Dual Action of the PN159/KLAL/MAP Peptide: Increase of Drug Penetration across Caco-2 Intestinal Barrier Model by Modulation of Tight Junctions and Plasma Membrane Permeability

 , , and
, , and 
Abstract
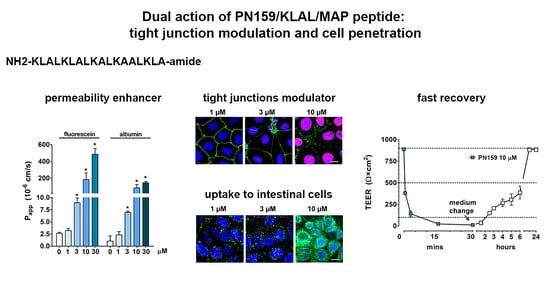
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Peptide Synthesis
2.3. Cell Culture
2.4. Peptide Treatment
2.5. MTT Assay
2.6. Impedance Measurement
2.7. Measurement of the Electrical Resistance of Caco-2 Cell Layers
2.8. Penetration of Marker and Drug Molecules across Caco-2 Cell Layers
2.9. High-Performance Liquid Chromatography (HPLC)Analytical Procedures
2.10. Electron Microscopy
2.11. Immunohistochemistry
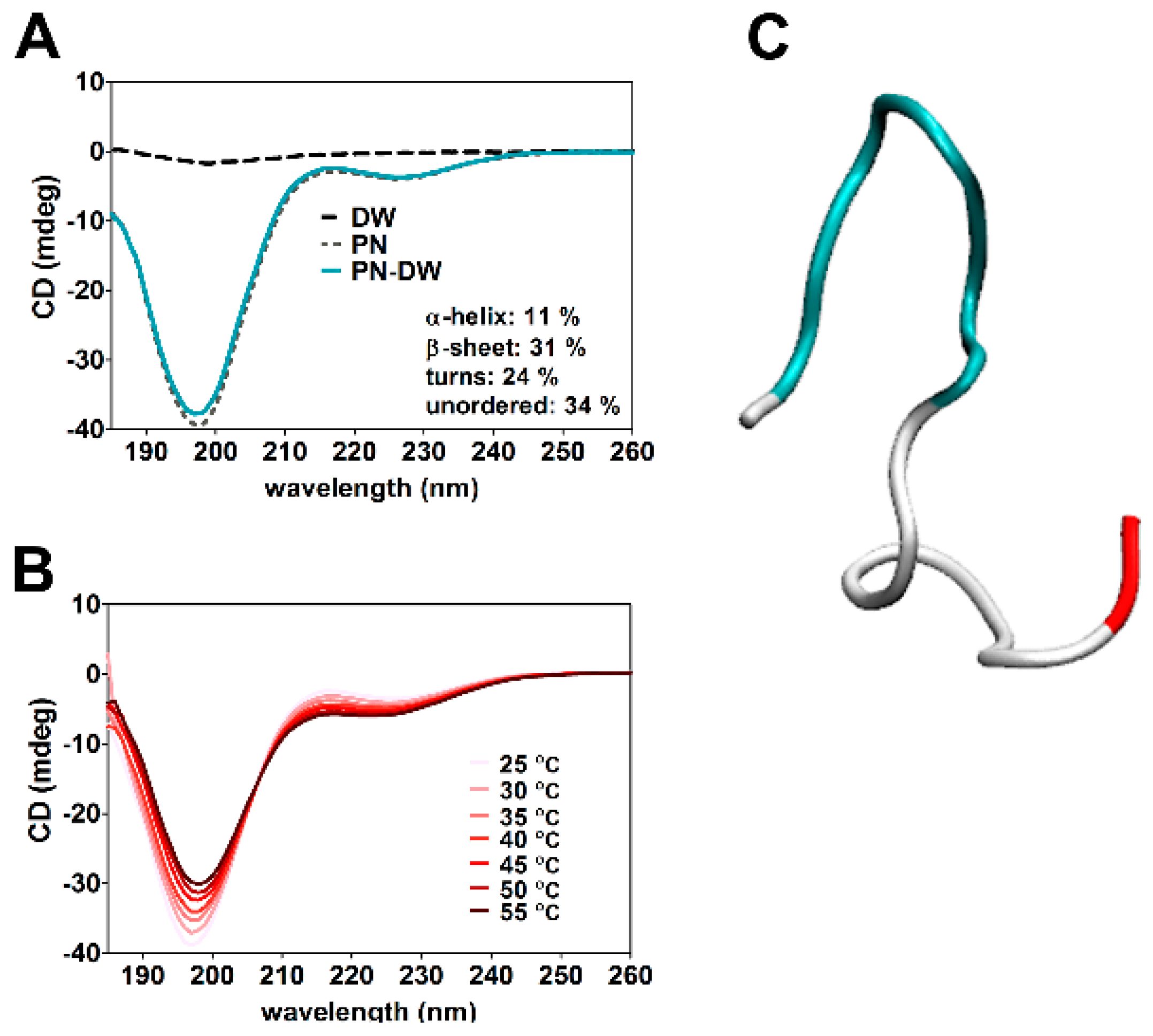
2.12. Circular Dichroism (CD) Spectroscopy
2.13. Molecular Modelling
2.14. Visualization of the Uptake of PN159 Peptide in Caco-2 Cells
2.15. Determination of Minimum Inhibitory Concentration (MIC) on Microbial Pathogens
2.16. Statistical Analysis
3. Results
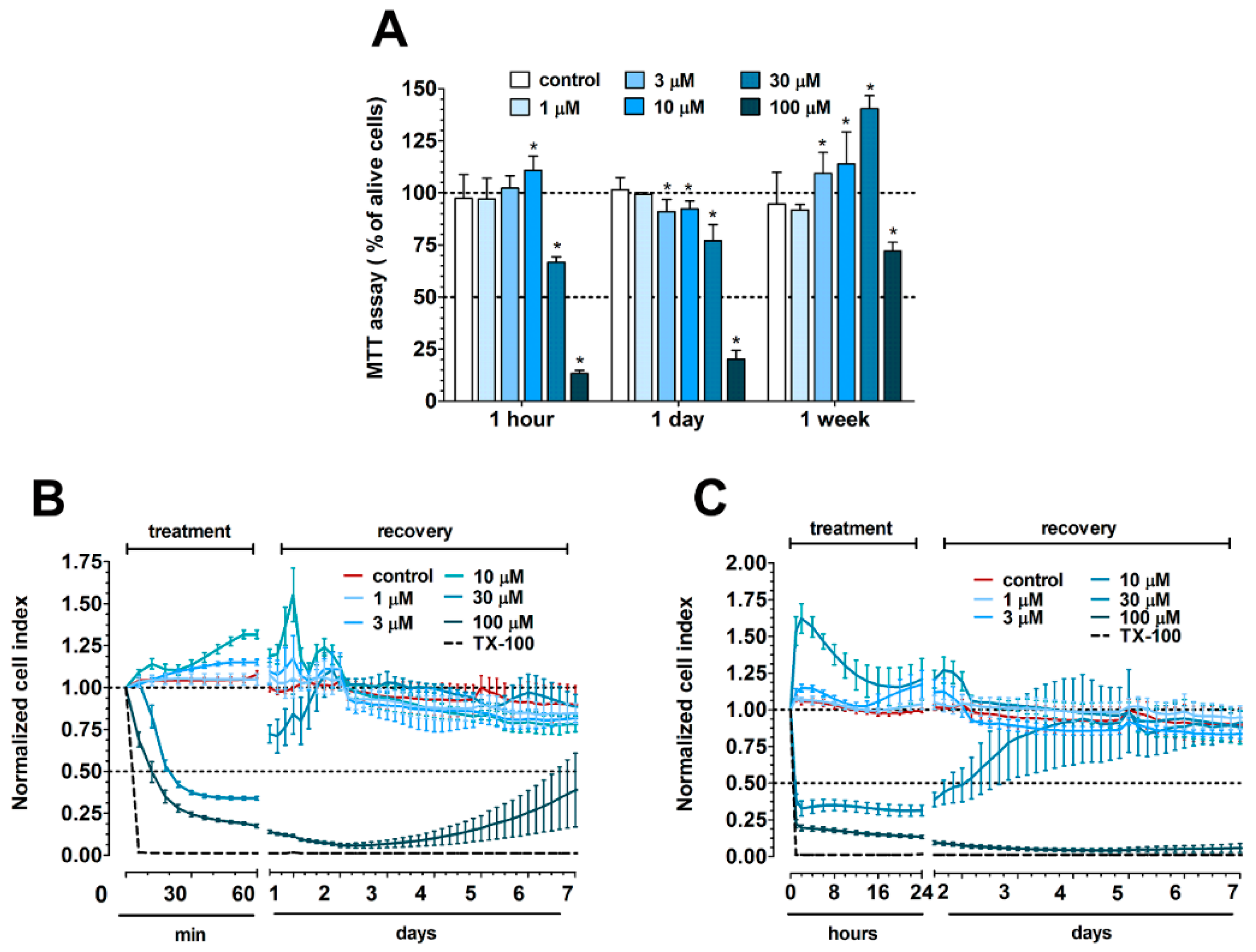
3.1. Concentration-Dependent Effect of PN159 Peptide on Epithelial Cell Viability
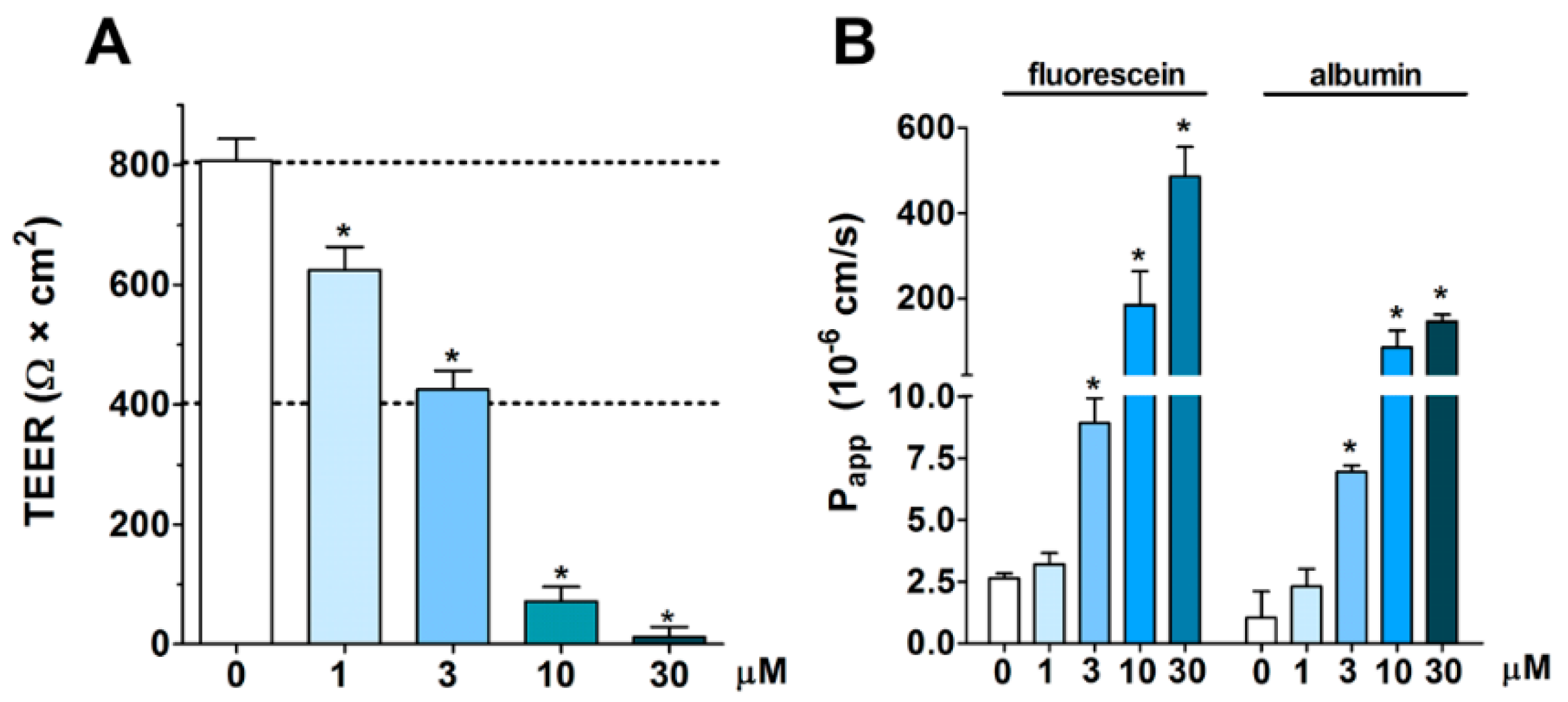
3.2. Concentration-Dependent Effect of PN159 Peptide on Intestinal Epithelial Barrier Integrity
3.3. Reversible Effect of PN159 Peptide on the Opening of the Paracellular Cleft
3.4. Effects of PN159 Peptide on the Penetration of Dextran Marker Molecules and Drugs
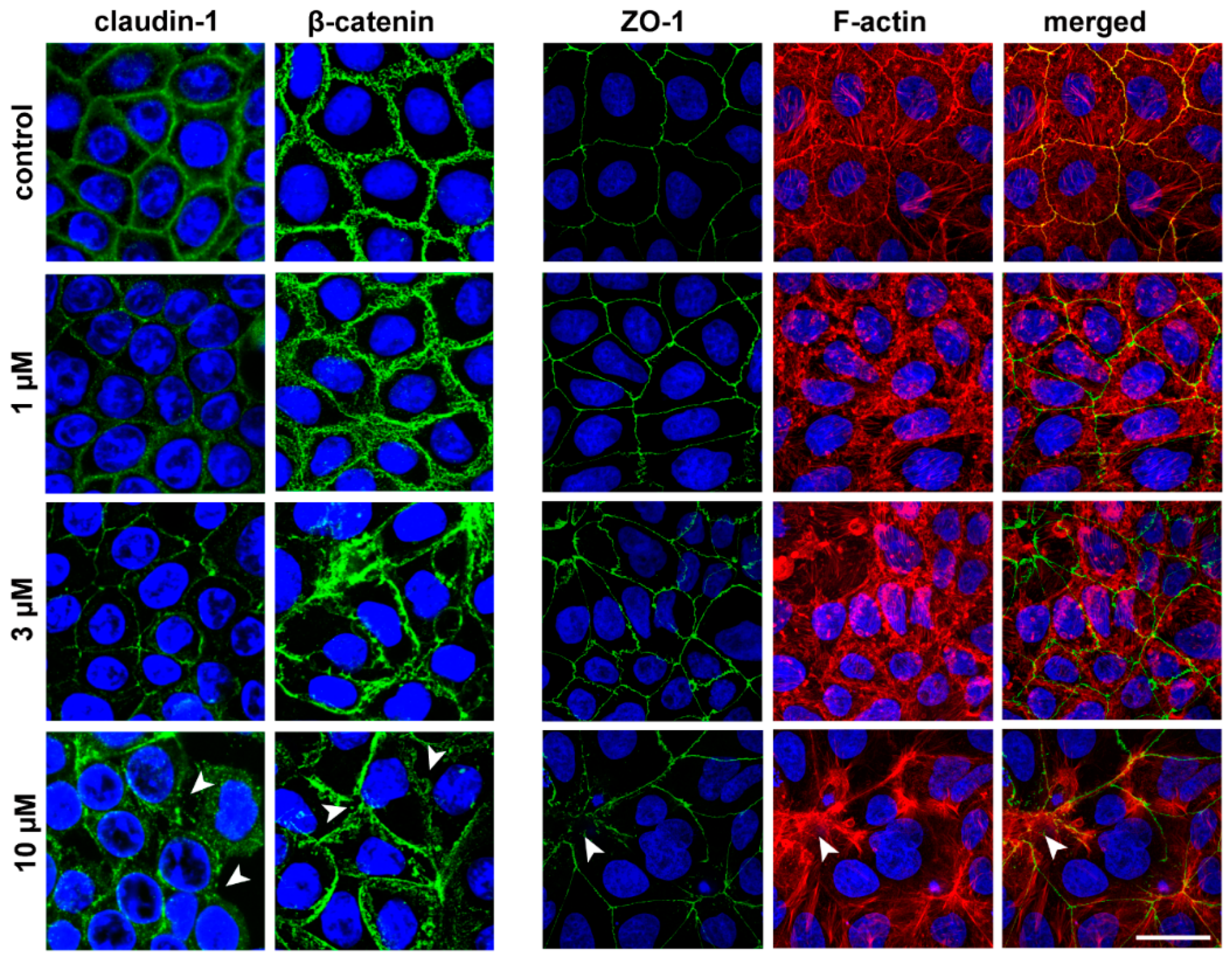
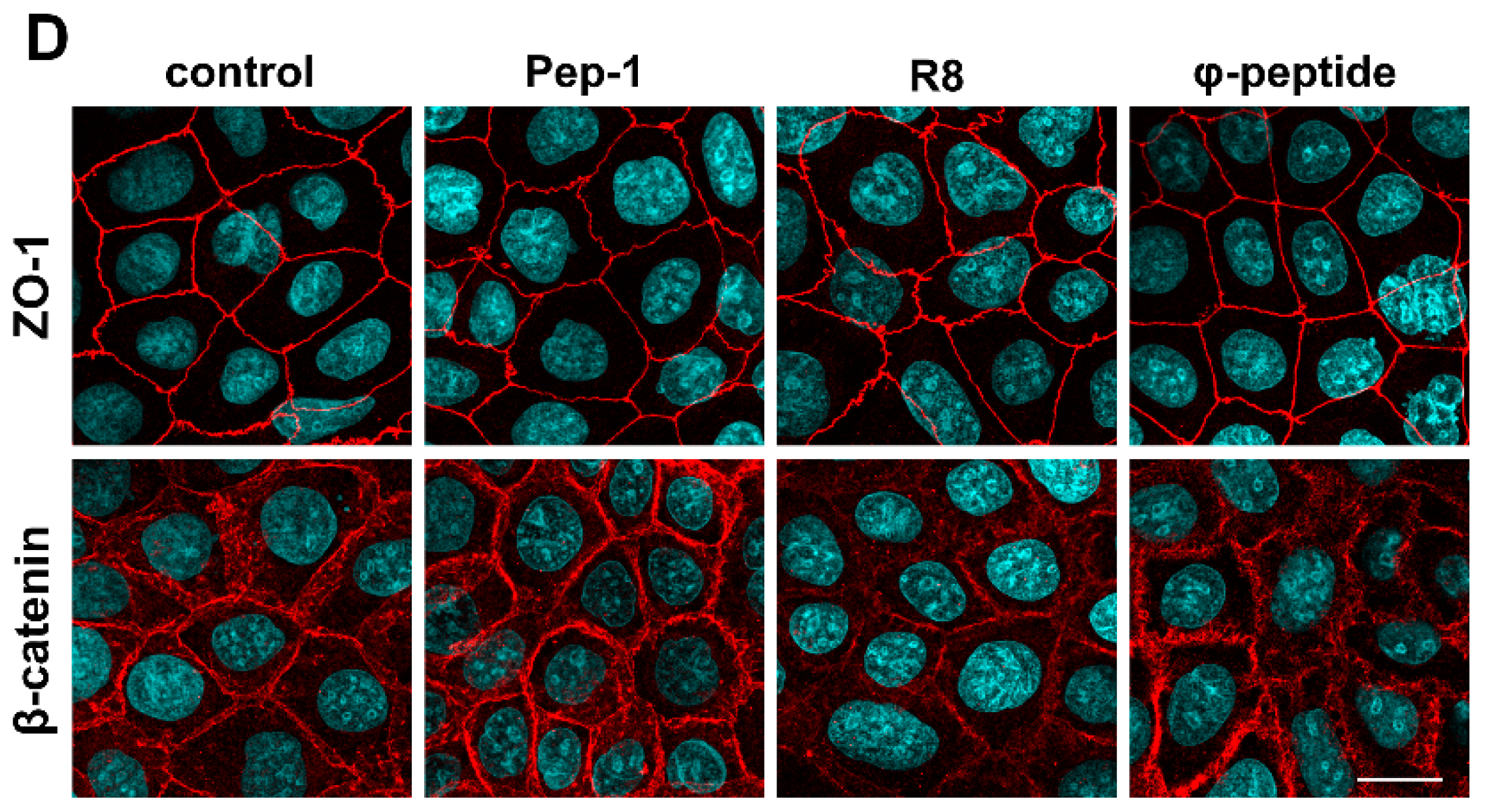
3.5. The Effect of PN159 Peptide on the Staining of Junctional Proteins and F-Actin
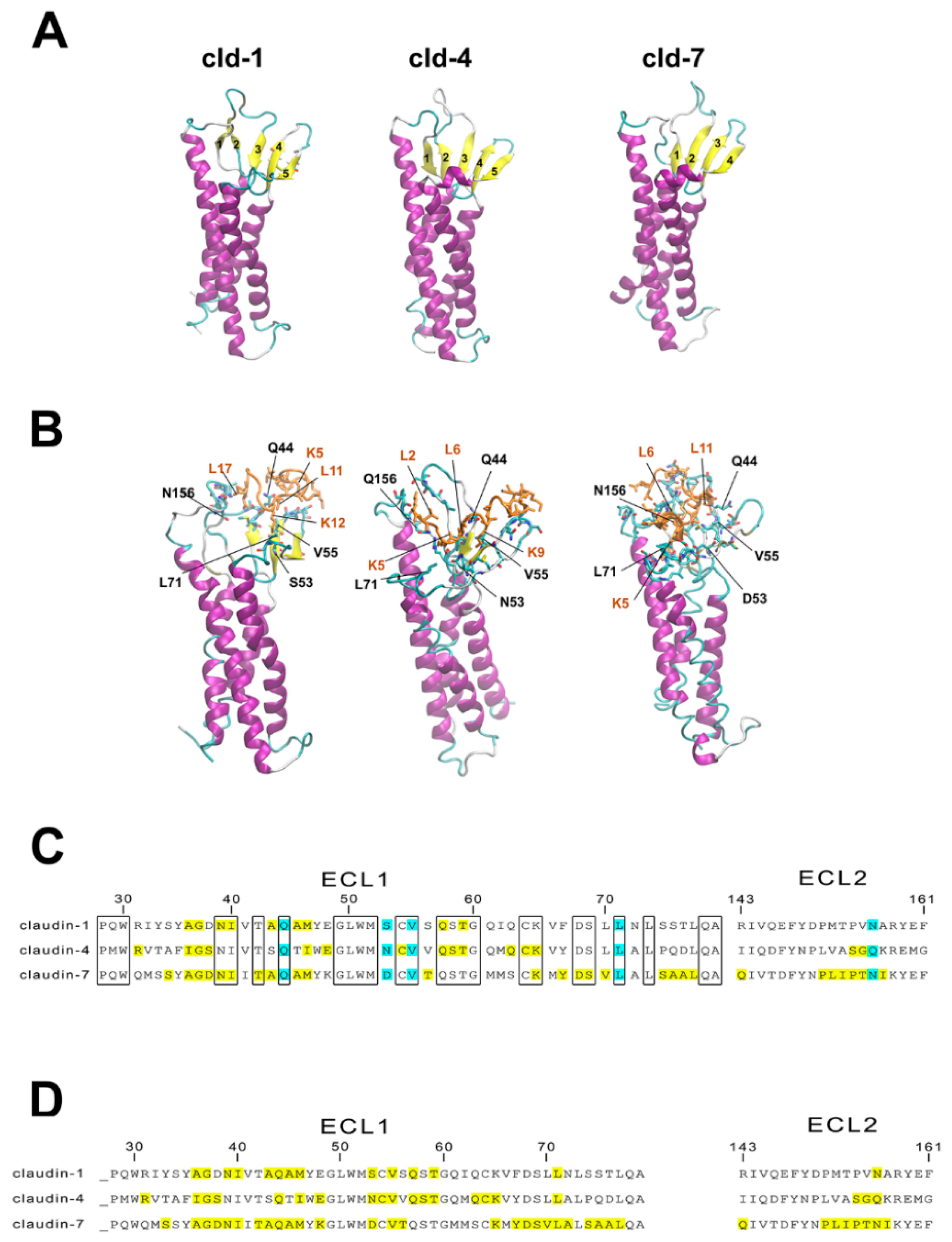
3.6. Molecular Modeling of Human Claudin Proteins and Docking of PN159 Peptide
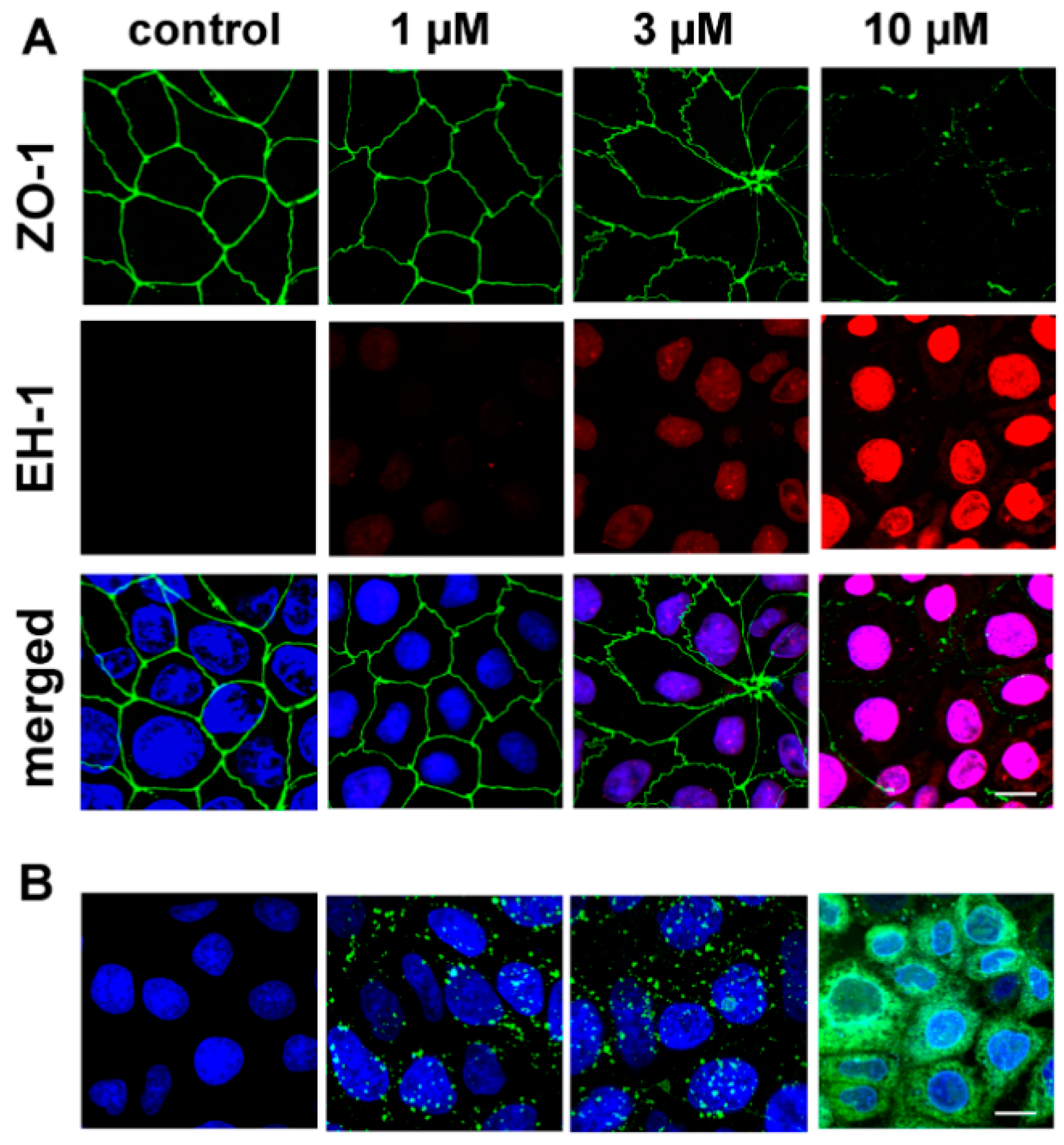
3.7. Cell-Penetrating Effect and Uptake of PN159 Peptide in Epithelial Cells
3.8. Antimicrobial Effect of PN159
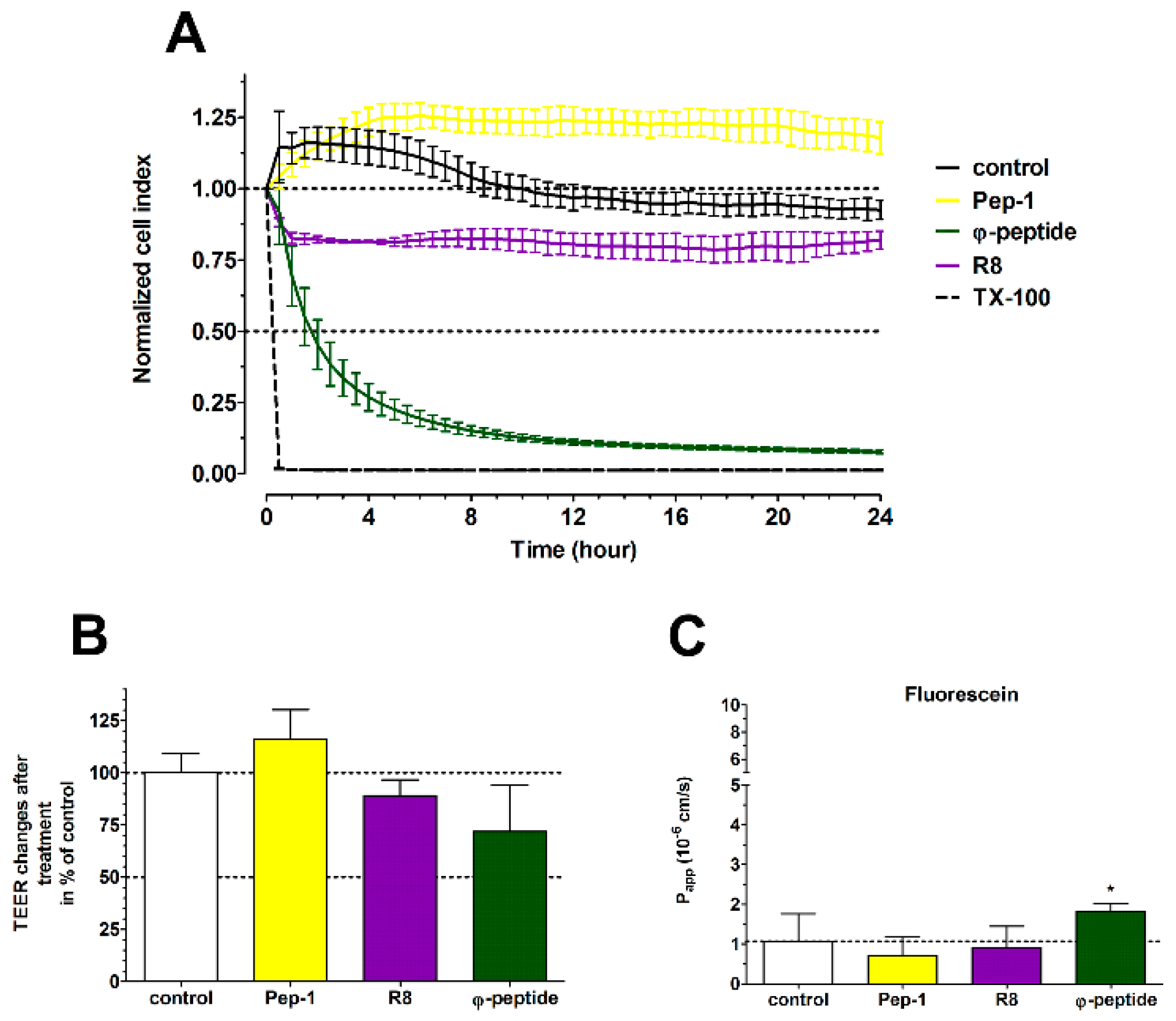
3.9. The Effect of Cell-Penetrating Peptides on Intestinal Barrier Integrity
4. Discussion
4.1. TJ Modulator Effect of PN159: Safety, Concentration Dependence and Reversibility
4.2. Tight Junction (TJ)Modulator Effect of PN159: Interaction with Claudins
4.3. Cell Penetration and Antimicrobial Effect of PN159
4.4. TJ Modulator Effect of Other CPPs?
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Deli, M.A. Potential use of tight junction modulators to reversibly open membranous barriers and improve drug delivery. A review. Biochim. Biophys. Acta 2009, 1788, 892–910. [Google Scholar] [CrossRef] [PubMed]
- Kiss, L.; Hellinger, É.; Pilbat, A.M.; Kittel, Á.; Török, Z.; Füredi, A.; Szakács, G.; Veszelka, S.; Sipos, P.; Ózsvári, B.; et al. Sucrose esters increase drug penetration, but do not inhibit p-glycoprotein in caco-2 intestinal epithelial cells. J. Pharm. Sci. 2014, 103, 3107–3119. [Google Scholar] [CrossRef] [PubMed]
- Perinelli, D.R.; Vllasaliu, D.; Bonacucina, G.; Come, B.; Pucciarelli, S.; Ricciutelli, M.; Cespi, M.; Itri, R.; Spinozzi, F.; Palmieri, G.F.; et al. Rhamnolipids as epithelial permeability enhancers for macromolecular therapeutics. Eur. J. Pharm. Biopharm. 2017, 119, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Johnson, P.H.; Quay, S.C. Advances in nasal drug delivery through tight junction technology. A review. Expert. Opin. Drug Deliv. 2005, 2, 281–298. [Google Scholar] [CrossRef] [PubMed]
- Chiba, H.; Osanai, M.; Murata, M.; Kojima, T.; Sawada, N. Transmembrane proteins of tight junctions. A review. Biochim. Biophys. Acta 2008, 1778, 588–600. [Google Scholar] [CrossRef] [PubMed]
- Krause, G.; Winkler, L.; Mueller, S.L.; Haseloff, R.F.; Piontek, J.; Blasig, I.E. Structure and function of claudins. A review. Biochim. Biophys. Acta 2008, 1778, 631–645. [Google Scholar] [CrossRef] [PubMed]
- Tscheik, C.; Blasig, I.E.; Winkler, L. Trends in drug delivery through tissue barriers containing tight junctions. Tissue Barriers 2013, 1, e24565. [Google Scholar] [CrossRef] [PubMed]
- Bocsik, A.; Walter, R.F.; Gyebrovszki, A.; Fülöp, L.; Blasig, I.; Dabrowski, S.; Ötvös, F.; Tóth, A.; Rákhely, G.; Veszelka, S.; et al. Reversible opening of intercellular junctions of intestinal epithelial and brain endothelial cells with tight junction modulator peptides. J. Pharm. Sci. 2016, 105, 754–765. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.C.; Eiting, K.; Cui, K.; Leonard, A.K.; Morris, D.; Li, C.Y.; Farber, K.; Sileno, A.P.; Houston, M.E., Jr.; Johnson, P.H.; et al. Therapeutic utility of a novel tight junction modulating peptide for enhancing intranasal drug delivery. J. Pharm. Sci. 2006, 95, 1364–1371. [Google Scholar] [CrossRef] [PubMed]
- Dathe, M.; Schümann, M.; Wieprecht, T.; Winkler, A.; Beyermann, M.; Krause, E.; Matsuzaki, K.; Murase, O.; Bienert, M. Peptide helicity and membrane surface charge modulate the balance of electrostatic and hydrophobic interactions with lipid bilayers and biological membranes. Biochemistry 1996, 35, 12612–12622. [Google Scholar] [CrossRef]
- Oehlke, J.; Scheller, A.; Wiesner, B.; Krause, E.; Beyermann, M.; Klauschenz, E.; Melzig, M.; Bienert, M. Cellular uptake of an alpha-helical amphipathic model peptide with the potential to deliver polar compounds into the cell interiornon-endocytically. Biochim. Biophys. Acta 1998, 1414, 127–139. [Google Scholar] [CrossRef]
- Hällbrink, M.; Florén, A.; Elmquist, A.; Pooga, M.; Bartfai, T.; Langel, U. Cargo delivery kinetics of cell-penetrating peptides. Biochim. Biophys. Acta 2001, 1515, 101–109. [Google Scholar] [CrossRef]
- Arouri, A.; Dathe, M.; Blume, A. The helical propensity of KLA amphipathic peptides enhances their binding to gel-state lipid membranes. Biophys. Chem. 2013, 180–181, 10–21. [Google Scholar] [CrossRef] [PubMed]
- Copolovici, D.M.; Langel, K.; Eriste, E.; Langel, Ü. Cell-penetrating peptides: Design, synthesis, and applications. A review. ACS Nano 2014, 8, 1972–1994. [Google Scholar] [CrossRef] [PubMed]
- Qian, Z.; LaRochelle, J.R.; Jiang, B.; Lian, W.; Hard, R.L.; Selner, N.G.; Luechapanichkul, R.; Barrios, A.M.; Pei, D. Early endosomal escape of a cyclic cell-penetrating peptide allows effective cytosolic cargo delivery. Biochemistry 2014, 53, 4034–4046. [Google Scholar] [CrossRef]
- Morris, M.C.; Deshayes, S.; Heitz, F.; Divita, G. Cell-penetrating peptides: From molecular mechanisms to therapeutics. A review. Biol. Cell 2008, 100, 201–217. [Google Scholar] [CrossRef]
- Guidotti, G.; Brambilla, L.; Rossi, D. Cell-Penetrating Peptides: From Basic Research to Clinics. A review. Trends Pharmacol. Sci. 2017, 38, 406–424. [Google Scholar] [CrossRef]
- He, L.; Sayers, E.J.; Watson, P.; Jones, A.T. Contrasting roles for actin in the cellular uptake of cell penetrating peptide conjugates. Sci. Rep. 2018, 8, 7318. [Google Scholar] [CrossRef]
- Palm, C.; Netzereab, S.; Hällbrink, M. Quantitatively determined uptake of cell-penetrating peptides in non-mammalian cells with an evaluation of degradation and antimicrobial effects. Peptides 2006, 27, 1710–1716. [Google Scholar] [CrossRef]
- Boucher, H.W.; Talbot, G.H.; Bradley, J.S.; Edwards, J.E.; Gilbert, D.; Rice, L.B.; Scheld, M.; Spellberg, B.; Bartlett, J. Bad bugs, no drugs: No ESKAPE! An update from the Infectious Diseases Society of America. A review. Clin. Infect. Dis. 2009, 48, 1–12. [Google Scholar] [CrossRef]
- Lázár, V.; Martins, A.; Spohn, R.; Daruka, L.; Grézal, G.; Fekete, G.; Számel, M.; Jangir, P.K.; Kintses, B.; Csörgő, B.; et al. Antibiotic-resistant bacteria show widespread collateral sensitivity to antimicrobial peptides. Nat. Microbiol. 2018, 3, 718–731. [Google Scholar] [CrossRef] [PubMed]
- Artursson, P.; Palm, K.; Luthman, K. Caco-2 monolayers in experimental and theoretical predictions of drug transport. Adv. Drug Deliv. Rev. 2001, 46, 27–43. [Google Scholar] [CrossRef]
- Hubatsch, I.; Ragnarsson, E.G.; Artursson, P. Determination of drug permeability and prediction of drug absorption in Caco-2 monolayers. Nat. Protoc. 2007, 2, 2111–2119. [Google Scholar] [CrossRef] [PubMed]
- Hellinger, E.; Veszelka, S.; Tóth, A.E.; Walter, F.; Kittel, A.; Bakk, M.L.; Tihanyi, K.; Háda, V.; Nakagawa, S.; Duy, T.D.; Niwa, M.; et al. Comparison of brain capillary endothelial cell-based and epithelial (MDCK-MDR1, Caco-2, and VB-Caco-2) cell-based surrogate blood-brain barrier penetration models. Eur. J. Pharm. Biopharm. 2012, 82, 340–351. [Google Scholar] [CrossRef] [PubMed]
- Roka, E.; Vecsernyes, M.; Bacskay, I.; Félix, C.; Rhimi, M.; Coleman, A.W.; Perret, F. para-Sulphonato-calix[n]arenes as selective activators for the passage of molecules across the Caco-2 model intestinal membrane. Chem. Commun. 2015, 51, 9374–9376. [Google Scholar] [CrossRef] [PubMed]
- Kürti, L.; Veszelka, S.; Bocsik, A.; Dung, N.T.; Ozsvári, B.; Puskás, L.G.; Kittel, A.; Szabó-Révész, P.; Deli, M.A. The effect of sucrose esters on a culture model of thenasal barrier. Toxicol. In Vitro 2012, 26, 445–454. [Google Scholar] [CrossRef] [PubMed]
- Dichroweb, on-line Analysis for Protein Circular Dichroism spectra. Available online: dichroweb.cryst.bbk.ac.uk/html/home.shtml (accessed on 10 December 2015).
- Sali, A.; Blundell, T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Nishizawa, T.; Tani, K.; Yamazaki, Y.; Tamura, A.; Ishitani, R.; Dohmae, N.; Tsukita, S.; Nureki, O.; Fujiyoshi, Y. Crystal structure of a claudin provides insight into the architecture of tight junctions. Science 2014, 344, 304–307. [Google Scholar] [CrossRef]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kalé, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Kurcinski, M.; Jamroz, M.; Blaszczyk, M.; Kolinski, A.; Kmiecik, S. CABS-dock web server for the flexible docking of peptides to proteins without prior knowledge of the binding site. Nucleic. Acids. Res. 2015, 43, W419–W424. [Google Scholar] [CrossRef] [PubMed]
- Wiegand, I.; Hilpert, K.; Hancock, R.E. Agar and broth dilution methods to determine the minimal inhibitory concentration (MIC) of antimicrobial substances. Nat. Protoc. 2008, 3, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Veszelka, S.; Tóth, A.; Walter, F.R.; Tóth, A.E.; Gróf, I.; Mészáros, M.; Bocsik, A.; Hellinger, É.; Vastag, M.; Rákhely, G.; Deli, M.A. Comparison of a Rat Primary Cell-Based Blood-Brain Barrier Model With Epithelial and Brain Endothelial Cell Lines: Gene Expression and Drug Transport. Front. Mol. Neurosci. 2018, 11, 166. [Google Scholar] [CrossRef] [PubMed]
- Erbe, A.; Kerth, A.; Dathe, M.; Blume, A. Interactions of KLA amphipathic model peptides with lipid monolayers. ChemBioChem 2009, 10, 2884–2892. [Google Scholar] [CrossRef] [PubMed]
- Staat, C.; Coisne, C.; Dabrowski, S.; Stamatovic, S.M.; Andjelkovic, A.V.; Wolburg, H.; Engelhardt, B.; Blasig, I.E. Mode of action of claudin peptidomimetics in the transient opening of cellular tight junction barriers. Biomaterials 2015, 54, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Dithmer, S.; Staat, C.; Müller, C.; Ku, M.C.; Pohlmann, A.; Niendorf, T.; Gehne, N.; Fallier-Becker, P.; Kittel, Á.; Walter, F.R.; Veszelka, S.; et al. Claudin peptidomimetics modulate tissue barriers for enhanced drug delivery. Ann. N. Y. Acad. Sci. 2017, 1397, 169–184. [Google Scholar] [CrossRef] [PubMed]
- Hellinger, E.; Bakk, M.L.; Pócza, P.; Tihanyi, K.; Vastag, M. Drug penetration model of vinblastine-treated Caco-2 cultures. Eur. J. Pharm. Sci. 2010, 41, 96–106. [Google Scholar] [CrossRef] [PubMed]
- Sonoda, N.; Furuse, M.; Sasaki, H.; Yonemura, S.; Katahira, J.; Horiguchi, Y.; Tsukita, S. Clostridium perfringens enterotoxin fragment removes specific claudins from tight junction strands: Evidence for direct involvement of claudins in tight junction barrier. J. Cell Biol. 1999, 147, 195–204. [Google Scholar] [CrossRef]
- Dabrowski, S.; Staat, C.; Zwanziger, D.; Sauer, R.S.; Bellmann, C.; Günther, R.; Krause, E.; Haseloff, R.F.; Rittner, H.; Blasig, I.E. Redox-sensitive structure and function of the first extracellular loop of the cell-cell contact protein claudin-1: Lessons from molecular structure to animals. Antioxid. Redox. Signal 2015, 22, 1–14. [Google Scholar] [CrossRef]
- Veshnyakova, A.; Piontek, J.; Protze, J.; Waziri, N.; Heise, I.; Krause, G. Mechanism of Clostridium perfringens enterotoxin interaction with claudin-3/-4 protein suggests structural modifications of the toxin to target specific claudins. J. Biol. Chem. 2012, 287, 1698–1708. [Google Scholar] [CrossRef]
- Mueller, J.; Kretzschmar, I.; Volkmer, R.; Boisguerin, P. Comparison of cellular uptake using 22 CPPs in 4 different cell lines. Bioconjug. Chem. 2008, 19, 2363–2374. [Google Scholar] [CrossRef] [PubMed]
- Bagheri, M.; Beyermann, M.; Dathe, M. Immobilization reduces the activity of surface-bound cationic antimicrobial peptides with no influence upon the activity spectrum. Antimicrob. Agents Chemother. 2009, 53, 1132–1141. [Google Scholar] [CrossRef] [PubMed]
- Maher, S.; McClean, S. Investigation of the cytotoxicity of eukaryotic and prokaryotic antimicrobial peptides in intestinal epithelial cells in vitro. Biochem. Pharmacol. 2006, 71, 1289–1298. [Google Scholar] [CrossRef] [PubMed]
- Lam, S.J.; O’Brien-Simpson, N.M.; Pantarat, N.; Sulistio, A.; Wong, E.H.; Chen, Y.Y.; Lenzo, J.C.; Holden, J.A.; Blencowe, A.; Reynolds, E.C.; et al. Combating multidrug-resistant Gram-negative bacteria with structurally nanoengineered antimicrobial peptide polymers. Nat. Microbiol. 2016, 1, 16162. [Google Scholar] [CrossRef] [PubMed]
- Maher, S.; Feighery, L.; Brayden, D.J.; McClean, S. Melittin as an epithelial permeability enhancer I: Investigation of its mechanism of action in Caco-2 monolayers. Pharm. Res. 2007, 24, 1336–1345. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CPPs | Amino Acid Sequence | References |
|---|---|---|
| PN159 (KLAL/MAP) | KLALKLALKALKAALKLA-amide | [4,10] |
| Pep-1 | KETWWETWWTEWSQPKKKRKV-amide | [16] |
| R8 | RRRRRRRR-amide | [18] |
| φ-peptide | cyclo[CGGFWRRRRGE(εAca)G] | [15] |
| Markers and Drug | Control | PN159 | Fold Change |
|---|---|---|---|
| FD-4 | 0.021 ± 0.001 | 4.2 ± 0.1 | 200.0 |
| FD-10 | 0.025 ± 0.010 | 4.8 ± 0.7 | 192.0 |
| FD-20 | 0.022 ± 0.002 | 3.5 ± 1.0 | 159.0 |
| FD-40 | 0.015 ± 0.003 | 6.0 ± 0.3 | 400.0 |
| atenolol | 1.2 ± 0.3 | 36.3 ± 1.9 | 30.0 |
| cimetidine | 0.9 ± 0.2 | 34.5 ± 4.5 | 38.3 |
| quinidine | 45.0 ± 13.3 | 72.6 ± 2.4 | 1.6 |
| verapamil | 46.2 ± 3.9 | 86.7 ± 18.8 | 1.9 |
| Energy | Claudin-1 | Claudin-3 | Claudin-4 | Claudin-7 |
| Elig | −34 | −17 | −61 | −32 |
| Eint | −63 | −59 | −79 | −186 |
| Etot | −1228 | −1163 | −1229 | −1798 |
| ESKAPE Pethogens (ATCC) | PN159 | Cefoxitin | Gentamicin | Ciprofloxacin |
|---|---|---|---|---|
| Acinetobacter baumannii (17978) | 3.6 | 222.6 | 0.6 | 0.94 |
| Enterococcus faecium (700221) | 4.4 | >222.6 | >143.9 | >30.18 |
| Staphylococcus aureus (29213) | 9.2 | 6.9 | 1.1 | 1.89 |
| Klebsiella pneumonia (10031) | 13.8 | 3.5 | 0.6 | 0.06 |
| Enterobacter cloacae (13047) | 31.0 | >222.6 | 1.1 | 0.06 |
| Pseudomonas aeruginosa (27853) | 46.4 | >222.6 | 2.3 | 1.89 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bocsik, A.; Gróf, I.; Kiss, L.; Ötvös, F.; Zsíros, O.; Daruka, L.; Fülöp, L.; Vastag, M.; Kittel, Á.; Imre, N.; et al. Dual Action of the PN159/KLAL/MAP Peptide: Increase of Drug Penetration across Caco-2 Intestinal Barrier Model by Modulation of Tight Junctions and Plasma Membrane Permeability. Pharmaceutics 2019, 11, 73. https://doi.org/10.3390/pharmaceutics11020073
Bocsik A, Gróf I, Kiss L, Ötvös F, Zsíros O, Daruka L, Fülöp L, Vastag M, Kittel Á, Imre N, et al. Dual Action of the PN159/KLAL/MAP Peptide: Increase of Drug Penetration across Caco-2 Intestinal Barrier Model by Modulation of Tight Junctions and Plasma Membrane Permeability. Pharmaceutics. 2019; 11(2):73. https://doi.org/10.3390/pharmaceutics11020073
Chicago/Turabian StyleBocsik, Alexandra, Ilona Gróf, Lóránd Kiss, Ferenc Ötvös, Ottó Zsíros, Lejla Daruka, Lívia Fülöp, Monika Vastag, Ágnes Kittel, Norbert Imre, and et al. 2019. "Dual Action of the PN159/KLAL/MAP Peptide: Increase of Drug Penetration across Caco-2 Intestinal Barrier Model by Modulation of Tight Junctions and Plasma Membrane Permeability" Pharmaceutics 11, no. 2: 73. https://doi.org/10.3390/pharmaceutics11020073