Repurposing Dihydropyridines for Treatment of Helicobacter pylori Infection

 ,
,  , , , and
, , , and
Abstract
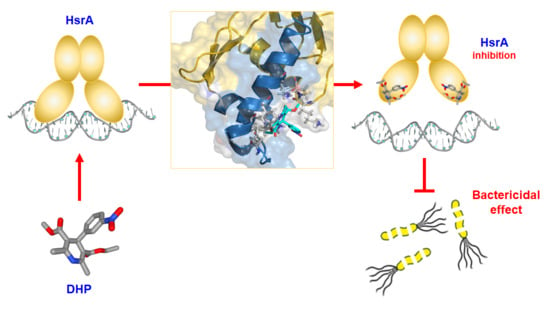
:
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Bacterial Strains, Culture Media and Growth Conditions
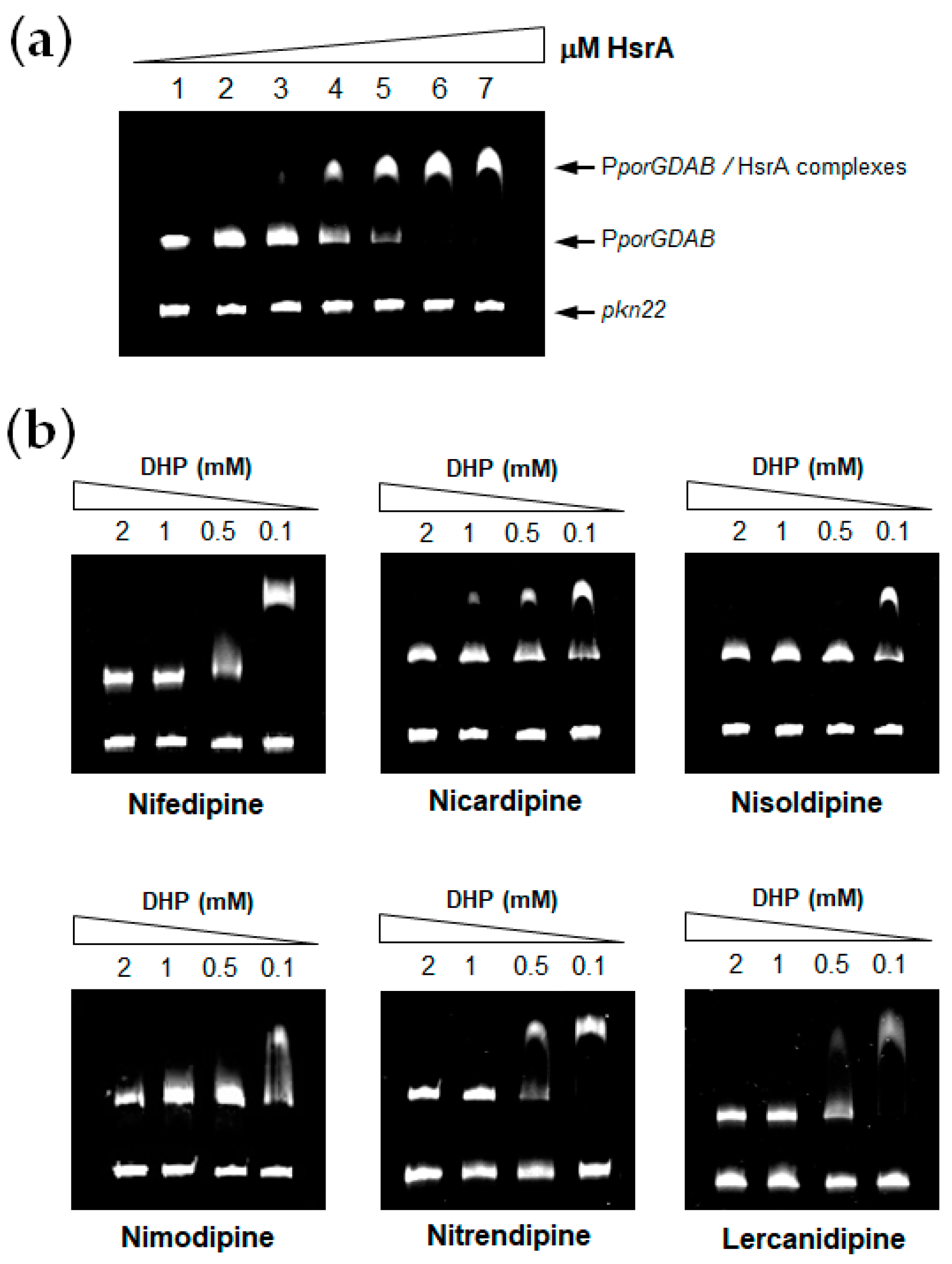
2.3. Electrophoretic Mobility Shift Assays
2.4. Minimal Inhibitory and Bactericidal Concentrations
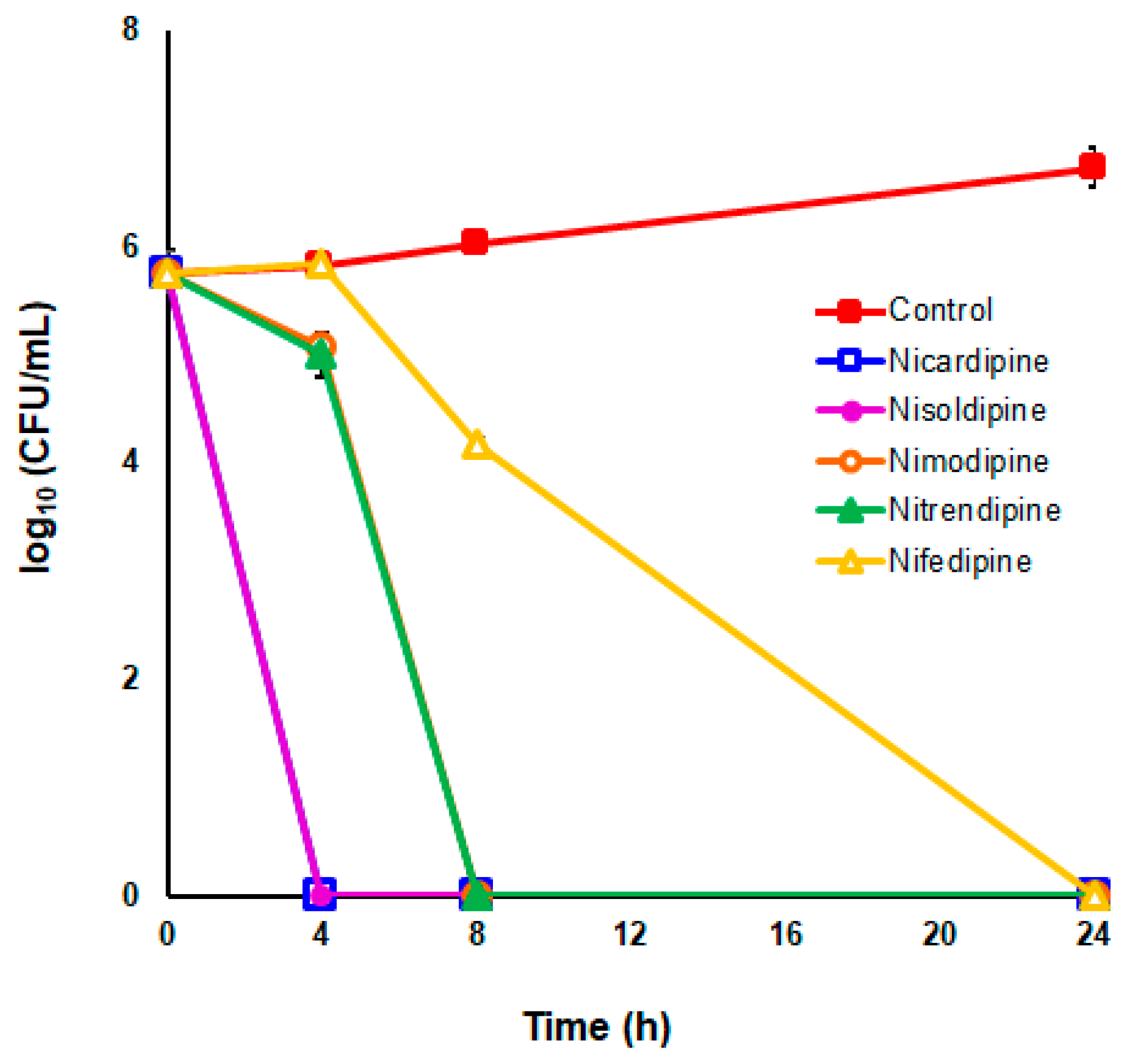
2.5. Time–kill Kinetics Assays
2.6. Checkerboard Assays
2.7. Isothermal Titration Calorimetry Assays
2.8. Molecular Docking
2.9. Mouse Infection and DHP Treatment
2.10. Bacterial Counts and qPCR
2.11. Statistical Analysis
3. Results
3.1. DHPs Inhibit the DNA Binding Activity of the H. pylori Essential Response Regulator HsrA
3.2. Analysis of the Molecular Interaction between HsrA and its DHP-Class Inhibitors
3.3. DHP-Class Inhibitors of HsrA Exhibit Strong Bactericidal Activities against H. pylori
3.4. Combinatory Effects of DHPs with Metronidazole and Clarithromycin against Antibiotic-Resistant H. pylori Strains
3.5. Nimodipine and Nitrendipine Significantly Reduced the H. pylori Gastric Colonization in Mice
4. Discussion
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hooi, J.K.Y.; Lai, W.Y.; Ng, W.K.; Suen, M.M.Y.; Underwood, F.E.; Tanyingoh, D.; Malfertheiner, P.; Graham, D.Y.; Wong, V.W.S.; Wu, J.C.Y.; et al. Global prevalence of Helicobacter pylori infection: Systematic review and meta-analysis. Gastroenterology 2017, 153, 420–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kusters, J.G.; van Vliet, A.H.; Kuipers, E.J. Pathogenesis of Helicobacter pylori infection. Clin. Microbiol. Rev. 2006, 19, 449–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamaoka, Y. Mechanisms of disease: Helicobacter pylori virulence factors. Nat. Rev. Gastroenterol. Hepatol. 2010, 7, 629–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moss, S.F. The clinical evidence linking Helicobacter pylori to gastric cancer. Cell Mol. Gastroenterol. Hepatol. 2017, 3, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Fallone, C.A.; Chiba, N.; van Zanten, S.V.; Fischbach, L.; Gisbert, J.P.; Hunt, R.H.; Jones, N.L.; Render, C.; Leontiadis, G.I.; Moayyedi, P.; et al. The Toronto consensus for the treatment of Helicobacter pylori infection in adults. Gastroenterology 2016, 151, 51–69. [Google Scholar] [CrossRef] [Green Version]
- Tacconelli, E.; Carrara, E.; Savoldi, A.; Harbarth, S.; Mendelson, M.; Monnet, D.L.; Pulcini, C.; Kahlmeter, G.; Kluytmans, J.; Carmeli, Y.; et al. Discovery, research, and development of new antibiotics: The WHO priority list of antibiotic-resistant bacteria and tuberculosis. Lancet Infect. Dis. 2018, 18, 318–327. [Google Scholar] [CrossRef]
- Ventola, C.L. The antibiotic resistance crisis: Part 1: Causes and threats. Pharm. Ther. 2015, 40, 277–283. [Google Scholar]
- Piddock, L.J. The crisis of no new antibiotics--what is the way forward? Lancet Infect. Dis. 2012, 12, 249–253. [Google Scholar] [CrossRef]
- Zheng, W.; Sun, W.; Simeonov, A. Drug repurposing screens and synergistic drug-combinations for infectious diseases. Br. J. Pharmacol. 2018, 175, 181–191. [Google Scholar] [CrossRef]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2019, 18, 41–58. [Google Scholar] [CrossRef]
- McDaniel, T.K.; Dewalt, K.C.; Salama, N.R.; Falkow, S. New approaches for validation of lethal phenotypes and genetic reversion in Helicobacter pylori. Helicobacter 2001, 6, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Muller, S.; Pflock, M.; Schar, J.; Kennard, S.; Beier, D. Regulation of expression of atypical orphan response regulators of Helicobacter pylori. Microbiol. Res. 2007, 162, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Olekhnovich, I.N.; Vitko, S.; Chertihin, O.; Hontecillas, R.; Viladomiu, M.; Bassaganya-Riera, J.; Hoffman, P.S. Mutations to essential orphan response regulator HP1043 of Helicobacter pylori result in growth-stage regulatory defects. Infect. Immunol. 2013, 81, 1439–1449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olekhnovich, I.N.; Vitko, S.; Valliere, M.; Hoffman, P.S. Response to metronidazole and oxidative stress is mediated through homeostatic regulator HsrA (HP1043) in Helicobacter pylori. J. Bacteriol. 2014, 196, 729–739. [Google Scholar] [CrossRef] [Green Version]
- Delany, I.; Spohn, G.; Rappuoli, R.; Scarlato, V. Growth phase-dependent regulation of target gene promoters for binding of the essential orphan response regulator HP1043 of Helicobacter pylori. J. Bacteriol. 2002, 184, 4800–4810. [Google Scholar] [CrossRef] [Green Version]
- Pelliciari, S.; Pinatel, E.; Vannini, A.; Peano, C.; Puccio, S.; De Bellis, G.; Danielli, A.; Scarlato, V.; Roncarati, D. Insight into the essential role of the Helicobacter pylori HP1043 orphan response regulator: Genome-wide identification and characterization of the DNA-binding sites. Sci. Rep. 2017, 7, 41063. [Google Scholar] [CrossRef]
- González, A.; Salillas, S.; Velázquez-Campoy, A.; Espinosa Angarica, V.; Fillat, M.F.; Sancho, J.; Lanas, Á. Identifying potential novel drugs against Helicobacter pylori by targeting the essential response regulator HsrA. Sci. Rep. 2019, 9, 11294. [Google Scholar] [CrossRef] [Green Version]
- Godfraind, T. Discovery and development of calcium channel blockers. Front. Pharmacol. 2017, 8, 286. [Google Scholar] [CrossRef] [Green Version]
- Coca, A.; Mazon, P.; Aranda, P.; Redon, J.; Divison, J.A.; Martinez, J.; Calvo, C.; Galceran, J.M.; Barrios, V.; Roca-Cusachs, I.C.A. Role of dihydropyridinic calcium channel blockers in the management of hypertension. Expert Rev. Cardiovasc. Ther. 2013, 11, 91–105. [Google Scholar] [CrossRef]
- Arnold, I.C.; Lee, J.Y.; Amieva, M.R.; Roers, A.; Flavell, R.A.; Sparwasser, T.; Muller, A. Tolerance rather than immunity protects from Helicobacter pylori-induced gastric preneoplasia. Gastroenterology 2011, 140, 199–209. [Google Scholar] [CrossRef] [Green Version]
- Dyer, V.; Bruggemann, H.; Sorensen, M.; Kuhl, A.A.; Hoffman, K.; Brinkmann, V.; Reines, M.D.M.; Zimmerman, S.; Meyer, T.F.; Koch, M. Genomic features of the Helicobacter pylori strain PMSS1 and its virulence attributes as deduced from its in vivo colonisation patterns. Mol. Microbiol. 2018, 110, 761–776. [Google Scholar] [CrossRef] [PubMed]
- Ducournau, A.; Benejat, L.; Sifre, E.; Bessede, E.; Lehours, P.; Megraud, F. Helicobacter pylori resistance to antibiotics in 2014 in France detected by phenotypic and genotypic methods. Clin. Microbiol. Infect. 2016, 22, 715–718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cremades, N.; Velázquez-Campoy, A.; Martínez-Júlvez, M.; Neira, J.L.; Pérez-Dorado, I.; Hermoso, J.; Jiménez, P.; Lanas, A.; Hoffman, P.S.; Sancho, J. Discovery of specific flavodoxin inhibitors as potential therapeutic agents against Helicobacter pylori infection. ACS Chem. Biol. 2009, 4, 928–938. [Google Scholar] [CrossRef] [PubMed]
- Salillas, S.; Alias, M.; Michel, V.; Mahia, A.; Lucia, A.; Rodrigues, L.; Bueno, J.; Galano-Frutos, J.J.; De Reuse, H.; Velázquez-Campoy, A.; et al. Design, synthesis, and efficacy testing of nitroethylene- and 7-nitrobenzoxadiazol-based flavodoxin Inhibitors against Helicobacter pylori drug-resistant clinical strains and in Helicobacter pylori-infected mice. J. Med. Chem. 2019, 62, 6102–6115. [Google Scholar] [CrossRef]
- Krzyzek, P.; Franiczek, R.; Krzyzanowska, B.; Laczmanski, L.; Migdal, P.; Gosciniak, G. In vitro activity of 3-Bromopyruvate, an anticancer compound, against antibiotic-susceptible and antibiotic-resistant Helicobacter pylori strains. Cancers 2019, 11, 229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Velázquez-Campoy, A.; Leavitt, S.A.; Freire, E. Characterization of protein-protein interactions by isothermal titration calorimetry. Methods Mol. Biol. 2015, 1278, 183–204. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Floch, P.; Izotte, J.; Guillemaud, J.; Sifre, E.; Costet, P.; Rousseau, B.; Laur, A.M.; Giese, A.; Korolik, V.; Megraud, F.; et al. A new animal model of gastric lymphomagenesis: APRIL transgenic mice Infected by Helicobacter species. Am. J. Pathol. 2017, 187, 1473–1484. [Google Scholar] [CrossRef] [Green Version]
- Oleastro, M.; Menard, A.; Santos, A.; Lamouliatte, H.; Monteiro, L.; Barthelemy, P.; Megraud, F. Real-time PCR assay for rapid and accurate detection of point mutations conferring resistance to clarithromycin in Helicobacter pylori. J. Clin. Microbiol. 2003, 41, 397–402. [Google Scholar] [CrossRef] [Green Version]
- Laur, A.M.; Floch, P.; Chambonnier, L.; Benejat, L.; Korolik, V.; Giese, A.; Dubus, P.; Megraud, F.; Bandeira, A.; Lehours, P. Regulatory T cells may participate in Helicobacter pylori persistence in gastric MALT lymphoma: Lessons from an animal model. Oncotarget 2016, 7, 3394–3402. [Google Scholar] [CrossRef]
- Testa, R.; Leonardi, A.; Tajana, A.; Riscassi, E.; Magliocca, R.; Sartani, A. Lercanidipine (Rec 15/2375): A novel 1,4-dihydropyridine calcium antagonist for hypertension. Cardiovasc. Drug Rev. 1997, 15, 187–219. [Google Scholar] [CrossRef]
- Loo, V.G.; Fallone, C.A.; De Souza, E.; Lavallee, J.; Barkun, A.N. In-vitro susceptibility of Helicobacter pylori to ampicillin, clarithromycin, metronidazole and omeprazole. J. Antimicrob. Chemother. 1997, 40, 881–883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, M.; Sharma, R.; Jain, D.K. Nanotechnology based approaches for enhancing oral bioavailability of poorly water soluble antihypertensive drugs. Scientifica 2016, 2016, 8525679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paul, S.M.; Mytelka, D.S.; Dunwiddie, C.T.; Persinger, C.C.; Munos, B.H.; Lindborg, S.R.; Schacht, A.L. How to improve R&D productivity: The pharmaceutical industry’s grand challenge. Nat. Rev. Drug Discov. 2010, 9, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Shim, J.S.; Liu, J.O. Recent advances in drug repositioning for the discovery of new anticancer drugs. Int. J. Biol. Sci. 2014, 10, 654–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turanli, B.; Grotli, M.; Boren, J.; Nielsen, J.; Uhlen, M.; Arga, K.Y.; Mardinoglu, A. Drug repositioning for effective prostate cancer treatment. Front. Physiol. 2018, 9, 500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corbett, A.; Pickett, J.; Burns, A.; Corcoran, J.; Dunnett, S.B.; Edison, P.; Hagan, J.J.; Holmes, C.; Jones, E.; Katona, C.; et al. Drug repositioning for Alzheimer’s disease. Nat. Rev. Drug Discov. 2012, 11, 833–846. [Google Scholar] [CrossRef]
- Bhattarai, D.; Singh, S.; Jang, Y.; Hyeon Han, S.; Lee, K.; Choi, Y. An insight into drug repositioning for the development of novel anti-cancer drugs. Curr. Top. Med. Chem. 2016, 16, 2156–2168. [Google Scholar] [CrossRef]
- Sampath, R.; Cummins, N.W.; Natesampillai, S.; Bren, G.D.; Chung, T.D.; Baker, J.; Henry, K.; Pagliuzza, A.; Badley, A.D. Increasing procaspase 8 expression using repurposed drugs to induce HIV infected cell death in ex vivo patient cells. PLoS ONE 2017, 12, e0179327. [Google Scholar] [CrossRef]
- Roder, C.; Thomson, M.J. Auranofin: Repurposing an old drug for a golden new age. Drugs R D 2015, 15, 13–20. [Google Scholar] [CrossRef] [Green Version]
- Peyclit, L.; Baron, S.A.; Rolain, J.M. Drug repurposing to fight colistin and carbapenem-resistant bacteria. Front. Cell Infect. Microbiol. 2019, 9, 193. [Google Scholar] [CrossRef] [PubMed]
- Miro-Canturri, A.; Ayerbe-Algaba, R.; Smani, Y. Drug repurposing for the treatment of bacterial and fungal infections. Front. Microbiol. 2019, 10, 41. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.S.; Sun, W.; Xu, M.; Shen, M.; Khraiwesh, M.; Sciotti, R.J.; Zheng, W. Repurposing screen identifies unconventional drugs with activity against multidrug resistant Acinetobacter baumannii. Front. Cell Infect. Microbiol. 2018, 8, 438. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.S.; Williamson, P.R.; Zheng, W. Improving therapy of severe infections through drug repurposing of synergistic combinations. Curr. Opin. Pharmacol. 2019, 48, 92–98. [Google Scholar] [CrossRef]
- Dutta, N.K.; Mazumdar, K.; DasGupta, A.; Dastidar, S.G. In vitro and in vivo efficacies of amlodipine against Listeria monocytogenes. Eur. J. Clin. Microbiol. Infect. Dis. 2009, 28, 849–853. [Google Scholar] [CrossRef]
- Sirisha, K.; Bikshapathi, D.; Achaiah, G.; Reddy, V.M. Synthesis, antibacterial and antimycobacterial activities of some new 4-aryl/heteroaryl-2,6-dimethyl-3,5-bis-N-(aryl)-carbamoyl-1,4-dihydropyridines. Eur. J. Med. Chem. 2011, 46, 1564–1571. [Google Scholar] [CrossRef]
- Olejnikova, P.; Svorc, L.; Olsovska, D.; Panakova, A.; Vihonska, Z.; Kovaryova, K.; Marchalin, S. Antimicrobial activity of novel C2-substituted 1,4-dihydropyridine analogues. Sci. Pharm. 2014, 82, 221–232. [Google Scholar] [CrossRef] [Green Version]
- Gunics, G.; Farkas, S.; Motohashi, N.; Shah, A.; Harsukh, G.; Kawase, M.; Molnar, J. Interaction between 3,5-diacetyl-1,4-dihydropyridines and ampicillin, and erythromycin on different E. coli strains. Int. J. Antimicrob. Agents 2002, 20, 227–229. [Google Scholar] [CrossRef] [Green Version]
- Ahamed, A.; Arif, I.A.; Mateen, M.; Surendra Kumar, R.; Idhayadhulla, A. Antimicrobial, anticoagulant, and cytotoxic evaluation of multidrug resistance of new 1,4-dihydropyridine derivatives. Saudi J. Biol. Sci. 2018, 25, 1227–1235. [Google Scholar] [CrossRef]
- Dasgupta, A.; Jeyaseeli, L.; Dutta, N.K.; Mazumdar, K.; Karak, P.; Dastidar, S.G.; Motohashi, N.; Shirataki, Y. Studies on the antimicrobial potential of the cardiovascular drug lacidipine. In Vivo 2007, 21, 847–850. [Google Scholar]
- Chhillar, A.K.; Arya, P.; Mukherjee, C.; Kumar, P.; Yadav, Y.; Sharma, A.K.; Yadav, V.; Gupta, J.; Dabur, R.; Jha, H.N.; et al. Microwave-assisted synthesis of antimicrobial dihydropyridines and tetrahydropyrimidin-2-ones: Novel compounds against aspergillosis. Bioorg. Med. Chem. 2006, 14, 973–981. [Google Scholar] [CrossRef] [PubMed]
- Palit, P.; Ali, N. Oral therapy with amlodipine and lacidipine, 1,4-dihydropyridine derivatives showing activity against experimental visceral leishmaniasis. Antimicrob. Agents Chemother. 2008, 52, 374–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reimao, J.Q.; Scotti, M.T.; Tempone, A.G. Anti-leishmanial and anti-trypanosomal activities of 1,4-dihydropyridines: In vitro evaluation and structure-activity relationship study. Bioorg. Med. Chem. 2010, 18, 8044–8053. [Google Scholar] [CrossRef] [PubMed]
- Maya, J.D.; Morello, A.; Repetto, Y.; Tellez, R.; Rodriguez, A.; Zelada, U.; Puebla, P.; Caballero, E.; Medarde, M.; Nunez-Vergara, L.J.; et al. Effects of 3-chloro-phenyl-1,4-dihydropyridine derivatives on Trypanosome cruzi epimastigotes. Comp. Biochem. Physiol. C Toxicol. Pharmacol. 2000, 125, 103–109. [Google Scholar] [CrossRef]
- Ghotaslou, R.; Leylabadlo, H.E.; Asl, Y.M. Prevalence of antibiotic resistance in Helicobacter pylori: A recent literature review. World J. Methodol. 2015, 5, 164–174. [Google Scholar] [CrossRef] [PubMed]
- Savoldi, A.; Carrara, E.; Graham, D.Y.; Conti, M.; Tacconelli, E. Prevalence of antibiotic resistance in Helicobacter pylori: A systematic review and meta-analysis in World Health Organization regions. Gastroenterology 2018, 155, 1372–1382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, Z. Potential of fumarate reductase as a novel therapeutic target in Helicobacter pylori infection. Expert Opin. Ther. Targets 2002, 6, 135–146. [Google Scholar] [CrossRef]
- Nishimori, I.; Onishi, S.; Takeuchi, H.; Supuran, C.T. The alpha and beta classes carbonic anhydrases from Helicobacter pylori as novel drug targets. Curr. Pharm. Des. 2008, 14, 622–630. [Google Scholar] [CrossRef]
- Duckworth, M.J.; Okoli, A.S.; Mendz, G.L. Novel Helicobacter pylori therapeutic targets: The unusual suspects. Expert Rev. Anti-Infect. Ther. 2009, 7, 835–867. [Google Scholar] [CrossRef]
- Debraekeleer, A.; Remaut, H. Future perspective for potential Helicobacter pylori eradication therapies. Future Microbiol. 2018, 13, 671–687. [Google Scholar] [CrossRef] [Green Version]
- Ohishi, T.; Inaoka, D.K.; Kita, K.; Kawada, M. Dihydroorotate dehydrogenase as a target for the development of novel Helicobacter pylori-specific antimicrobials. Chem. Pharm. Bull. 2018, 66, 239–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karkare, S.; Yousafzai, F.; Mitchenall, L.A.; Maxwell, A. The role of Ca2+ in the activity of Mycobacterium tuberculosis DNA gyrase. Nucleic Acids Res. 2012, 40, 9774–9787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uesawa, Y.; Mohri, K. Relationship between lipophilicities of 1,4-dihydropyridine derivatives and pharmacokinetic interaction strengths with grapefruit juice. Yakugaku Zasshi 2008, 128, 117–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asok Kumar, K.; Mazumdar, K.; Dutta, N.K.; Karak, P.; Dastidar, S.G.; Ray, R. Evaluation of synergism between the aminoglycoside antibiotic streptomycin and the cardiovascular agent amlodipine. Biol. Pharm. Bull. 2004, 27, 1116–1120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dasgupta, A.; Chaki, S.; Mukherjee, S.; Lourduraja, J.; Mazumdar, K.; Dutta, N.K.; Dastidar, S.G. Experimental analyses of synergistic combinations of antibiotics with a recently recognised antibacterial agent, lacidipine. Eur. J. Clin. Microbiol. Infect. Dis. 2010, 29, 239–243. [Google Scholar] [CrossRef]
- Philpott, D.J.; Belaid, D.; Troubadour, P.; Thiberge, J.M.; Tankovic, J.; Labigne, A.; Ferrero, R.L. Reduced activation of inflammatory responses in host cells by mouse-adapted Helicobacter pylory isolates. Cell Microbiol. 2002, 4, 285–296. [Google Scholar] [CrossRef]
- Nair, A.B.; Jacob, S. A simple practice guide for dose conversion between animals and human. J. Basic Clin. Pharm. 2016, 7, 27–31. [Google Scholar] [CrossRef] [Green Version]
- Grasso, G.; Alafaci, C.; Macdonald, R.L. Management of aneurysmal subarachnoid hemorrhage: State of the art and future perspectives. Surg. Neurol. Int. 2017, 8, 11. [Google Scholar] [CrossRef]
- Ishikawa, M.; Hashimoto, Y. Improvement in aqueous solubility in small molecule drug discovery programs by disruption of molecular planarity and symmetry. J. Med. Chem. 2011, 54, 1539–1554. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug (Brand Name) | DHP Generation | Molecular Formula | Chemical Structure | Mouse LD50 (mg/kg, oral) * |
|---|---|---|---|---|
| Nifedipine (Adalat) | first | C17H18N2O6 |  | 202 |
| Nicardipine (Cardene) | second | C26H29N3O6 |  | 305 |
| Nisoldipine (Sular) | second | C20H24N2O6 |  | 411 |
| Nimodipine (Nimotop) | second | C21H26N2O7 |  | 940 |
| Nitrendipine (Baypress) | third | C18H20N2O6 |  | 2540 |
| Lercanidipine (Zanidip) | fourth | C36H41N3O6 |  | 622 |
| DHP Drug | ITC a | Molecular Docking b | |||
|---|---|---|---|---|---|
| n | Kd (µM) | ΔH (kcal/mol) | ΔG (kcal/mol) | Interacting Residues | |
| Nifedipine | 0.74 | 15 | −5.0 | −6.6 | T206, R192, Y212, R208, K165 |
| Nicardipine | 0.72 | 3.0 | −2.0 | −7.5 | T153, E133, F149, R157, S68 |
| Nisoldipine | 0.70 | 11 | −2.2 | −6.8 | R157, T153, E133, P130, D131 |
| Nimodipine | 0.71 | 4.1 | −2.1 | −7.3 | P198, M195, I135, P148, K145, K194, V144 |
| Nitrendipine | 0.75 | 9.0 | −2.6 | −6.9 | P148, K147, Q190, V186, V183, V179, W173 |
| Lercanidipine | 0.81 | 20 | −4.8 | −6.4 | P148, A187, Q190, V183, W173 |
| DHP Drug | MIC (MBC), mg/L | ||
|---|---|---|---|
| ATCC 700392 | ATCC 43504 (MTZ-R) | ATCC 700684 (CLR-R) | |
| Nifedipine | 8 (8) | 16 (16) | 8 (8) |
| Nicardipine | 8 (8) | 8 (8) | 8 (8) |
| Nisoldipine | 4 (4) | 16 (16) | 4 (4) |
| Nimodipine | 8 (8) | 16 (32) | 4 (4) |
| Nitrendipine | 8 (8) | 16 (16) | 8 (8) |
| Lercanidipine | 8 (8) | 32 (64) | 8 (8) |
| Metronidazole | 1 (2) | 64 (128) | 1 (2) |
| Clarithromycin | ≤0.12 (≤0.12) | ≤0.12 (≤0.12) | 16 (32) |
| Strain | Combination Tested | FICantibiotic | FICDHP | FICI a | Interaction b |
|---|---|---|---|---|---|
| ATCC 43504 (MTZ-R) | MTZ + Nifedipine | 0.25 | 0.5 | 0.75 | additive |
| MTZ + Nicardipine | 1 | 1 | 2 | no interaction | |
| MTZ + Nisoldipine | 0.5 | 0.5 | 1 | additive | |
| MTZ + Nimodipine | 0.125 | 0.5 | 0.62 | additive | |
| MTZ + Nitrendipine | 1 | 1 | 2 | no interaction | |
| MTZ + Lercanidipine | 1 | 1 | 2 | no interaction | |
| ATCC 700684 (CLR-R) | CLR + Nifedipine | 0.5 | 0.5 | 1 | additive |
| CLR + Nicardipine | 0.25 | 0.5 | 0.75 | additive | |
| CLR + Nisoldipine | 0.5 | 0.5 | 1 | additive | |
| CLR + Nimodipine | 0.5 | 0.5 | 1 | additive | |
| CLR + Nitrendipine | 1 | 1 | 2 | no interaction | |
| CLR + Lercanidipine | 0.0625 | 0.5 | 0.56 | additive |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
González, A.; Casado, J.; Chueca, E.; Salillas, S.; Velázquez-Campoy, A.; Espinosa Angarica, V.; Bénejat, L.; Guignard, J.; Giese, A.; Sancho, J.; et al. Repurposing Dihydropyridines for Treatment of Helicobacter pylori Infection. Pharmaceutics 2019, 11, 681. https://doi.org/10.3390/pharmaceutics11120681
González A, Casado J, Chueca E, Salillas S, Velázquez-Campoy A, Espinosa Angarica V, Bénejat L, Guignard J, Giese A, Sancho J, et al. Repurposing Dihydropyridines for Treatment of Helicobacter pylori Infection. Pharmaceutics. 2019; 11(12):681. https://doi.org/10.3390/pharmaceutics11120681
Chicago/Turabian StyleGonzález, Andrés, Javier Casado, Eduardo Chueca, Sandra Salillas, Adrián Velázquez-Campoy, Vladimir Espinosa Angarica, Lucie Bénejat, Jérome Guignard, Alban Giese, Javier Sancho, and et al. 2019. "Repurposing Dihydropyridines for Treatment of Helicobacter pylori Infection" Pharmaceutics 11, no. 12: 681. https://doi.org/10.3390/pharmaceutics11120681