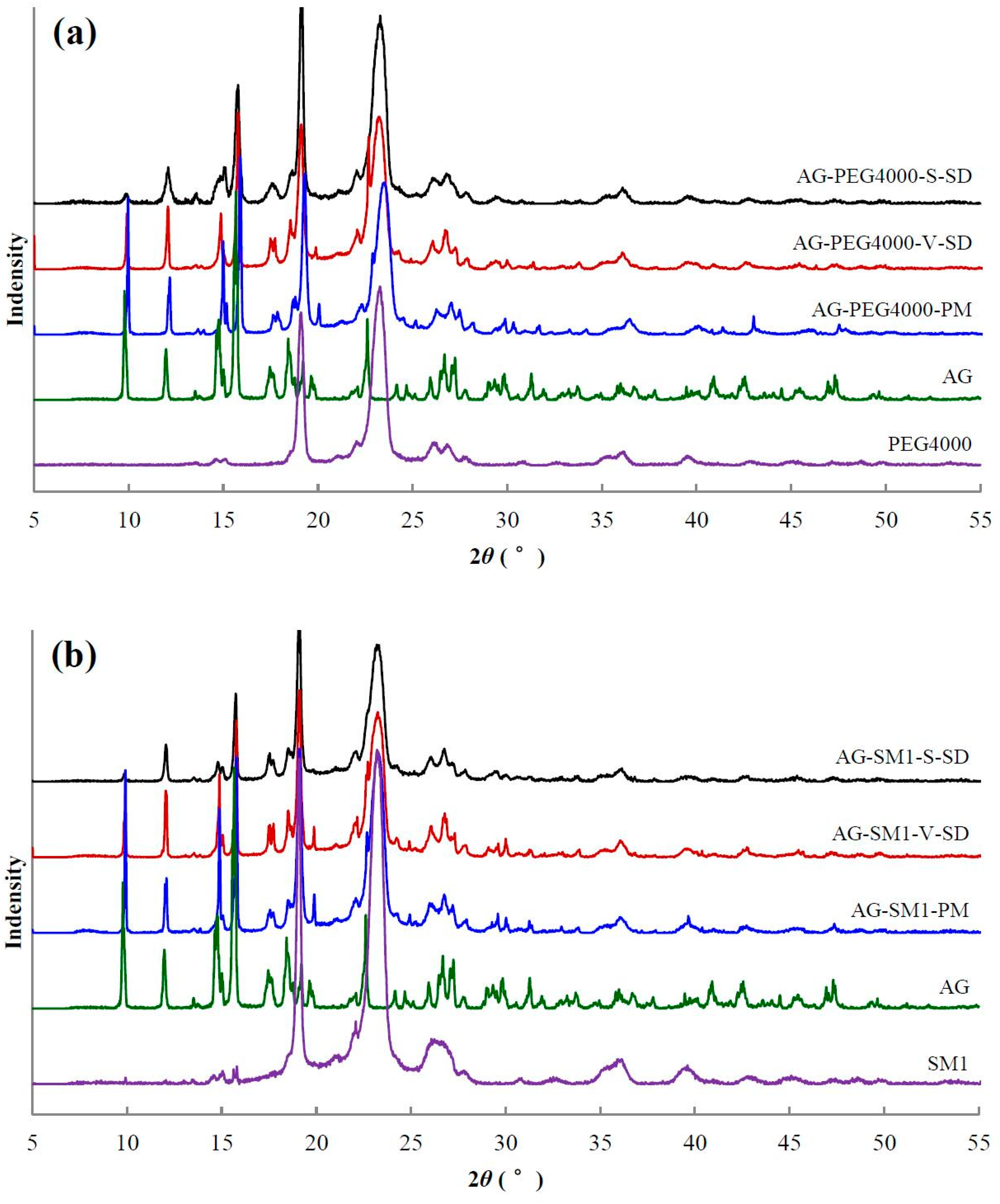
3.4. Solid State Characterization by XRD
3.5. Morphological Evaluation
3.6. Specific Surface Area, Pore Volume, and Particle Size
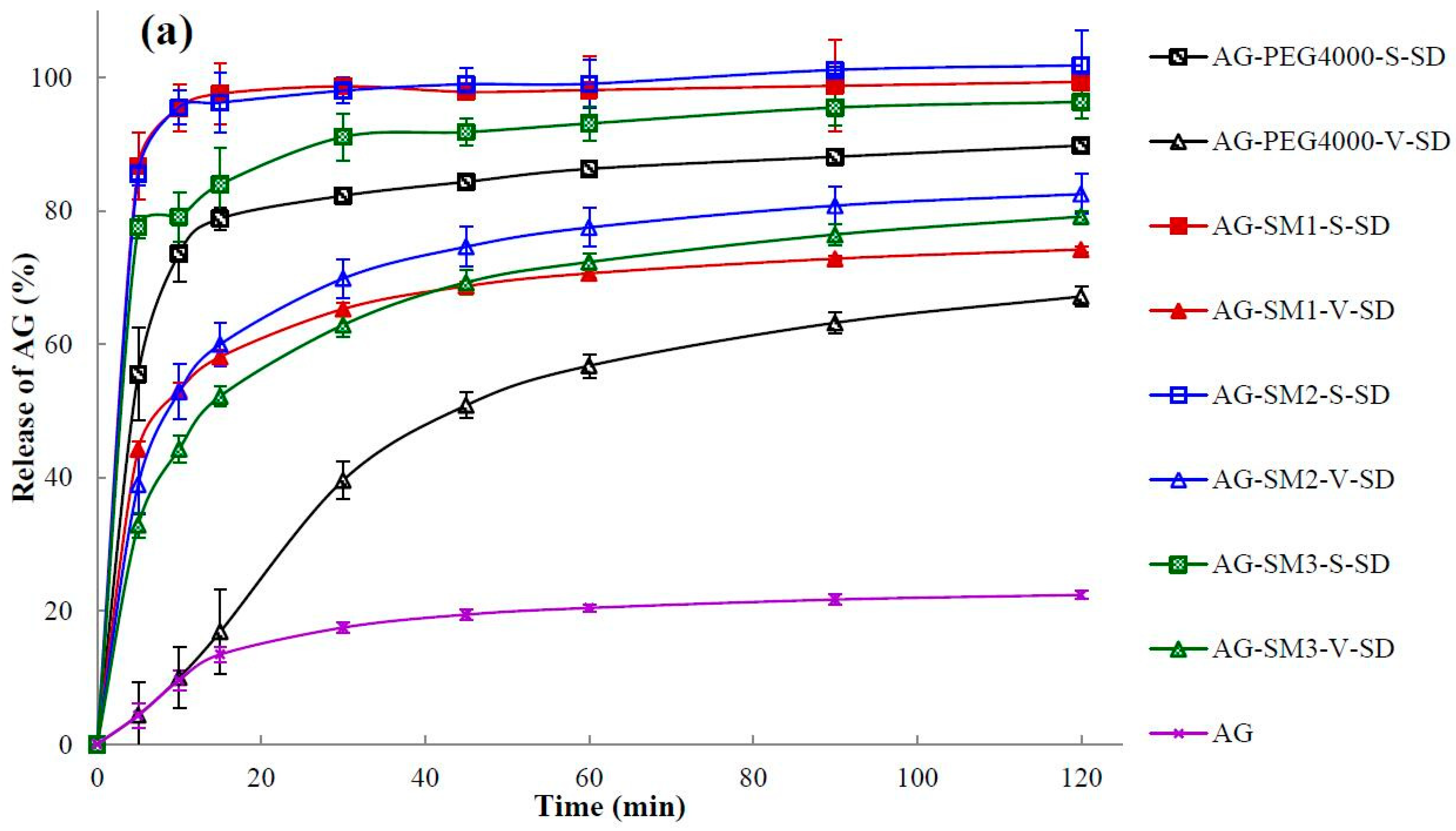
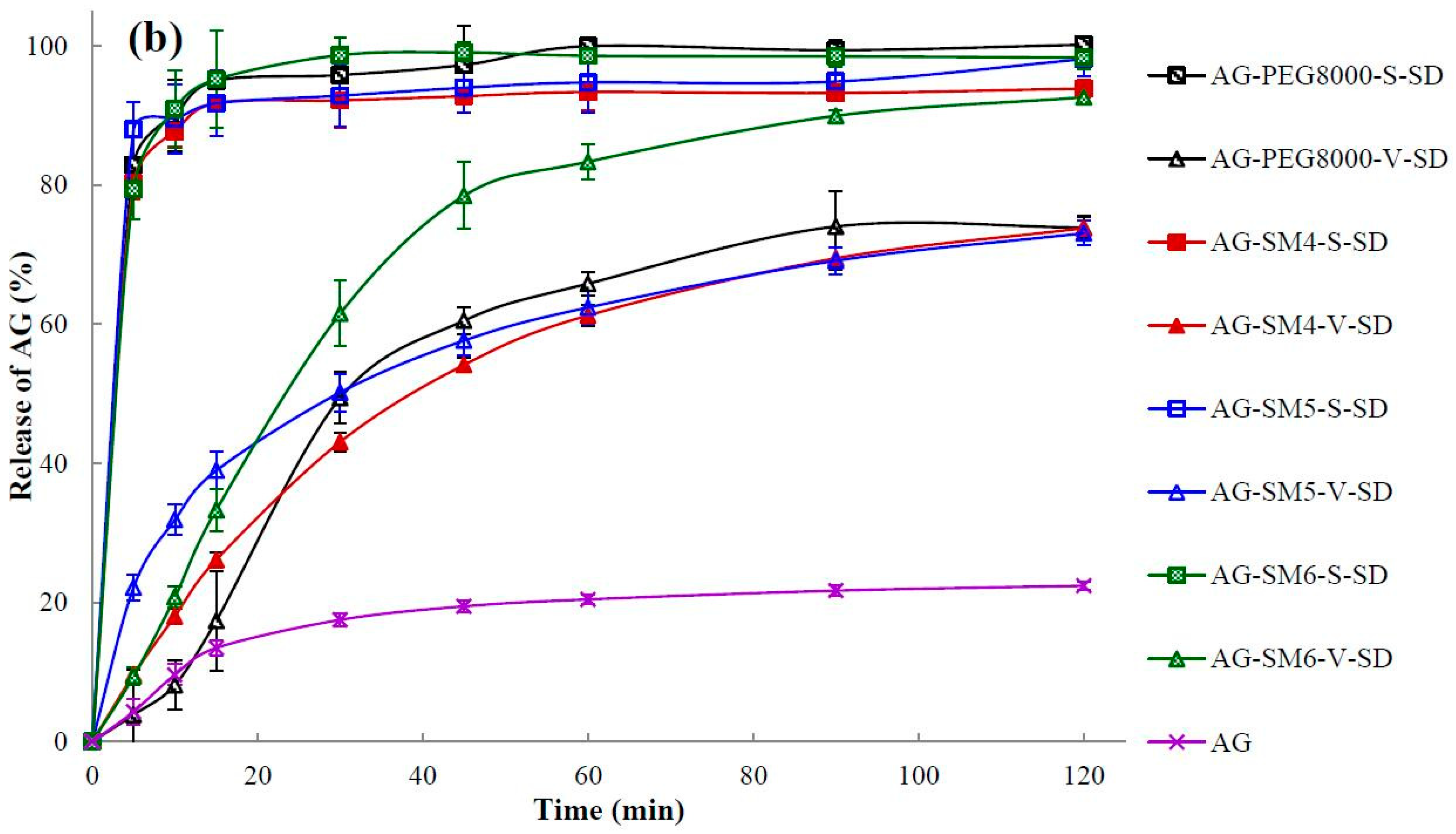
3.7. Dissolution Testing
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- FDA. Waiver of in vivo bioavailability and bioequivalence studies for immediate-release solid oral dosage forms based on a biopharmaceutics classification system. In Guidance for Industry; U. S. Food and Drug Administration: Rockwell, MD, USA, 2000. [Google Scholar]
- Kawabata, Y.; Wada, K.; Nakatani, M.; Yamada, S.; Onoue, S. Formulation design for poorly water-soluble drugs based on biopharmaceutics classification system: Basic approaches and practical applications. Int. J. Pharm. 2011, 420, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Vasconcelos, T.; Sarmento, B.; Costa, P. Solid dispersions as strategy to improve oral bioavailability of poor water soluble drugs. Drug Discov. Today 2007, 12, 1068–1075. [Google Scholar] [CrossRef] [PubMed]
- Vo, C.L.; Park, C.; Lee, B.J. Current trends and future perspectives of solid dispersions containing poorly water-soluble drugs. Eur. J. Pharm. Biopharm. 2013, 85, 799–813. [Google Scholar] [CrossRef]
- Chokshi, R.J.; Zia, H.; Sandhu, H.K.; Shah, N.H.; Malick, W.A. Improving the dissolution rate of poorly water soluble drug by solid dispersion and solid solution: Pros and cons. Drug Deliv. 2007, 14, 33–45. [Google Scholar] [CrossRef] [PubMed]
- Shi, N.-Q.; Lei, Y.-S.; Song, L.-M.; Yao, J.; Zhang, X.-B.; Wang, X.-L. Impact of amorphous and semicrystalline polymers on the dissolution and crystallization inhibition of pioglitazone solid dispersions. Powder Technol. 2013, 247, 211–221. [Google Scholar] [CrossRef]
- Jo, K.; Cho, J.M.; Lee, H.; Kim, E.K.; Kim, H.C.; Kim, H.; Lee, J. Enhancement of Aqueous Solubility and Dissolution of Celecoxib through Phosphatidylcholine-Based Dispersion Systems Solidified with Adsorbent Carriers. Pharmaceutics 2018, 11. [Google Scholar] [CrossRef]
- Leuner, C. Improving drug solubility for oral delivery using solid dispersions. Eur. J. Pharm. Biopharm. 2000, 50, 47–60. [Google Scholar] [CrossRef]
- Barmpalexis, P.; Koutsidis, I.; Karavas, E.; Louka, D.; Papadimitriou, S.A.; Bikiaris, D.N. Development of PVP/PEG mixtures as appropriate carriers for the preparation of drug solid dispersions by melt mixing technique and optimization of dissolution using artificial neural networks. Eur. J. Pharm. Biopharm. 2013, 85, 1219–1231. [Google Scholar] [CrossRef] [PubMed]
- Mahmah, O.; Tabbakh, R.; Kelly, A.; Paradkar, A. A comparative study of the effect of spray drying and hot-melt extrusion on the properties of amorphous solid dispersions containing felodipine. J. Pharm. Pharmacol. 2014, 66, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Thenmozhi, K.; Yoo, Y.J. Enhanced solubility of piperine using hydrophilic carrier-based potent solid dispersion systems. Drug Dev. Ind. Pharm. 2017, 43, 1501–1509. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, T.; Aso, Y.; Yoshioka, S.; Kawanishi, T. Differences in crystallization rate of nitrendipine enantiomers in amorphous solid dispersions with HPMC and HPMCP. Int. J. Pharm. 2011, 407, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Vo, A.Q.; Feng, X.; Zhang, J.; Zhang, F.; Repka, M.A. Dual mechanism of microenvironmental pH modulation and foam melt extrusion to enhance performance of HPMCAS based amorphous solid dispersion. Int. J. Pharm. 2018, 550, 216–228. [Google Scholar] [CrossRef] [PubMed]
- Lehmkemper, K.; Kyeremateng, S.O.; Heinzerling, O.; Degenhardt, M.; Sadowski, G. Long-Term Physical Stability of PVP-and PVPVA-Amorphous Solid Dispersions. Mol. Pharm. 2017, 14, 157–171. [Google Scholar] [CrossRef] [PubMed]
- Marsac, P.; Li, T.; Taylor, L. Estimation of Drug-Polymer Miscibility and Solubility in Amorphous Solid Dispersions Using Experimentally Determined Interaction Parameters. Pharm. Res.-Dordr. 2009, 26, 139–151. [Google Scholar] [CrossRef] [PubMed]
- Marsac, P.J.; Shamblin, S.L.; Taylor, L.S. Theoretical and practical approaches for prediction of drug-polymer miscibility and solubility. Pharm. Res. 2006, 23, 2417–2426. [Google Scholar] [CrossRef]
- Marsac, P.J.; Konno, H.; Rumondor, A.C.; Taylor, L.S. Recrystallization of nifedipine and felodipine from amorphous molecular level solid dispersions containing poly(vinylpyrrolidone) and sorbed water. Pharm. Res. 2008, 25, 647–656. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, H.; Hui-Gu, C.; Atef, E. Correlating the behavior of polymers in solution as precipitation inhibitor to its amorphous stabilization ability in solid dispersions. J. Pharm. Sci. 2013, 102, 1924–1935. [Google Scholar] [CrossRef]
- Bialleck, S.; Rein, H. Preparation of starch-based pellets by hot-melt extrusion. Eur. J. Pharm. Biopharm. 2011, 79, 440–448. [Google Scholar] [CrossRef]
- Eloy, J.O.; Marchetti, J.M. Solid dispersions containing ursolic acid in Poloxamer 407 and PEG 6000: A comparative study of fusion and solvent methods. Powder Technol. 2014, 253, 98–106. [Google Scholar] [CrossRef]
- Matsuda, T.; Kuroyanagi, M.; Sugiyama, S.; Umehara, K.; Ueno, A.; Nishi, K. Cell differentiation-inducing diterpenes from Andrographis paniculata Nees. Chem. Pharm. Bull. (Tokyo) 1994, 42, 1216–1225. [Google Scholar] [CrossRef]
- Calabrese, C.; Berman, S.H.; Babish, J.G.; Ma, X.; Shinto, L.; Dorr, M.; Wells, K.; Wenner, C.A.; Standish, L.J. A phase I trial of andrographolide in HIV positive patients and normal volunteers. Phytother. Res. 2000, 14, 333–338. [Google Scholar] [CrossRef]
- Zhang, X.F.; Tan, B.K. Antihyperglycaemic and anti-oxidant properties of Andrographis paniculata in normal and diabetic rats. Clin. Exp. Pharmacol. Physiol. 2000, 27, 358–363. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.C.; Chen, C.F.; Chiou, W.F. Andrographolide prevents oxygen radical production by human neutrophils: Possible mechanism(s) involved in its anti-inflammatory effect. Br. J. Pharmacol. 2002, 135, 399–406. [Google Scholar] [CrossRef] [PubMed]
- Singha, P.K.; Roy, S.; Dey, S. Protective activity of andrographolide and arabinogalactan proteins from Andrographis paniculata Nees. against ethanol-induced toxicity in mice. J. Ethnopharmacol. 2007, 111, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Sermkaew, N.; Ketjinda, W.; Boonme, P.; Phadoongsombut, N.; Wiwattanapatapee, R. Liquid and solid self-microemulsifying drug delivery systems for improving the oral bioavailability of andrographolide from a crude extract of Andrographis paniculata. Eur. J. Pharm. Sci. 2013, 50, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Wen, L.; Xia, N.; Chen, X.; Li, Y.; Hong, Y.; Liu, Y.; Wang, Z.; Liu, Y. Activity of antibacterial, antiviral, anti-inflammatory in compounds andrographolide salt. Eur. J. Pharmacol. 2014, 740, 421–427. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Wang, F.; Xu, H.; Liu, H.; Meng, Q.; Liu, W. Development of andrographolide loaded PLGA microspheres: optimization, characterization and in vitro-in vivo correlation. Int. J. Pharm. 2014, 475, 475–484. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Hu, X.; Liu, X.; Dandan, Y.; Di, D.; Yin, T.; Zhang, S.; Tang, X. Dry state microcrystals stabilized by an HPMC film to improve the bioavailability of andrographolide. Int. J. Pharm. 2015, 493, 214–223. [Google Scholar] [CrossRef]
- Bhardwaj, V.; Trasi, N.S.; Zemlyanov, D.Y.; Taylor, L.S. Surface area normalized dissolution to study differences in itraconazole-copovidone solid dispersions prepared by spray-drying and hot melt extrusion. Int. J. Pharm. 2018, 540, 106–119. [Google Scholar] [CrossRef]
- Marasini, N.; Tran, T.H.; Poudel, B.K.; Cho, H.J.; Choi, Y.K.; Chi, S.C.; Choi, H.G.; Yong, C.S.; Kim, J.O. Fabrication and evaluation of pH-modulated solid dispersion for telmisartan by spray-drying technique. Int. J. Pharm. 2013, 441, 424–432. [Google Scholar] [CrossRef]
- Huo, T.; Tao, C.; Zhang, M.; Liu, Q.; Lin, B.; Liu, Z.; Zhang, J.; Zhang, M.; Yang, H.; Wu, J.; et al. Preparation and comparison of tacrolimus-loaded solid dispersion and self-microemulsifying drug delivery system by in vitro/in vivo evaluation. Eur. J. Pharm. Sci. 2018, 114, 74–83. [Google Scholar] [CrossRef] [PubMed]
- Shamma, R.N.; Basha, M. Solupluse (R): A novel polymeric solubilizer for optimization of Carvedilol solid dispersions: Formulation design and effect of method of preparation. Powder Technol. 2013, 237, 406–414. [Google Scholar] [CrossRef]
- Zhao, G.W.; Zhang, S.D.; Liang, X.L.; Zhong, Y.Q.; Liao, Z.G. Preparation and in vitro evaluation of Andrographolide-polyethylene glycol solid dispersion. Chin. J. Pharm. 2017, 48, 200–203. [Google Scholar]
- Sousa, J.J.; Sousa, A.; Podczeck, F.; Newton, J.M. Factors influencing the physical characteristics of pellets obtained by extrusion-spheronization. Int.J. Pharm. 2002, 232, 91–106. [Google Scholar] [CrossRef]
- Wegiel, L.A.; Mauer, L.J.; Edgar, K.J.; Taylor, L.S. Mid-infrared spectroscopy as a polymer selection tool for formulating amorphous solid dispersions. J. Pharm. Pharmacol. 2014, 66, 244–255. [Google Scholar] [CrossRef] [PubMed]
- Albadarin, A.B.; Potter, C.B.; Davis, M.T.; Iqbal, J.; Korde, S.; Pagire, S.; Paradkar, A.; Walker, G. Development of stability-enhanced ternary solid dispersions via combinations of HPMCP and Soluplus((R)) processed by hot melt extrusion. Int. J. Pharm. 2017, 532, 603–611. [Google Scholar] [CrossRef] [PubMed]
- Veronez, I.P.; Daniel, J.S.P.; Júnior, C.E.C.; Garcia, J.S.; Trevisan, M.G. Development, characterization, and stability studies of ethinyl estradiol solid dispersion. J. Therm. Anal. Calorim. 2015, 120, 573–581. [Google Scholar] [CrossRef]
- Lim, R.T.Y.; Ng, W.K.; Tan, R.B.H. Dissolution enhancement of indomethacin via amorphization using co-milling and supercritical co-precipitation processing. Powder Technol. 2013, 240, 79–87. [Google Scholar] [CrossRef]
- Zhou, D.; Zhang, G.G.; Law, D.; Grant, D.J.; Schmitt, E.A. Thermodynamics, molecular mobility and crystallization kinetics of amorphous griseofulvin. Mol. Pharm. 2008, 5, 927–936. [Google Scholar] [CrossRef]
- Mohammadi, G.; Barzegar-Jalali, M.; Valizadeh, H.; Nazemiyeh, H.; Barzegar-Jalali, A.; Siahi Shadbad, M.R.; Adibkia, K.; Zare, M. Reciprocal powered time model for release kinetic analysis of ibuprofen solid dispersions in oleaster powder, microcrystalline cellulose and crospovidone. J. Pharm. Pharm. Sci. 2010, 13, 152–161. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
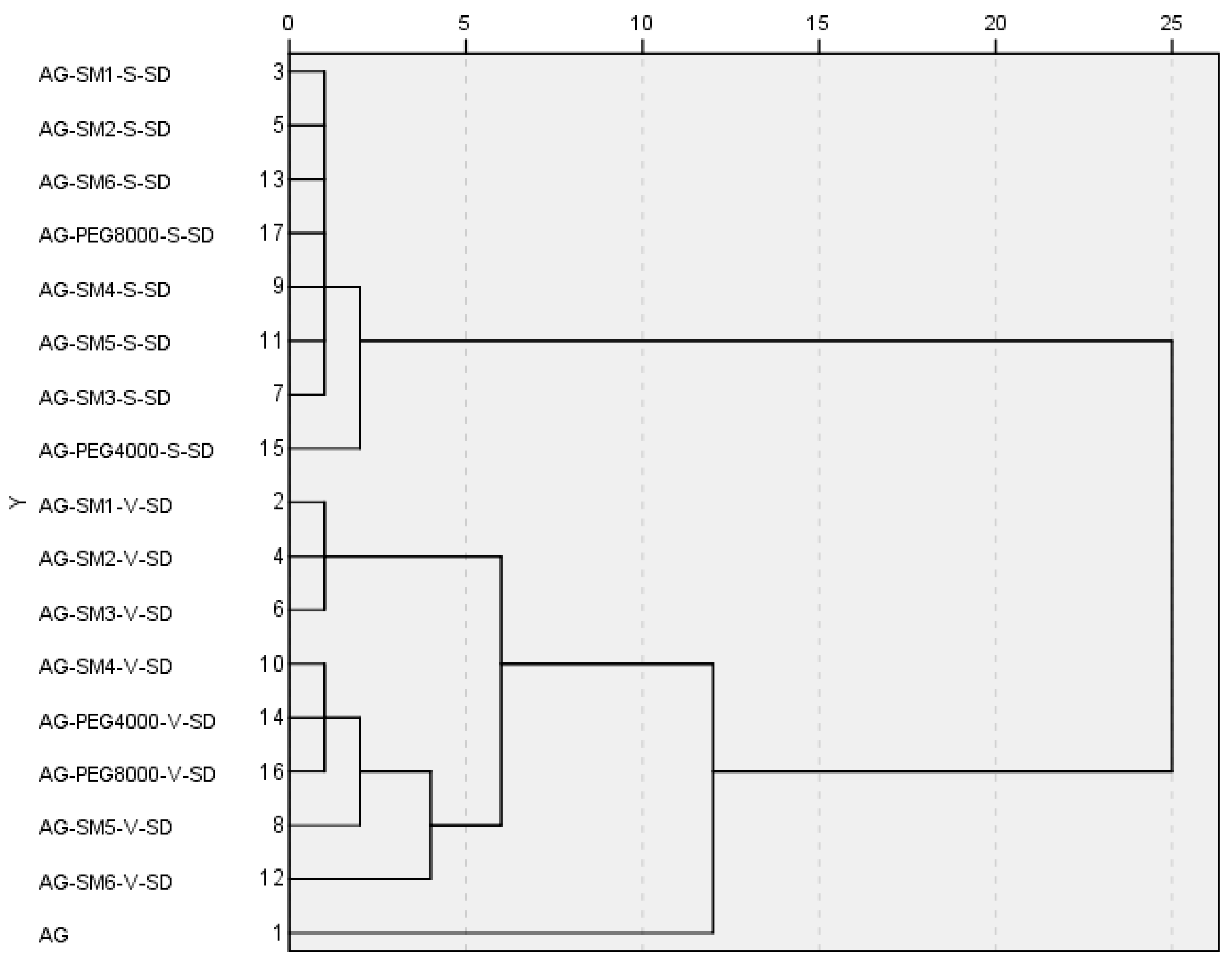{kind=link}
| Sample | Peak Position (cm−1) | Sample | Peak Position (cm−1) |
|---|---|---|---|
| AG | 3317.0 ± 1.4 | ||
| AG–PEG4000–S–SD | 3323.9 ± 0.9 | AG–PEG4000–V–SD | 3332.4 ± 1.7 |
| AG–PEG8000–S–SD | 3321.0 ± 1.0 | AG–PEG8000–V–SD | 3331.3 ± 0.5 |
| AG–SM1–S–SD | 3328.3 ± 0.4 | AG–SM1–V–SD | 3327.4 ± 0.8 |
| AG–SM2–S–SD | 3328.7 ± 1.3 | AG–SM2–V–SD | 3324.3 ± 0.8 |
| AG–SM3–S–SD | 3322.3 ± 1.6 | AG–SM3–V–SD | 3329.9 ± 1.3 |
| AG–SM4–S–SD | 3327.7 ± 0.3 | AG–SM4–V–SD | 3322.7 ± 2.5 |
| AG–SM5–S–SD | 3328.4 ± 1.3 | AG–SM5–V–SD | 3325.4 ± 1.2 |
| AG–SM6–S–SD | 3329.1 ± 0.2 | AG–SM6–V–SD | 3330.4 ± 2.6 |
| Sample | Ti(°C) | Tm(°C) | Sample | Ti(°C) | Tm(°C) | Sample | Ti(°C) |
|---|---|---|---|---|---|---|---|
| AG | 272 | 243 | |||||
| AG–PEG4000 –S–SD | 335 | 206 | AG–PEG4000 –V–SD | 345 | 210 | AG–PEG4000 –PM | 346 |
| AG–PEG8000 –S–SD | 336 | 209 | AG–PEG8000 –V–SD | 348 | 217 | AG–PEG8000 –PM | 345 |
| AG–SM1–S–SD | 298 | 220 | AG–SM1–V–SD | 318 | 220 | AG–SM1–PM | 304 |
| AG–SM2–S–SD | 297 | 221 | AG–SM2–V–SD | 317 | 229 | AG–SM2–PM | 318 |
| AG–SM3–S–SD | 319 | 221 | AG–SM3–V–SD | 304 | 230 | AG–SM3–PM | 317 |
| AG–SM4–S–SD | 311 | 218 | AG–SM4–V–SD | 308 | 231 | AG–SM4–PM | 329 |
| AG–SM5–S–SD | 327 | 218 | AG–SM5–V–SD | 305 | 227 | AG–SM5–PM | 336 |
| AG–SM6–S–SD | 308 | 219 | AG–SM6–V–SD | 323 | 221 | AG–SM6–PM | 323 |
| Sample | Specific Surface Area (m2/g) | Pore Volume (×103, m3/g) | d0.5 (μm) | Span | Sample | Specific Surface Area (m2/g) | Pore Volume (×103, m3/g) | d0.5 (μm) | Span |
|---|---|---|---|---|---|---|---|---|---|
| AG–PEG4000 –S–SD | 0.5641 | 0.964 | 42.8 | 2.748 | AG–PEG4000 –V–SD | 0.2969 | 1.650 | 109.4 | 7.036 |
| AG–PEG8000 –S–SD | 0.1943 | 0.148 | 33.1 | 4.524 | AG–PEG8000 –V–SD | 0.9994 | 1.745 | 113.3 | 4.425 |
| AG–SM1–S–SD | 0.5871 | 0.689 | 52.7 | 2.538 | AG–SM1–V–SD | 0.3739 | 3.546 | 131.3 | 6.747 |
| AG–SM2–S–SD | 0.2857 | 0.932 | 79.6 | 3.920 | AG–SM2–V–SD | 0.2765 | 1.761 | 96.0 | 8.696 |
| AG–SM3–S–SD | 0.1126 | 0.656 | 64.3 | 2.946 | AG–SM3–V–SD | 0.0337 | 3.405 | 125.4 | 5.621 |
| AG–SM4–S–SD | 0.3863 | 1.094 | 48.6 | 3.302 | AG–SM4–V–SD | 0.2478 | 3.411 | 127.2 | 5.198 |
| AG–SM5–S–SD | 0.3056 | 0.965 | 52.4 | 2.813 | AG–SM5–V–SD | 0.6194 | 1.542 | 99.8 | 5.199 |
| AG–SM6–S–SD | 0.5387 | 0.957 | 48.5 | 2.158 | AG–SM6–V–SD | 0.4387 | 2.160 | 144.7 | 5.020 |
| Sample | Q5min (%) | t85% (min) | Sample | Q5min (%) | t85% (min) |
|---|---|---|---|---|---|
| AG | 4.32 ± 1.84 | >120 | |||
| AG–PEG4000–S–SD | 55.49 ± 6.95 | 50 | AG–PEG4000–V–SD | 4.37 ± 2.50 | >120 |
| AG–PEG8000–S–SD | 82.86 ± 3.90 | 6 | AG–PEG8000–V–SD | 3.86 ± 1.90 | >120 |
| AG–SM1–S–SD | 86.72 ± 5.10 | 5 | AG–SM1–V–SD | 44.22 ± 1.15 | >120 |
| AG–SM2–S–SD | 85.61 ± 1.85 | 5 | AG–SM2–V–SD | 38.97 ± 4.44 | >120 |
| AG–SM3–S–SD | 77.57 ± 1.66 | 17 | AG–SM3–V–SD | 32.85 ± 1.80 | >120 |
| AG–SM4–S–SD | 80.24 ± 2.31 | 7 | AG–SM4–V–SD | 19.64 ± 0.75 | >120 |
| AG–SM5–S–SD | 87.97 ± 4.02 | 5 | AG–SM5–V–SD | 22.17 ± 1.87 | >120 |
| AG–SM6–S–SD | 79.40 ± 4.35 | 6 | AG–SM6–V–SD | 19.40 ± 1.31 | 67 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, G.; Zeng, Q.; Zhang, S.; Zhong, Y.; Wang, C.; Chen, Y.; Ou, L.; Liao, Z. Effect of Carrier Lipophilicity and Preparation Method on the Properties of Andrographolide–Solid Dispersion. Pharmaceutics 2019, 11, 74. https://doi.org/10.3390/pharmaceutics11020074
Zhao G, Zeng Q, Zhang S, Zhong Y, Wang C, Chen Y, Ou L, Liao Z. Effect of Carrier Lipophilicity and Preparation Method on the Properties of Andrographolide–Solid Dispersion. Pharmaceutics. 2019; 11(2):74. https://doi.org/10.3390/pharmaceutics11020074
Chicago/Turabian StyleZhao, Guowei, Qingyun Zeng, Shoude Zhang, Youquan Zhong, Changhao Wang, Yashao Chen, Liquan Ou, and Zhenggen Liao. 2019. "Effect of Carrier Lipophilicity and Preparation Method on the Properties of Andrographolide–Solid Dispersion" Pharmaceutics 11, no. 2: 74. https://doi.org/10.3390/pharmaceutics11020074