Strategies for Delivery of siRNAs to Ovarian Cancer Cells

 ,
,  , , , , ,
, , , , ,  ,
,
Abstract
:1. Introduction
1.1. Ovarian Carcinoma
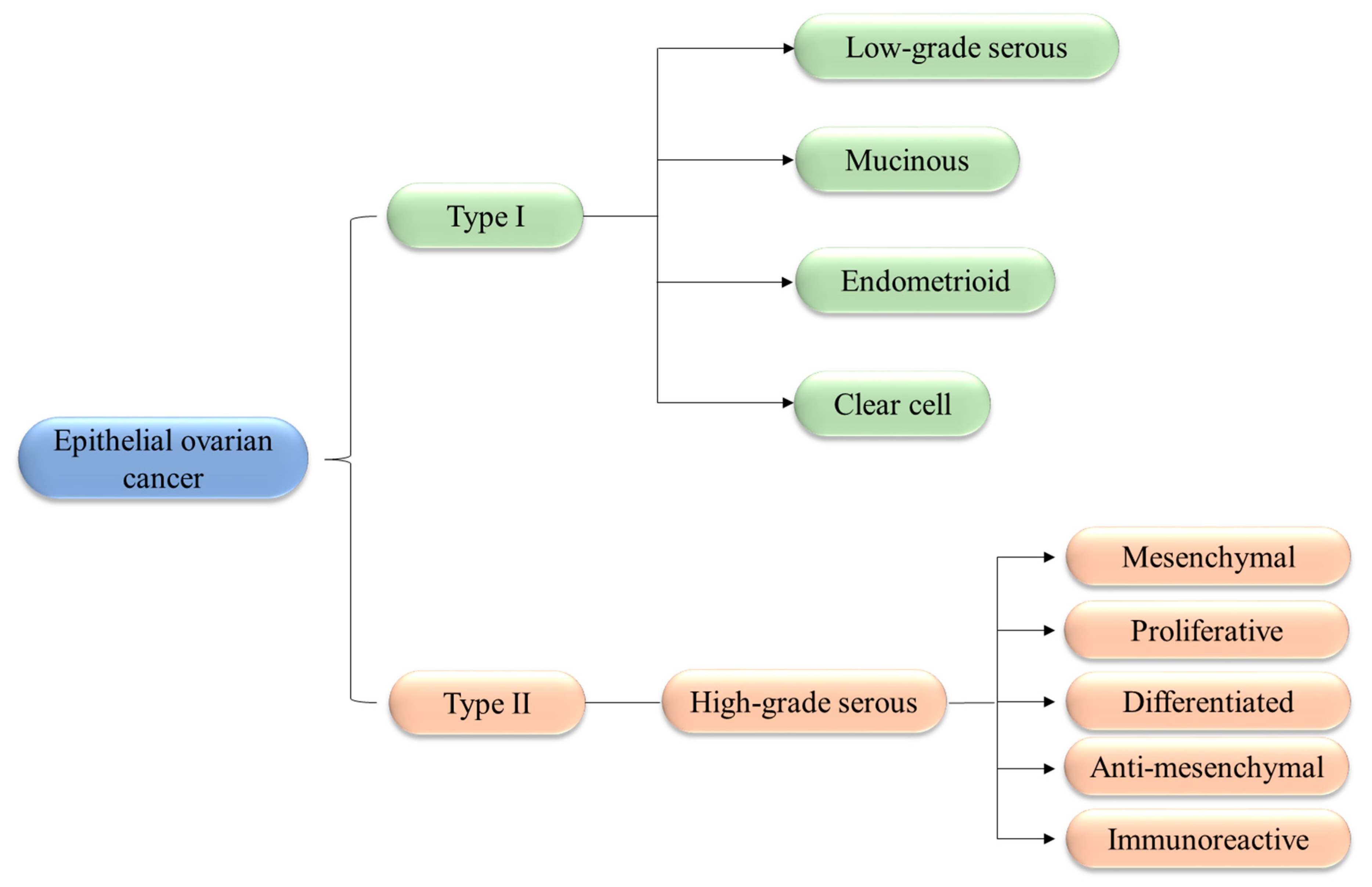
1.2. Classification and Biology of OCs
1.3. Current Therapies
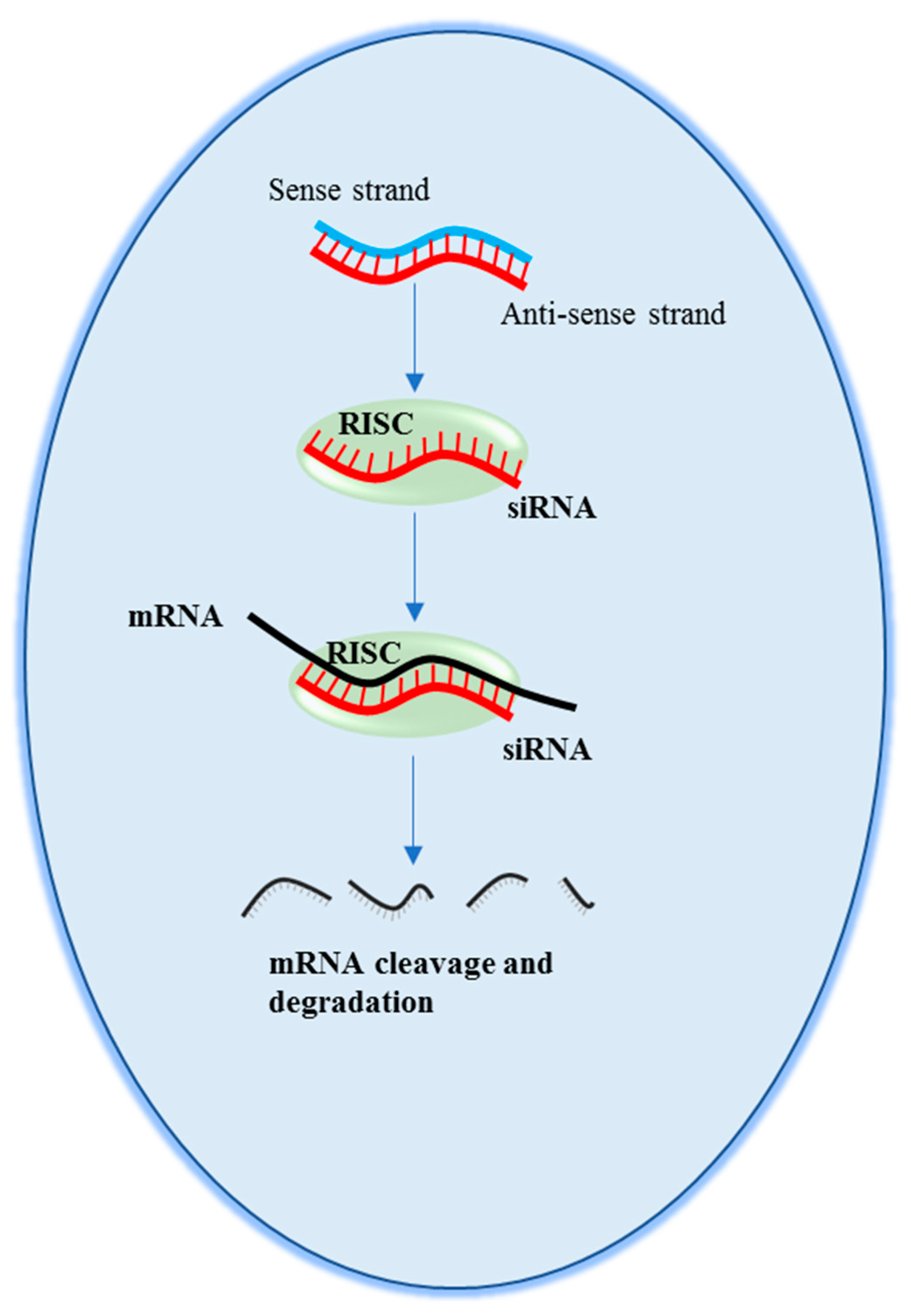
2. Small Interfering RNAs
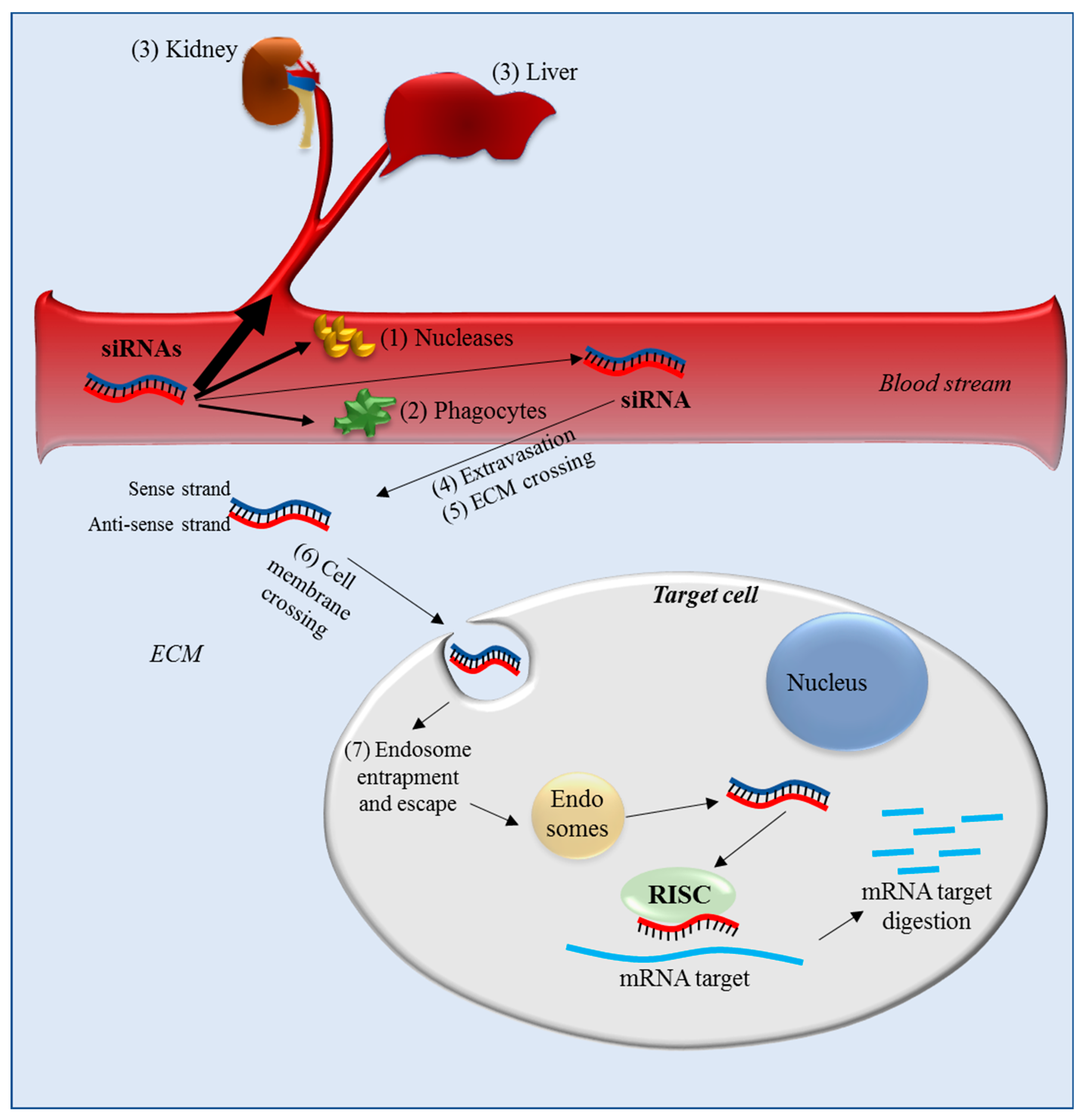
2.1. Delivery Barriers for Systemic Administration
2.2. Delivery Barriers for IP Administration
3. Strategies to Optimize siRNA Delivery
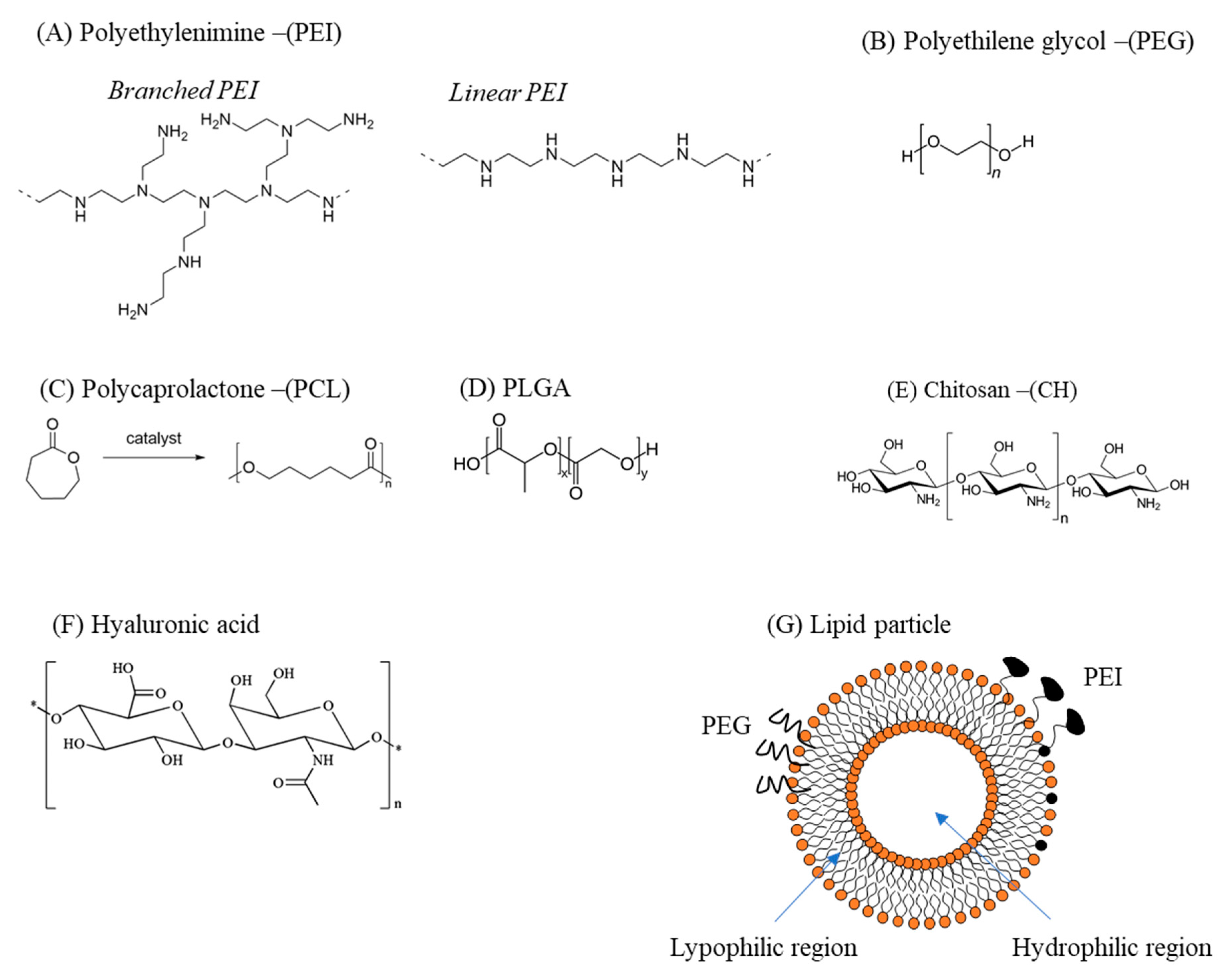
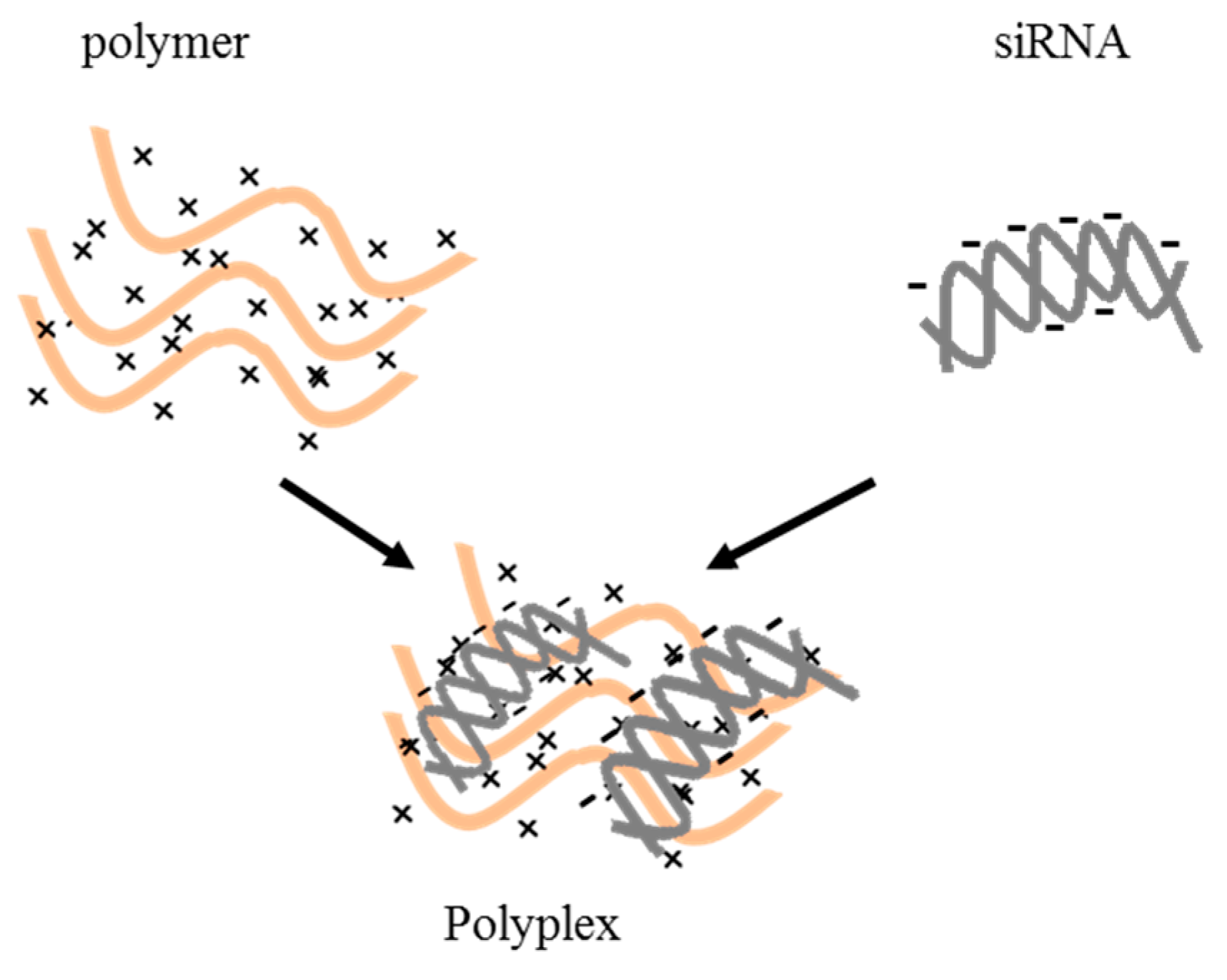
3.1. Polymer-Based Delivery Vectors
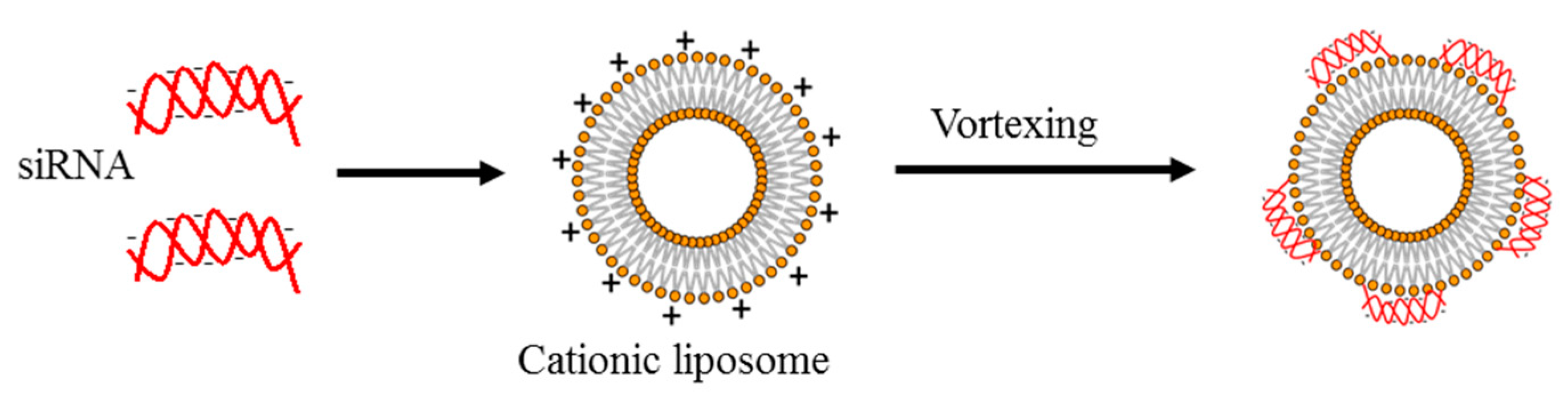
3.2. Lipid-Based Delivery Vectors
4. Targeting OC: Delivery Systems and siRNA Targets
4.1. OC-Targeting Molecules
4.2. siRNA Targets in OC
4.2.1. Molecular Targets Implicated in Cell Growth/Migration
4.2.2. Molecular Targets Implicated in Angiogenesis
4.2.3. Molecular Targets Implicated in Drug Resistance
5. Experimental Models of OC
5.1. Cellular Model
5.2. Animal Models
5.3. Mathematical Models
6. siRNA Delivery to OC Cells
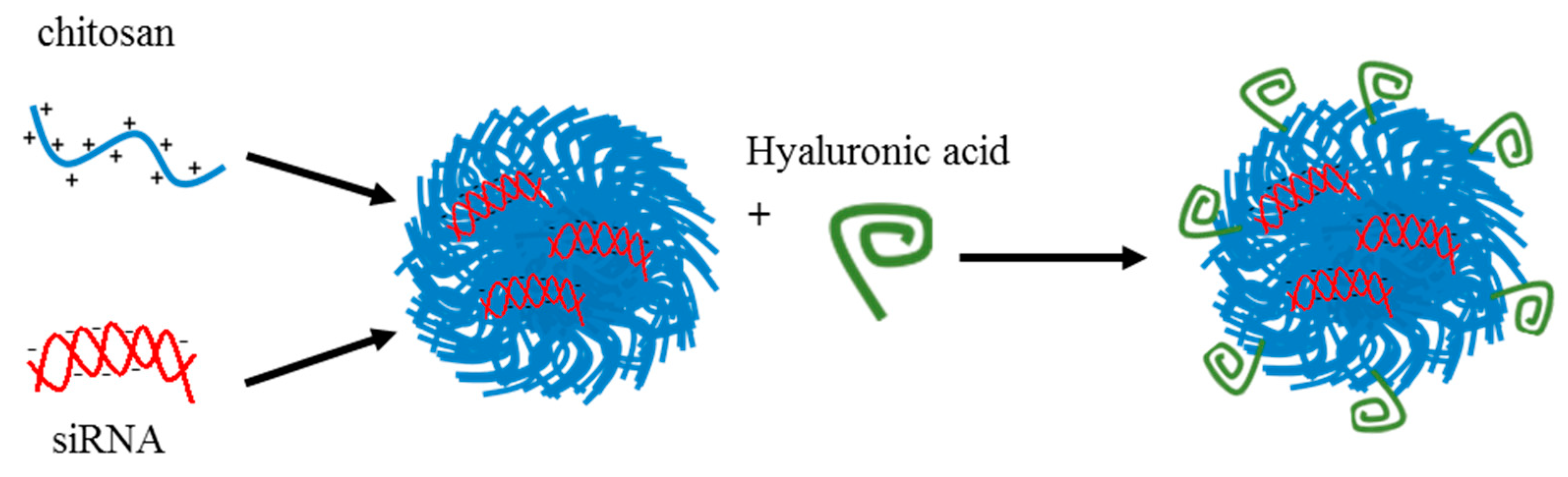
6.1. Polymeric Delivery Systems
6.1.1. Non-Targeted Delivery Systems
6.1.2. Targeted Delivery Systems
6.2. Lipid-Based Delivery Systems
7. Final Considerations
Author Contributions
Funding
Conflicts of Interest
References
- Farra, R.; Dapas, B.; Pozzato, G.; Scaggiante, B.; Agostini, F.; Zennaro, C.; Grassi, M.; Rosso, N.; Giansante, C.; Fiotti, N.; et al. Effects of E2F1–cyclin E1–E2 circuit down regulation in hepatocellular carcinoma cells. Dig. Liver Dis. 2011, 43, 1006–1014. [Google Scholar] [CrossRef]
- Grassi, M.; Cavallaro, G.; Scirè, S.; Scaggiante, B.; Daps, B.; Farra, R.; Baiz, D.; Giansante, C.; Guarnieri, G.; Perin, D.; et al. Current Strategies to Improve the Efficacy and the Delivery of Nucleic Acid Based Drugs. Curr. Signal Transduct. Ther. 2010, 5, 92–120. [Google Scholar] [CrossRef]
- Grassi, G.; Dawson, P.; Guarnieri, G.; Kandolf, R.; Grassi, M. Therapeutic potential of hammerhead ribozymes in the treatment of hyper-proliferative diseases. Curr. Pharm. Biotechnol. 2004, 5, 369–386. [Google Scholar] [CrossRef]
- Grassi, G.; Marini, J.C. Ribozymes: Structure, function, and potential therapy for dominant genetic disorders. Ann. Med. 1996, 28, 499–510. [Google Scholar] [CrossRef]
- Grassi, G.; Schneider, A.; Engel, S.; Racchi, G.; Kandolf, R.; Kuhn, A. Hammerhead ribozymes targeted against cyclin E and E2F1 cooperate to down-regulate coronary smooth muscle cell proliferation. J. Gene Med. 2005, 7, 1223–1234. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, M.S. siRNA delivery for the treatment of ovarian cancer. Methods 2013, 63, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Van den Brand, D.; Mertens, V.; Massuger, L.F.A.G.; Brock, R. siRNA in ovarian cancer—Delivery strategies and targets for therapy. J. Control. Release 2018, 283, 45–58. [Google Scholar] [CrossRef] [PubMed]
- Aghamiri, S.; Mehrjardi, K.F.; Shabani, S.; Keshavarz-Fathi, M.; Kargar, S.; Rezaei, N. Nanoparticle-siRNA: A potential strategy for ovarian cancer therapy? Nanomedicine 2019, 14, 2083–2100. [Google Scholar] [CrossRef] [PubMed]
- Halbur, C.; Choudhury, N.; Chen, M.; Kim, J.H.; Chung, E.J. siRNA-Conjugated Nanoparticles to Treat Ovarian Cancer. SLAS Technol. 2019, 24, 137–150. [Google Scholar] [CrossRef]
- Webb, P.M.; Jordan, S.J. Epidemiology of epithelial ovarian cancer. Best Pract. Res. Clin. Obstet. Gynaecol. 2017, 41, 3–14. [Google Scholar] [CrossRef] [Green Version]
- Reid, B.M.; Permuth, J.B.; Sellers, T.A. Epidemiology of ovarian cancer: A review. Cancer Biol. Med. 2017, 14, 9–32. [Google Scholar] [PubMed]
- Singh, A.; Gupta, S.; Sachan, M. Epigenetic Biomarkers in the Management of Ovarian Cancer: Current Prospectives. Front. Cell Dev. Biol. 2019, 7, 182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2016. CA Cancer J. Clin. 2016, 66, 7–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, S.J.; Hodeib, M.; Chang, J.; Bristow, R.E. Survival impact of complete cytoreduction to no gross residual disease for advanced-stage ovarian cancer: A meta-analysis. Gynecol. Oncol. 2013, 130, 493–498. [Google Scholar] [CrossRef] [PubMed]
- Shih, I.; Kurman, R.J. Ovarian tumorigenesis: A proposed model based on morphological and molecular genetic analysis. Am. J. Pathol. 2004, 164, 1511–1518. [Google Scholar] [CrossRef]
- Verhaak, R.G.; Tamayo, P.; Yang, J.Y.; Hubbard, D.; Zhang, H.; Creighton, C.J.; Fereday, S.; Lawrence, M.; Carter, S.L.; Mermel, C.H.; et al. Prognostically relevant gene signatures of high-grade serous ovarian carcinoma. J. Clin. Investig. 2013, 123, 517–525. [Google Scholar] [CrossRef]
- Konecny, G.E.; Wang, C.; Hamidi, H.; Winterhoff, B.; Kalli, K.R.; Dering, J.; Ginther, C.; Chen, H.W.; Dowdy, S.; Cliby, W.; et al. Prognostic and therapeutic relevance of molecular subtypes in high-grade serous ovarian cancer. J. Natl. Cancer Inst. 2014, 106. [Google Scholar] [CrossRef]
- Wang, C.; Armasu, S.M.; Kalli, K.R.; Maurer, M.J.; Heinzen, E.P.; Keeney, G.L.; Cliby, W.A.; Oberg, A.L.; Kaufmann, S.H.; Goode, E.L. Pooled Clustering of High-Grade Serous Ovarian Cancer Gene Expression Leads to Novel Consensus Subtypes Associated with Survival and Surgical Outcomes. Clin. Cancer Res. 2017, 23, 4077–4085. [Google Scholar] [CrossRef] [Green Version]
- Lisio, M.A.; Fu, L.; Goyeneche, A.; Gao, Z.H.; Telleria, C. High-Grade Serous Ovarian Cancer: Basic Sciences, Clinical and Therapeutic Standpoints. Int. J. Mol. Sci. 2019, 20, 952. [Google Scholar] [CrossRef]
- Narod, S. Can advanced-stage ovarian cancer be cured? Nat. Rev. Clin. Oncol. 2016, 13, 255–261. [Google Scholar] [CrossRef]
- Nieman, K.M.; Kenny, H.A.; Penicka, C.V.; Ladanyi, A.; Buell-Gutbrod, R.; Zillhardt, M.R.; Romero, I.L.; Carey, M.S.; Mills, G.B.; Hotamisligil, G.S.; et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat. Med. 2011, 17, 1498–1503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matulonis, U.A.; Sood, A.K.; Fallowfield, L.; Howitt, B.E.; Sehouli, J.; Karlan, B.Y. Ovarian cancer. Nat. Rev. Dis. Primers 2016, 2, 16061. [Google Scholar] [CrossRef]
- Gonzalez-Martin, A.; Sanchez-Lorenzo, L.; Bratos, R.; Marquez, R.; Chiva, L. First-line and maintenance therapy for ovarian cancer: Current status and future directions. Drugs 2014, 74, 879–889. [Google Scholar] [CrossRef] [PubMed]
- Markman, M. Optimizing primary chemotherapy in ovarian cancer. Hematol. Oncol. Clin. N. Am. 2003, 17, 957–968. [Google Scholar] [CrossRef]
- Marchetti, C.; De Felice, F.; Perniola, G.; Palaia, I.; Musella, A.; Di Donato, V.; Cascialli, G.; Muzii, L.; Tombolini, V.; Cascialli, G.; et al. Role of intraperitoneal chemotherapy in ovarian cancer in the platinum-taxane-based era: A meta-analysis. Crit. Rev. Oncol. Hematol. 2019, 136, 64–69. [Google Scholar] [CrossRef]
- Bast, R.C., Jr.; Hennessy, B.; Mills, G.B. The biology of ovarian cancer: New opportunities for translation. Nat. Rev. Cancer 2009, 9, 415–428. [Google Scholar] [CrossRef]
- Papa, A.; Caruso, D.; Strudel, M.; Tomao, S.; Tomao, F. Update on Poly-ADP-ribose polymerase inhibition for ovarian cancer treatment. J. Transl. Med. 2016, 14, 267. [Google Scholar] [CrossRef]
- Napoli, C.; Lemieux, C.; Jorgensen, R. Introduction of a Chimeric Chalcone Synthase Gene into Petunia Results in Reversible Co-Suppression of Homologous Genes in trans. Plant Cell 1990, 2, 279–289. [Google Scholar] [CrossRef]
- Fire, A.; Xu, S.; Montgomery, M.K.; Kostas, S.A.; Driver, S.E.; Mello, C.C. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 1998, 391, 806–811. [Google Scholar] [CrossRef]
- Elbashir, S.M.; Harborth, J.; Lendeckel, W.; Yalcin, A.; Weber, K.; Tuschl, T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 2001, 411, 494–498. [Google Scholar] [CrossRef]
- Lang, C.; Sauter, M.; Szalay, G.; Racchi, G.; Grassi, G.; Rainaldi, G.; Mercatanti, A.; Lang, F.; Kandolf, R.; Klingel, K. Connective tissue growth factor: A crucial cytokine-mediating cardiac fibrosis in ongoing enterovirus myocarditis. J. Mol. Med. 2008, 86, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Grassi, G.; Pozzato, G.; Moretti, M.; Giacca, M. Quantitative analysis of hepatitis C virus RNA in liver biopsies by competitive reverse transcription and polymerase chain reaction. J. Hepatol. 1995, 23, 403–411. [Google Scholar] [CrossRef]
- Scaggiante, B.; Dapas, B.; Farra, R.; Grassi, M.; Pozzato, G.; Giansante, C.; Fiotti, N.; Grassi, G. Improving siRNA bio-distribution and minimizing side effects. Curr. Drug Metab. 2011, 12, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Farra, R.; Grassi, M.; Grassi, G.; Dapas, B. Therapeutic potential of small interfering RNAs/micro interfering RNA in hepatocellular carcinoma. World J. Gastroenterol. 2015, 21, 8994–9001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grassi, G.; Scaggiante, B.; Dapas, B.; Farra, R.; Tonon, F.; Lamberti, G.; Barba, A.; Fiorentino, S.; Fiotti, N.; Zanconati, F.; et al. Therapeutic potential of nucleic acid-based drugs in coronary hyper- proliferative vascular diseases. Curr. Med. Chem. 2013, 20, 3515–3538. [Google Scholar] [CrossRef] [PubMed]
- Agostini, F.; Dapas, B.; Farra, R.; Grassi, M.; Racchi, G.; Klingel, K.; Kandolf, R.; Heidenreich, O.; Mercatahnti, A.; Rainaldi, G.; et al. Potential applications of small interfering RNAs in the cardiovascular field. Drug Future 2006, 31, 513–525. [Google Scholar] [CrossRef]
- Huang, Y.; Hong, J.; Zheng, S.; Ding, Y.; Guo, S.; Zhang, H.; Zhang, X.; Du, Q.; Liang, Z. Elimination pathways of systemically delivered siRNA. Mol. Ther. 2011, 19, 381–385. [Google Scholar] [CrossRef]
- Jackson, A.L.; Linsley, P.S. Recognizing and avoiding siRNA off-target effects for target identification and therapeutic application. Nat. Rev. Drug Discov. 2010, 9, 57–67. [Google Scholar] [CrossRef]
- Sarfarazi, A.; Lee, G.; Mirjalili, S.A.; Phillips, A.R.J.; Windsor, J.A.; Trevaskis, N.L. Therapeutic delivery to the peritoneal lymphatics: Current understanding, potential treatment benefits and future prospects. Int. J. Pharm. 2019, 567, 118456. [Google Scholar] [CrossRef]
- Trevaskis, N.L.; Kaminskas, L.M.; Porter, C.J. From sewer to saviour—Targeting the lymphatic system to promote drug exposure and activity. Nat. Rev. Drug Discov. 2015, 14, 781–803. [Google Scholar] [CrossRef]
- Mirahmadi, N.; Babaei, M.H.; Vali, A.M.; Dadashzadeh, S. Effect of liposome size on peritoneal retention and organ distribution after intraperitoneal injection in mice. Int. J. Pharm. 2010, 383, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Lengyel, E. Ovarian cancer development and metastasis. Am. J. Pathol. 2010, 177, 1053–1064. [Google Scholar] [CrossRef] [PubMed]
- Luft, C.; Ketteler, R. Electroporation Knows No Boundaries: The Use of Electrostimulation for siRNA Delivery in Cells and Tissues. J. Biomol. Screen. 2015, 20, 932–942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leonetti, J.P.; Degols, G.; Lebleu, B. Biological activity of oligonucleotide-poly(l-lysine) conjugates: Mechanism of cell uptake. Bioconjug. Chem. 1990, 1, 149–153. [Google Scholar] [CrossRef] [PubMed]
- Sardo, C.; Farra, R.; Licciardi, M.; Dapas, B.; Scialabba, C.; Giammona, G.; Grassi, M.; Grassi, G.; Cavallaro, G. Development of a simple, biocompatible and cost-effective Inulin-Diethylenetriamine based siRNA delivery system. Eur. J. Pharm. Sci. 2015, 75, 60–71. [Google Scholar] [CrossRef] [PubMed]
- Cavallaro, G.; Licciardi, M.; Amato, G.; Sardo, C.; Giammona, G.; Farra, R.; Dapas, B.; Grassi, M.; Grassi, G. Synthesis and characterization of polyaspartamide copolymers obtained by ATRP for nucleic acid delivery. Int. J. Pharm. 2014, 466, 246–257. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Wang, J. Delivery systems for siRNA drug development in cancer therapy. Asian J. Pharm. Sci. 2015, 10, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Hobel, S.; Aigner, A. Polyethylenimines for siRNA and miRNA delivery in vivo. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2013, 5, 484–501. [Google Scholar] [CrossRef]
- Liu, L.; Zheng, M.; Librizzi, D.; Renette, T.; Merkel, O.M.; Kissel, T. Efficient and Tumor Targeted siRNA Delivery by Polyethylenimine-graft-polycaprolactone-block-poly(ethylene glycol)-folate (PEI-PCL-PEG-Fol). Mol. Pharm. 2016, 13, 134–143. [Google Scholar] [CrossRef]
- Roberts, M.J.; Bentley, M.D.; Harris, J.M. Chemistry for peptide and protein PEGylation. Adv. Drug Deliv. Rev. 2002, 54, 459–476. [Google Scholar] [CrossRef]
- Bao, Y.; Jin, Y.; Chivukula, P.; Zhang, J.; Liu, Y.; Liu, J.; Clamme, J.P.; Mahato, R.I.; Ng, D.; Ying, W.; et al. Effect of PEGylation on biodistribution and gene silencing of siRNA/lipid nanoparticle complexes. Pharm. Res. 2013, 30, 342–351. [Google Scholar] [CrossRef] [PubMed]
- Muralidharan, P.; Mallory, E.; Malapit, M.; Hayes, D., Jr.; Mansour, H.M. Inhalable PEGylated Phospholipid Nanocarriers and PEGylated Therapeutics for Respiratory Delivery as Aerosolized Colloidal Dispersions and Dry Powder Inhalers. Pharmaceutics 2014, 6, 333–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azimi, B.; Nourpanak, P.; Rabiee, M.; Arab, S. Poly (ε-caprolactone) Fiber: An Overview. J. Eng. Fibers Fabr. 2014, 9, 74–90. [Google Scholar]
- Xu, Z.; Wang, D.; Cheng, Y.; Yang, M.; Wu, L.P. Polyester based nanovehicles for siRNA delivery. Mater. Sci. Eng. C Mater. Biol. Appl. 2018, 92, 1006–1015. [Google Scholar] [CrossRef] [PubMed]
- Posocco, B.; Dreussi, E.; de Santa, J.; Toffoli, G.; Abrami, M.; Musiani, F.; Grassi, M.; Farra, R.; Tonon, F.; Grassi, G.; et al. Polysaccharides for the Delivery of Antitumor Drugs. Materials 2015, 8, 2569–2615. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, T.; Aljaeid, B. Preparation characterization and potential application of chitosan, chitosan derivates, and chitosan metal nanoparticles in pharmaceutical drug delivery. Drug Des. Dev. Ther. 2016, 10, 483–507. [Google Scholar] [CrossRef] [PubMed]
- Khan, W.; Hosseinkhani, H.; Ickowicz, D.; Hong, P.D.; Yu, D.S.; Domb, A.J. Polysaccharide gene transfection agents. Acta Biomater. 2012, 8, 4224–4232. [Google Scholar] [CrossRef]
- Oh, E.J.; Park, K.; Kim, K.S.; Kim, J.; Yang, J.A.; Kong, J.H.; Lee, M.Y.; Hoffman, A.S.; Hahn, S.K. Target specific and long-acting delivery of protein, peptide, and nucleotide therapeutics using hyaluronic acid derivatives. J. Control. Release 2010, 141, 2–12. [Google Scholar] [CrossRef]
- Barba, A.A.; Lamberti, G.; Sardo, C.; Dapas, B.; Abrami, M.; Grassi, M.; Farra, R.; Tonon, F.; Forte, G.; Musiani, F.; et al. Novel Lipid and Polymeric Materials as Delivery Systems for Nucleic Acid Based Drugs. Curr. Drug Metab. 2015, 16, 427–452. [Google Scholar] [CrossRef]
- Bochicchio, S.; Dalmoro, A.; Barba, A.A.; Grassi, G.; Lamberti, G. Liposomes as siRNA delivery vectors. Curr. Drug Metab. 2014, 15, 882–892. [Google Scholar] [CrossRef]
- Ayen, A.; Jimenez, M.Y.; Marchal, J.A.; Boulaiz, H. Recent Progress in Gene Therapy for Ovarian Cancer. Int. J. Mol. Sci. 2018, 19, 1930. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.; Jablons, D.; Yang, C.; You, L. Epidermal Growth Factor Receptor (EGFR) Pathway, Yes-Associated Protein (YAP) and the Regulation of Programmed Death-Ligand 1 (PD-L1) in Non-Small Cell Lung Cancer (NSCLC). Int. J. Mol. Sci. 2019, 20, 3821. [Google Scholar] [CrossRef] [PubMed]
- Shepard, H.M.; Brdlik, C.M.; Schreiber, H. Signal integration: A framework for understanding the efficacy of therapeutics targeting the human EGFR family. J. Clin. Investig. 2008, 118, 3574–3581. [Google Scholar] [CrossRef] [PubMed]
- Dickerson, E.B.; Blackburn, W.H.; Smith, M.H.; Kapa, L.B.; Lyon, L.A.; McDonald, J.F. Chemosensitization of cancer cells by siRNA using targeted nanogel delivery. BMC Cancer 2010, 10, 10. [Google Scholar] [CrossRef]
- Zhou, Y.; Sakurai, H. Emerging and Diverse Functions of the EphA2 Noncanonical Pathway in Cancer Progression. Biol. Pharm. Bull. 2017, 40, 1616–1624. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, Y.; Hamasaki, M.; Aoki, M.; Koga, K.; Koshikawa, N.; Miyamoto, S.; Nabeshima, K. Activated EphA2 Processing by MT1-MMP Is Involved in Malignant Transformation of Ovarian Tumours In Vivo. Anticancer Res. 2018, 38, 4257–4266. [Google Scholar] [CrossRef]
- Cortez, A.J.; Tudrej, P.; Kujawa, K.A.; Lisowska, K.M. Advances in ovarian cancer therapy. Cancer Chemother. Pharmacol. 2018, 81, 17–38. [Google Scholar] [CrossRef]
- Cheung, A.; Bax, H.J.; Josephs, D.H.; Ilieva, K.M.; Pellizzari, G.; Opzoomer, J.; Bloomfield, J.; Fittall, M.; Grigoriadis, A.; Figini, M.; et al. Targeting folate receptor alpha for cancer treatment. Oncotarget 2016, 7, 52553–52574. [Google Scholar] [CrossRef]
- Zong, X.; Nephew, K.P. Ovarian Cancer Stem Cells: Role in Metastasis and Opportunity for Therapeutic Targeting. Cancers 2019, 11, 934. [Google Scholar] [CrossRef]
- Bartakova, A.; Michalova, K.; Presl, J.; Vlasak, P.; Kostun, J.; Bouda, J. CD44 as a cancer stem cell marker and its prognostic value in patients with ovarian carcinoma. J. Obstet. Gynaecol. 2018, 38, 110–114. [Google Scholar] [CrossRef]
- Shah, V.; Taratula, O.; Garbuzenko, O.B.; Taratula, O.R.; Rodriguez-Rodriguez, L.; Minko, T. Targeted nanomedicine for suppression of CD44 and simultaneous cell death induction in ovarian cancer: An optimal delivery of siRNA and anticancer drug. Clin. Cancer Res. 2013, 19, 6193–6204. [Google Scholar] [CrossRef] [PubMed]
- Skubitz, A.P.; Taras, E.P.; Boylan, K.L.; Waldron, N.N.; Oh, S.; Panoskaltsis-Mortari, A.; Vallera, D.A. Targeting CD133 in an in vivo ovarian cancer model reduces ovarian cancer progression. Gynecol. Oncol. 2013, 130, 579–587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, B.; Yan, X.; Liu, L.; Jiang, C.; Hou, S. Overexpression of the cancer stem cell marker CD117 predicts poor prognosis in epithelial ovarian cancer patients: Evidence from meta-analysis. Onco Targets Ther. 2017, 10, 2951–2961. [Google Scholar] [CrossRef] [PubMed]
- Shaw, T.J.; Vanderhyden, B.C. AKT mediates the pro-survival effects of KIT in ovarian cancer cells and is a determinant of sensitivity to imatinib mesylate. Gynecol. Oncol. 2007, 105, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Schilder, R.J.; Sill, M.W.; Lee, R.B.; Shaw, T.J.; Senterman, M.K.; Klein-Szanto, A.J.; Miner, Z.; Vanderhyden, B.C. Phase II evaluation of imatinib mesylate in the treatment of recurrent or persistent epithelial ovarian or primary peritoneal carcinoma: A Gynecologic Oncology Group Study. J. Clin. Oncol. 2008, 26, 3418–3425. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Terai, Y.; Tanabe, A.; Ono, Y.J.; Hayashi, M.; Maeda, K.; Fujiwara, S.; Ashihara, K.; Nakamura, M.; Tanaka, Y.; et al. CD24 expression is a marker for predicting clinical outcome and regulates the epithelial-mesenchymal transition in ovarian cancer via both the Akt and ERK pathways. Oncol. Rep. 2017, 37, 3189–3200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, R.; Shi, H.; Chen, Z.; Wu, Q.; Ren, F.; Huang, H. Effects of metastasis-associated in colon cancer 1 inhibition by small hairpin RNA on ovarian carcinoma OVCAR-3 cells. J. Exp. Clin. Cancer Res. 2011, 30, 83. [Google Scholar] [CrossRef]
- Rao, Y.; Ji, M.; Chen, C.; Shi, H. Effect of siRNA targeting MTA1 on metastasis malignant phenotype of ovarian cancer A2780 cells. J. Huazhong Univ. Sci. Technol. Med. Sci. 2013, 33, 266–271. [Google Scholar] [CrossRef]
- Huo, X.; Ren, L.; Shang, L.; Wang, X.; Wang, J. Effect of WT1 antisense mRNA on the induction of apoptosis in ovarian carcinoma SKOV3 cells. Eur. J. Gynaecol. Oncol. 2011, 32, 651–656. [Google Scholar]
- De, P.; Aske, J.; Dey, N. RAC1 Takes the Lead in Solid Tumors. Cells 2019, 8, 382. [Google Scholar] [CrossRef]
- Hudson, L.; Gillette, J.; Kang, H.; Rivera, M.; Wandinger-Ness, A. Ovarian Tumor Microenvironment Signaling:Convergence on the Rac1 GTPase. Cancers 2018, 10, 358. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Yu, X. The balance of Polo-like kinase 1 in tumorigenesis. Cell Div. 2009, 4, 4. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Shi, H.; Ren, F.; Liu, H.; Zhang, M.; Deng, Y.; Li, X. Misregulation of polo-like protein kinase 1, P53 and P21WAF1 in epithelial ovarian cancer suggests poor prognosis. Oncol. Rep. 2015, 33, 1235–1242. [Google Scholar] [CrossRef] [PubMed]
- Farra, R.; Dapas, B.; Grassi, M.; Benedetti, F.; Grassi, G. E2F1 as a molecular drug target in ovarian cancer. Expert Opin. Ther. Targets 2019, 23, 161–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oku, Y.; Nishiya, N.; Tazawa, T.; Kobayashi, T.; Umezawa, N.; Sugawara, Y.; Uehara, Y. Augmentation of the therapeutic efficacy of WEE1 kinase inhibitor AZD1775 by inhibiting the YAP-E2F1-DNA damage response pathway axis. FEBS Open Bio 2018, 8, 1001–1012. [Google Scholar] [CrossRef] [PubMed]
- Ryo, A.; Liou, Y.C.; Wulf, G.; Nakamura, M.; Lee, S.W.; Lu, K.P. PIN1 is an E2F target gene essential for Neu/Ras-induced transformation of mammary epithelial cells. Mol. Cell. Biol. 2002, 22, 5281–5295. [Google Scholar] [CrossRef] [PubMed]
- Liou, Y.C.; Zhou, X.Z.; Lu, K.P. Prolyl isomerase Pin1 as a molecular switch to determine the fate of phosphoproteins. Trends Biochem. Sci. 2011, 36, 501–514. [Google Scholar] [CrossRef] [Green Version]
- Russo, S.C.; De, S.L.; Palazzolo, S.; Salis, B.; Granchi, C.; Minutolo, F.; Tuccinardi, T.; Fratamico, R.; Crotti, S.; D’Aronco, S.; et al. Liposomal delivery of a Pin1 inhibitor complexed with cyclodextrins as new therapy for high-grade serous ovarian cancer. J. Control. Release 2018, 281, 1–10. [Google Scholar] [CrossRef]
- Russo, S.C.; De, S.L.; Poli, G.; Granchi, C.; El, B.M.; Ecca, F.; Grassi, G.; Grassi, M.; Canzonieri, V.; Giordano, A.; et al. Virtual screening identifies a PIN1 inhibitor with possible antiovarian cancer effects. J. Cell. Physiol. 2019. [Google Scholar] [CrossRef]
- Van Beijnum, J.R.; Petersen, K.; Griffioen, A.W. Tumor endothelium is characterized by a matrix remodeling signature. Front. Biosci. (Sch. Ed.) 2009, 1, 216–225. [Google Scholar] [CrossRef]
- Ren, F.; Shen, J.; Shi, H.; Hornicek, F.J.; Kan, Q.; Duan, Z. Novel mechanisms and approaches to overcome multidrug resistance in the treatment of ovarian cancer. Biochim. Biophys. Acta 2016, 1866, 266–275. [Google Scholar] [CrossRef] [PubMed]
- Trnski, D.; Gregoric, M.; Levanat, S.; Ozretic, P.; Rincic, N.; Vidakovic, T.M.; Kalafatic, D.; Maurac, I.; Oreskovic, S.; Sabol, M.; et al. Regulation of Survivin Isoform Expression by GLI Proteins in Ovarian Cancer. Cells 2019, 8, 128. [Google Scholar] [CrossRef] [PubMed]
- Togashi, K.; Okada, M.; Yamamoto, M.; Suzuki, S.; Sanomachi, T.; Seino, S.; Yamashita, H.; Kitanaka, C. A Small-molecule Kinase Inhibitor, CEP-1347, Inhibits Survivin Expression and Sensitizes Ovarian Cancer Stem Cells to Paclitaxel. Anticancer Res. 2018, 38, 4535–4542. [Google Scholar] [CrossRef]
- Levy, A.; Alhazzani, K.; Dondapati, P.; Alaseem, A.; Cheema, K.; Thallapureddy, K.; Kaur, P.; Alobid, S.; Rathinavelu, A. Focal Adhesion Kinase in Ovarian Cancer: A Potential Therapeutic Target for Platinum and Taxane-Resistant Tumors. Curr. Cancer Drug Targets 2019, 19, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Li, S.; Sun, Y.; Zhang, D.; Zhao, Z.; Liu, L. Reversing platinum resistance in ovarian cancer multicellular spheroids by targeting Bcl-2. Onco Targets Ther. 2019, 12, 897–906. [Google Scholar] [CrossRef] [PubMed]
- Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [CrossRef] [PubMed]
- Domcke, S.; Sinha, R.; Levine, D.A.; Sander, C.; Schultz, N. Evaluating cell lines as tumour models by comparison of genomic profiles. Nat. Commun. 2013, 4, 2126. [Google Scholar] [CrossRef]
- Cho, K.R.; Shih, I. Ovarian cancer. Annu. Rev. Pathol. 2009, 4, 287–313. [Google Scholar] [CrossRef]
- Clevers, H. Modeling Development and Disease with Organoids. Cell 2016, 165, 1586–1597. [Google Scholar] [CrossRef] [Green Version]
- Jabs, J.; Zickgraf, F.M.; Park, J.; Wagner, S.; Jiang, X.; Jechow, K.; Kleinheinz, K.; Toprak, U.H.; Schneider, M.A.; Meister, M.; et al. Screening drug effects in patient-derived cancer cells links organoid responses to genome alterations. Mol. Syst. Biol. 2017, 13, 955. [Google Scholar] [CrossRef]
- Kopper, O.; de Witte, C.J.; Lohmussaar, K.; Valle-Inc, J.E.; Hami, N.; Kester, L.; Balgobind, A.V.; Korving, J.; Proost, N.; Begthel, H.; et al. An organoid platform for ovarian cancer captures intra- and interpatient heterogeneity. Nat. Med. 2019, 25, 838–849. [Google Scholar] [CrossRef] [PubMed]
- Maru, Y.; Tanaka, N.; Itami, M.; Hippo, Y. Efficient use of patient-derived organoids as a preclinical model for gynecologic tumors. Gynecol. Oncol. 2019, 154, 189–198. [Google Scholar] [CrossRef] [PubMed]
- McCloskey, C.W.; Rodriguez, G.M.; Galpin, K.J.C.; Vanderhyden, B.C. Ovarian Cancer Immunotherapy: Preclinical Models and Emerging Therapeutics. Cancers 2018, 10, 244. [Google Scholar] [CrossRef] [PubMed]
- Bobbs, A.S.; Cole, J.M.; Cowden Dahl, K.D. Emerging and Evolving Ovarian Cancer Animal Models. Cancer Growth Metastasis 2015, 8, 29–36. [Google Scholar] [CrossRef]
- Scott, C.L.; Becker, M.A.; Haluska, P.; Samimi, G. Patient-derived xenograft models to improve targeted therapy in epithelial ovarian cancer treatment. Front. Oncol. 2013, 3, 295. [Google Scholar] [CrossRef]
- Roby, K.F.; Taylor, C.C.; Sweetwood, J.P.; Cheng, Y.; Pace, J.L.; Tawfik, O.; Persons, D.L.; Smith, P.G.; Terranova, P.F. Development of a syngeneic mouse model for events related to ovarian cancer. Carcinogenesis 2000, 21, 585–591. [Google Scholar] [CrossRef]
- McCloskey, C.W.; Goldberg, R.L.; Carter, L.E.; Gamwell, L.F.; Al-Hujaily, E.M.; Collins, O.; Macdonald, E.A.; Garson, K.; Daneshmand, M.; Carmona, E.; et al. A new spontaneously transformed syngeneic model of high-grade serous ovarian cancer with a tumor-initiating cell population. Front. Oncol. 2014, 4, 53. [Google Scholar] [CrossRef]
- Barba, A.A.; Cascone, S.; Caccavo, D.; Lamberti, G.; Chiarappa, G.; Abrami, M.; Grassi, G.; Grassi, M.; Tomaiuolo, G.; Guido, S.; et al. Engineering approaches in siRNA delivery. Int. J. Pharm. 2017, 525, 343–358. [Google Scholar] [CrossRef]
- Chiarappa, G.; Abrami, M.; Dapas, B.; Farra, R.; Trebez, F.; Musiani, F.; Musiani, F.; Grassi, G.; Grassi, M. Mathematical Modeling of Drug Release from Natural Polysaccharides Based Matrices. Nat. Prod. Commun. 2017, 12, 873–880. [Google Scholar] [CrossRef]
- Bartlett, D.W.; Davis, M.E. Insights into the kinetics of siRNA-mediated gene silencing from live-cell and live-animal bioluminescent imaging. Nucleic Acids Res. 2006, 34, 322–333. [Google Scholar] [CrossRef]
- Polyak, D.; Krivitsky, A.; Scomparin, A.; Eliyahu, S.; Kalinski, H.; Avkin-Nachum, S.; Satchi-Fainaro, R. Systemic delivery of siRNA by aminated poly(α)glutamate for the treatment of solid tumors. J. Control. Release 2017, 257, 132–143. [Google Scholar] [CrossRef] [PubMed]
- Risnayanti, C.; Jang, Y.S.; Lee, J.; Ahn, H.J. PLGA nanoparticles co-delivering MDR1 and BCL2 siRNA for overcoming resistance of paclitaxel and cisplatin in recurrent or advanced ovarian cancer. Sci. Rep. 2018, 8, 7498. [Google Scholar] [CrossRef] [PubMed]
- Hazekawa, M.; Nishinakagawa, T.; Kawakubo-Yasukochi, T.; Nakashima, M. Glypican-3 gene silencing for ovarian cancer using siRNA-PLGA hybrid micelles in a murine peritoneal dissemination model. J. Pharmacol. Sci. 2019, 139, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Stadlmann, S.; Gueth, U.; Baumhoer, D.; Moch, H.; Terracciano, L.; Singer, G. Glypican-3 expression in primary and recurrent ovarian carcinomas. Int. J. Gynecol. Pathol. 2007, 26, 341–344. [Google Scholar] [CrossRef] [PubMed]
- Lou, B.; Beztsinna, N.; Mountrichas, G.; van den Dikkenberg, J.B.; Pispas, S.; Hennink, W.E. Small nanosized poly(vinyl benzyl trimethylammonium chloride) based polyplexes for siRNA delivery. Int. J. Pharm. 2017, 525, 388–396. [Google Scholar] [CrossRef] [PubMed]
- Leung, C.S.; Yeung, T.L.; Yip, K.P.; Wong, K.K.; Ho, S.Y.; Mangala, L.S.; Sood, A.K.; Lopez-Berestein, G.; Sheng, J.; Wong, S.T.; et al. Cancer-associated fibroblasts regulate endothelial adhesion protein LPP to promote ovarian cancer chemoresistance. J. Clin. Investig. 2018, 128, 589–606. [Google Scholar] [CrossRef]
- Kim, G.H.; Won, J.E.; Byeon, Y.; Kim, M.G.; Wi, T.I.; Lee, J.M.; Park, Y.Y.; Lee, J.W.; Kang, T.H.; Jung, I.D.; et al. Selective delivery of PLXDC1 small interfering RNA to endothelial cells for anti-angiogenesis tumor therapy using CD44-targeted chitosan nanoparticles for epithelial ovarian cancer. Drug Deliv. 2018, 25, 1394–1402. [Google Scholar] [CrossRef]
- Byeon, Y.; Lee, J.W.; Choi, W.S.; Won, J.E.; Kim, G.H.; Kim, M.G.; Wi, T.I.; Lee, J.M.; Kang, T.H.; Jung, I.D.; et al. CD44-Targeting PLGA Nanoparticles Incorporating Paclitaxel and FAK siRNA Overcome Chemoresistance in Epithelial Ovarian Cancer. Cancer Res. 2018, 78, 6247–6256. [Google Scholar]
- Hong, S.S.; Zhang, M.X.; Zhang, M.; Yu, Y.; Chen, J.; Zhang, X.Y.; Xu, C.J. Follicle-stimulating hormone peptide-conjugated nanoparticles for targeted shRNA delivery lead to effective gro-alpha silencing and antitumor activity against ovarian cancer. Drug Deliv. 2018, 25, 576–584. [Google Scholar] [CrossRef]
- Zhang, X.Y.; Chen, J.; Zheng, Y.F.; Gao, X.L.; Kang, Y.; Liu, J.C.; Cheng, M.J.; Sun, H.; Xu, C.J. Follicle-stimulating hormone peptide can facilitate paclitaxel nanoparticles to target ovarian carcinoma in vivo. Cancer Res. 2009, 69, 6506–6514. [Google Scholar] [CrossRef]
- Yang, G.; Rosen, D.G.; Zhang, Z.; Bast, R.C., Jr.; Mills, G.B.; Colacino, J.A.; Mercado-Uribe, I.; Liu, J. The chemokine growth-regulated oncogene 1 (Gro-1) links RAS signaling to the senescence of stromal fibroblasts and ovarian tumorigenesis. Proc. Natl. Acad. Sci. USA 2006, 103, 16472–16477. [Google Scholar] [CrossRef] [Green Version]
- Jones, S.K.; Douglas, K.; Shields, A.F.; Merkel, O.M. Correlating quantitative tumor accumulation and gene knockdown using SPECT/CT and bioluminescence imaging within an orthotopic ovarian cancer model. Biomaterials 2018, 178, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Ahn, H.J. PEGylated DC-Chol/DOPE cationic liposomes containing KSP siRNA as a systemic siRNA delivery Carrier for ovarian cancer therapy. Biochem. Biophys. Res. Commun. 2018, 503, 1716–1722. [Google Scholar] [CrossRef] [PubMed]
- She, Z.Y.; Yang, W.X. Molecular mechanisms of kinesin-14 motors in spindle assembly and chromosome segregation. J. Cell Sci. 2017, 130, 2097–2110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujiwaki, R.; Hata, K.; Nakayama, K.; Fukumoto, M.; Miyazaki, K. Thymidylate synthase expression in epithelial ovarian cancer: Relationship with thymidine phosphorylase expression and prognosis. Oncology 2000, 59, 152–157. [Google Scholar] [CrossRef] [PubMed]
- Iizuka, K.; Jin, C.; Eshima, K.; Hong, M.H.; Eshima, K.; Fukushima, M. Anticancer activity of the intraperitoneal-delivered DFP-10825, the cationic liposome-conjugated RNAi molecule targeting thymidylate synthase, on peritoneal disseminated ovarian cancer xenograft model. Drug Des. Dev. Ther. 2018, 12, 673–683. [Google Scholar] [CrossRef] [PubMed]
- Mendes, L.P.; Sarisozen, C.; Luther, E.; Pan, J.; Torchilin, V.P. Surface-engineered polyethyleneimine-modified liposomes as novel carrier of siRNA and chemotherapeutics for combination treatment of drug-resistant cancers. Drug Deliv. 2019, 26, 443–458. [Google Scholar] [CrossRef] [Green Version]
- Blanco, A.; Giger-Pabst, U.; Solass, W.; Zieren, J.; Reymond, M.A. Renal and hepatic toxicities after pressurized intraperitoneal aerosol chemotherapy (PIPAC). Ann. Surg. Oncol. 2013, 20, 2311–2316. [Google Scholar] [CrossRef]
- Minnaert, A.K.; Dakwar, G.R.; Benito, J.M.; Garcia Fernandez, J.M.; Ceelen, W.; De Smedt, S.C.; Remaut, K. High-Pressure Nebulization as Application Route for the Peritoneal Administration of siRNA Complexes. Macromol. Biosci. 2017, 17, 1700024. [Google Scholar] [CrossRef]
- Dykxhoorn, D.M.; Palliser, D.; Lieberman, J. The silent treatment: siRNAs as small molecule drugs. Gene Ther. 2006, 13, 541–552. [Google Scholar] [CrossRef] [Green Version]
- Cejka, D.; Losert, D.; Wacheck, V. Short interfering RNA (siRNA): Tool or therapeutic? Clin. Sci. 2006, 110, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Bochicchio, S.; Dapas, B.; Russo, I.; Ciacci, C.; Piazza, O.; De Smedt, S.; Pottie, E.; Barba, A.A.; Grassi, G. In vitro and ex vivo delivery of tailored siRNA-nanoliposomes for E2F1 silencing as a potential therapy for colorectal cancer. Int. J. Pharm. 2017, 2, 377–387. [Google Scholar] [CrossRef] [PubMed]
- TKM 080301 for Primary or Secondary Liver Cancer. Available online: https://clinicaltrials.gov/ct2/show/record/NCT01437007?term= siRNA&cond= Ovarian+Cancer&rank=1 (accessed on 3 August 2018).
- D’Apolito, R.; Tomaiuolo, G.; Taraballi, F.; Minardi, S.; Kirui, D.; Liu, X.; Cevenini, A.; Palomba, R.; Ferrari, M.; Salvatore, F.; et al. Red blood cells affect the margination of microparticles in synthetic microcapillaries and intravital microcirculation as a function of their size and shape. J. Control 2015, 217, 263–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Extended Name | Abbreviation | References |
|---|---|---|
| Epidermal growth factor receptor | EGFR | [61,62,63,64] |
| Erythropoietin-producing hepatocellular receptor A2 | EphA2 | [65,66] |
| Folic acid receptor | FR | [67,68] |
| CD44 surface transmembrane glycoprotein | CD44 | [69,70,71] |
| CD133 glycosylated transmembrane protein | CD133 | [72] |
| CD117 (also known as c-kit) receptor tyrosine kinase | CD117 | [73,74,75] |
| CD24 | CD24 | [76] |
| Extended Name | Abbreviation | References |
|---|---|---|
| Epidermal growth factor receptor | EGFR | [64] |
| Metastasis associated in colon cancer 1 | MACC1 | [77] |
| Metastasis-associated gene 1 | MTA1 | [78] |
| Wilms tumor gene | WT1 | [79] |
| Rac1 Rho family small GTPases | Rac1 | [80,81] |
| Polo-like kinase 1 | Plk1 | [82,83] |
| Notch1 Claudin3 Nin one binding protein Cyclooxigenase-2 | Notch1 CLDN3 NOB1p COX-2 | [61] |
| E2 promoter binding factor 1 | E2F1 | [84,85,86,87] |
| Peptidylprolyl cis–trans isomerase, NIMA-interacting 1 | PIN1 | [88,89] |
| Extended Name | Abbreviation | References |
|---|---|---|
| Vascular endothelial growth factors | VEGFs | [61] |
| VEGFs tyrosine kinase receptors | VEGFR-1/Ftl-1, VEGFR-2, VEGFR-3/Ftl-4 | [61] |
| Plexin domain containing 1 | PLXDC1 | [90] |
| Extended Name | Abbreviation | References |
|---|---|---|
| Multidrug resistance gene 1 | MDR1 | [91] |
| Survivin | SVV | [92,93] |
| Focal adhesion kinase | FAK | [94] |
| B-cell lymphoma 2 | BCL2 | [95] |
| First Author | Target | Delivery Material | Cell Model | siRNA Delivery Route | Animal Model | References |
|---|---|---|---|---|---|---|
| Polyak | Rac1; Plk1 | Poly(α)glutamate; no specific targeting; 160 ± 20 nm (Particle size) | SKOV3 | Intra-peritoneal (nine every other day intraperitoneal injections 8 mg/kg siRNA) | Orthotopic SKOV3 cells in athymic nude female mice (intra-peritoneal tumor cell injection) | [111] |
| Risnayanti | MDR1; BCL2 | PLGA–PLL; 197.8 ± 5.2 nm (Particle size) | SKOV3-TR and A2780-CP20 | - | - | [112] |
| Hazekawa | Gpc3 | PLGA–PEI; 108.5 ± 2.5 nm (Particle size) | HM-1 | Intra-peritoneal (100 pmol of siRNA on Day 1) | Syngeneic orthotopic intra-peritoneal HM-1 injection in syngeneic B6C3F1 mouse strain | [113] |
| Lou | Luciferase | POEGMA/PVTC; PVTC; 8–25 nm (particle sizes) | SKOV-3-luciferase | - | - | [115] |
| Leung | LPP | CHITOSAN | Luciferase-labeled OVCA432 | Intravenous (twice-weekly tail-vein 5 μg siRNA in combination with weekly i.p. injections of Paclitaxel, 3.5 mg/kg for 6 weeks) | Orthotopic luciferase-labeled OVCA432 in female nude mice (intra-peritoneal tumor cell injection) | [116] |
| First Author | Target | Delivery Material | Cell Model | siRNA Delivery Route | Animal Model | References |
|---|---|---|---|---|---|---|
| Kim | PLXDC1 | HA–CHITOSAN (targeting to CD44) 200 ± 10 nm (particle size) | HUVEC; MOEC; A2780; HeyA8 | Intravenous (150 mg/kg twice per week) | Orthotopic A2780, HeyA8- Female BALB/c nude mice (intra-peritoneal tumor cell injection) | [117] |
| Byeon | FAK, surviving | HA–PLGA (targeting to CD44) 200–220 nm (particle size) | HeyA8; SKOV3; HeyA8-MDR; SKOV3-TR | Intravenous (200 mg/kg siFAK and 1.4 mg/kg PTX) | Orthotopic HeyA8-MDR, SKOV3-TR, patient-derived cells in female BALB/c nude mice (intra-peritoneal tumor cell injection) | [118] |
| Hong | Gro-α | PEG–PEI–FSH (targeting FSHR) 142.0 ± 3.8 nm | Hey | Intravenous (5 mg/kg) | Subcutaneous xenograft Hey BALB/c nude mice | [119] |
| Jones | Luciferase | hyPEI-g-PCL-b-PEG with and without FA 150 nm (particle size) | SKOV3 | Intravenous (35 μg siRNA) Intra-peritoneal (35 μg siRNA) | Orthotopic SKOV3female nude mice (intra-peritoneal tumor cell injection) | [122] |
| First Author | Target | Delivery Material | Cell Model | siRNA Delivery Route | Animal Model | References |
|---|---|---|---|---|---|---|
| Lee | KSP | DC–Chol–DOPE–PEG 90–110 nm (Particle size) | SKOV3 | Intravenous (1 mg/kg every other day for a total of eight injections) | Subcutaneous xenograft mice model Balb/c nude mice | [123] |
| Iizuka | TS | DOPC–DOPE–DC 395 ± 32 nm (Particle size) | - | Intra-peritoneal (0.5–2 mg/kg every third day) | Orthotopic SKOV3-luc cell in male SOD/SCID mice (intra-peritoneal tumor cell injection) | [126] |
| Mendes | MDR1 | PC–Chol–NGPE–PEI 161 ± 9.4 nm (Particle size) | A2780-ADR and SKOV3-TR | Intravenous (total doses in multiple administration: 66 mg/kg and 9.6 mg/kg of PTX and siMDR1, respectively) | Subcutaneous xenograft mice (athymic nude mice) model using A2780-ADR | [127] |
| Minnaert | luciferase | Commercial lipid 193 ± 8 nm (Particle size) Following nebulization | SKOV3-luc | - | - | [129] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Farra, R.; Maruna, M.; Perrone, F.; Grassi, M.; Benedetti, F.; Maddaloni, M.; El Boustani, M.; Parisi, S.; Rizzolio, F.; Forte, G.; et al. Strategies for Delivery of siRNAs to Ovarian Cancer Cells. Pharmaceutics 2019, 11, 547. https://doi.org/10.3390/pharmaceutics11100547
Farra R, Maruna M, Perrone F, Grassi M, Benedetti F, Maddaloni M, El Boustani M, Parisi S, Rizzolio F, Forte G, et al. Strategies for Delivery of siRNAs to Ovarian Cancer Cells. Pharmaceutics. 2019; 11(10):547. https://doi.org/10.3390/pharmaceutics11100547
Chicago/Turabian StyleFarra, Rossella, Matea Maruna, Francesca Perrone, Mario Grassi, Fabio Benedetti, Marianna Maddaloni, Maguie El Boustani, Salvo Parisi, Flavio Rizzolio, Giancarlo Forte, and et al. 2019. "Strategies for Delivery of siRNAs to Ovarian Cancer Cells" Pharmaceutics 11, no. 10: 547. https://doi.org/10.3390/pharmaceutics11100547