Structure and Dynamics of a Site-Specific Labeled Fc Fragment with Altered Effector Functions


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Protein Production
DKTHTCPPCPAPELLGGPSCVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVKFNWYVDGVEVHNAKTKPREEQYNSTYRVVSVLTVLHQDWLNGKEYKCKVSNKALPAPIEKTISKAKGQPREPQVYTLPPSREEMTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPVLDSDGSFFLYSKLTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLSLSPGK
2.2. Sample Preparation for Crystallography
2.3. Crystallography and Structure Analysis
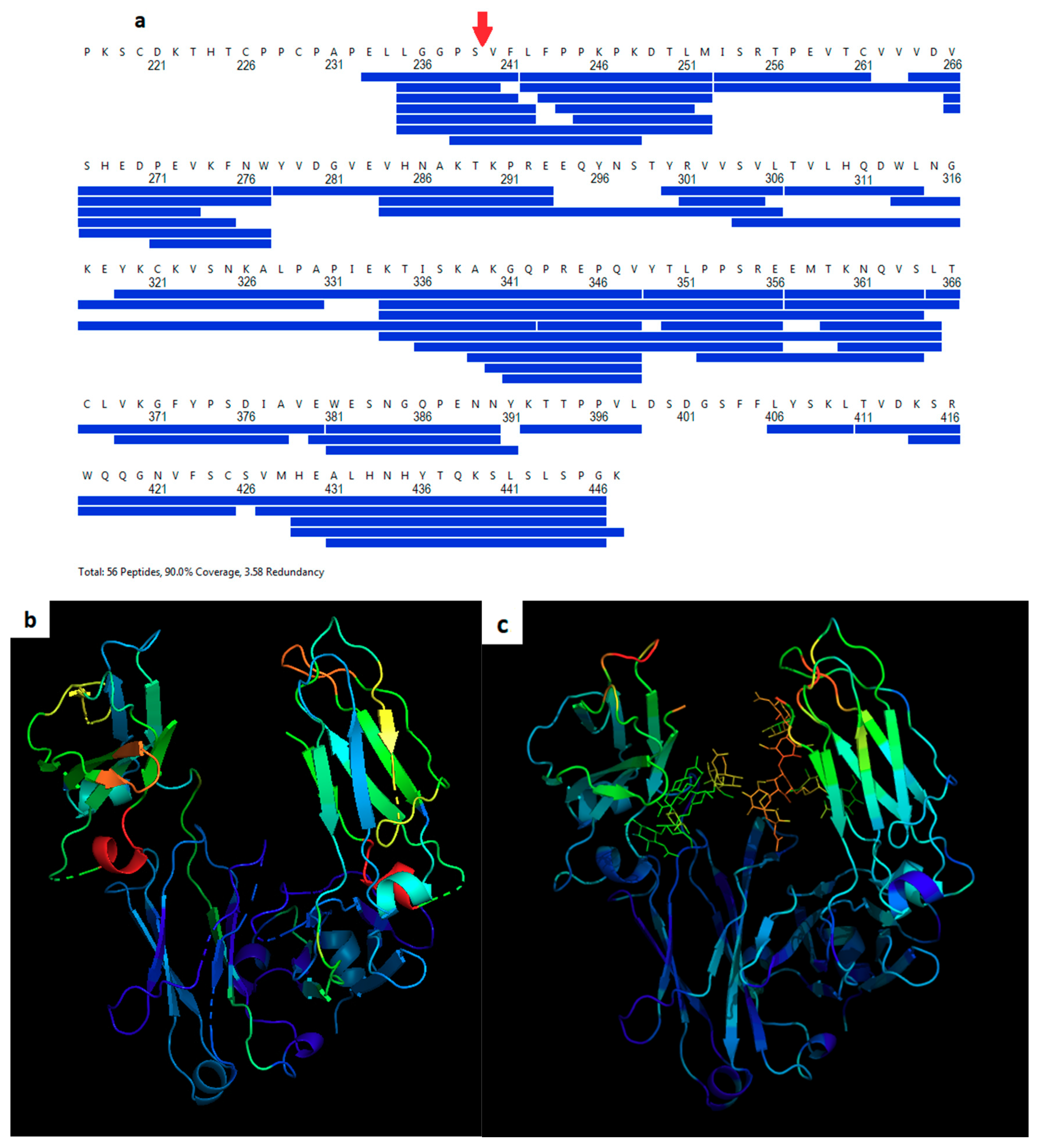
2.4. Hydrogen Deuterium Exchange Mass Spectrometry
3. Results
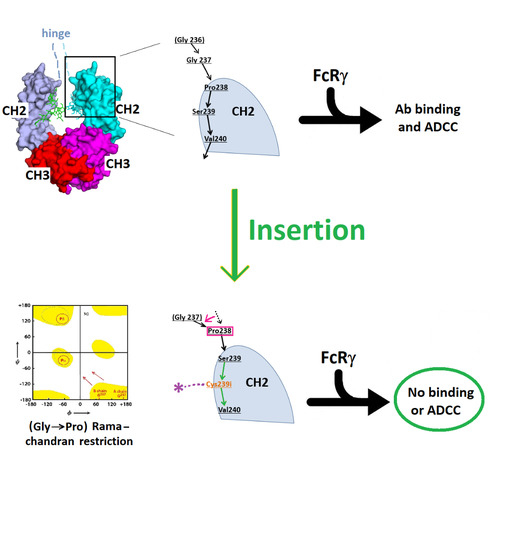
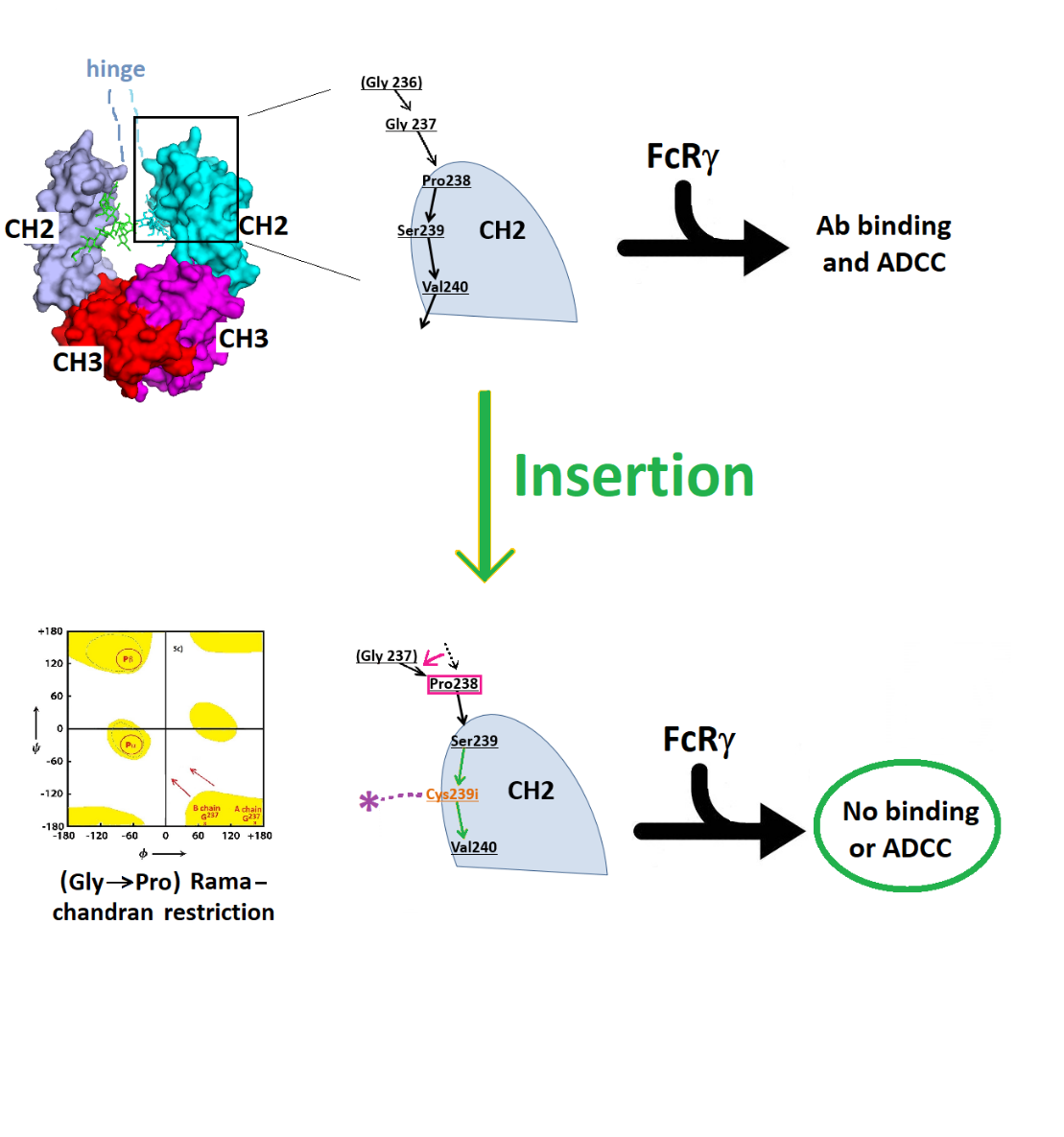
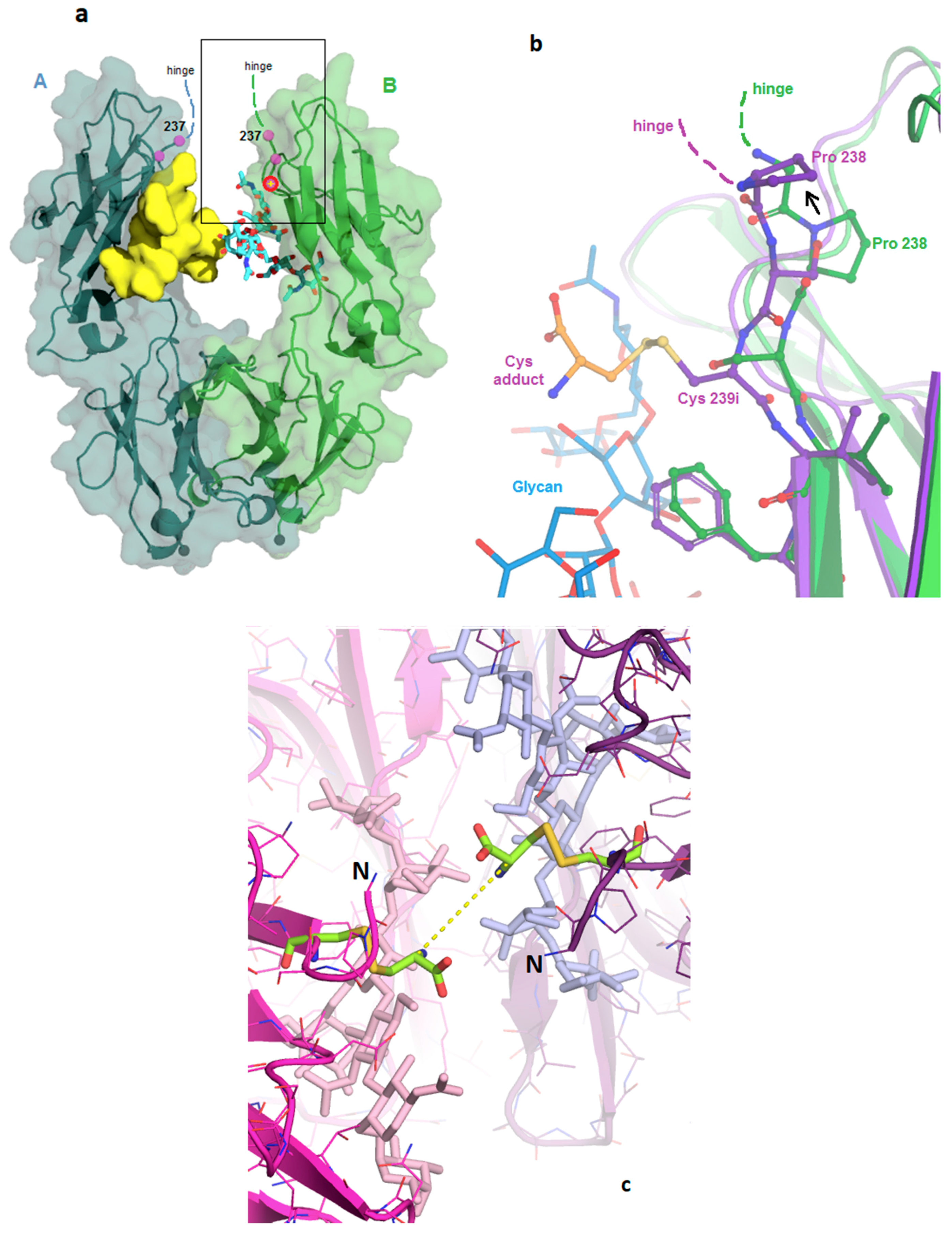
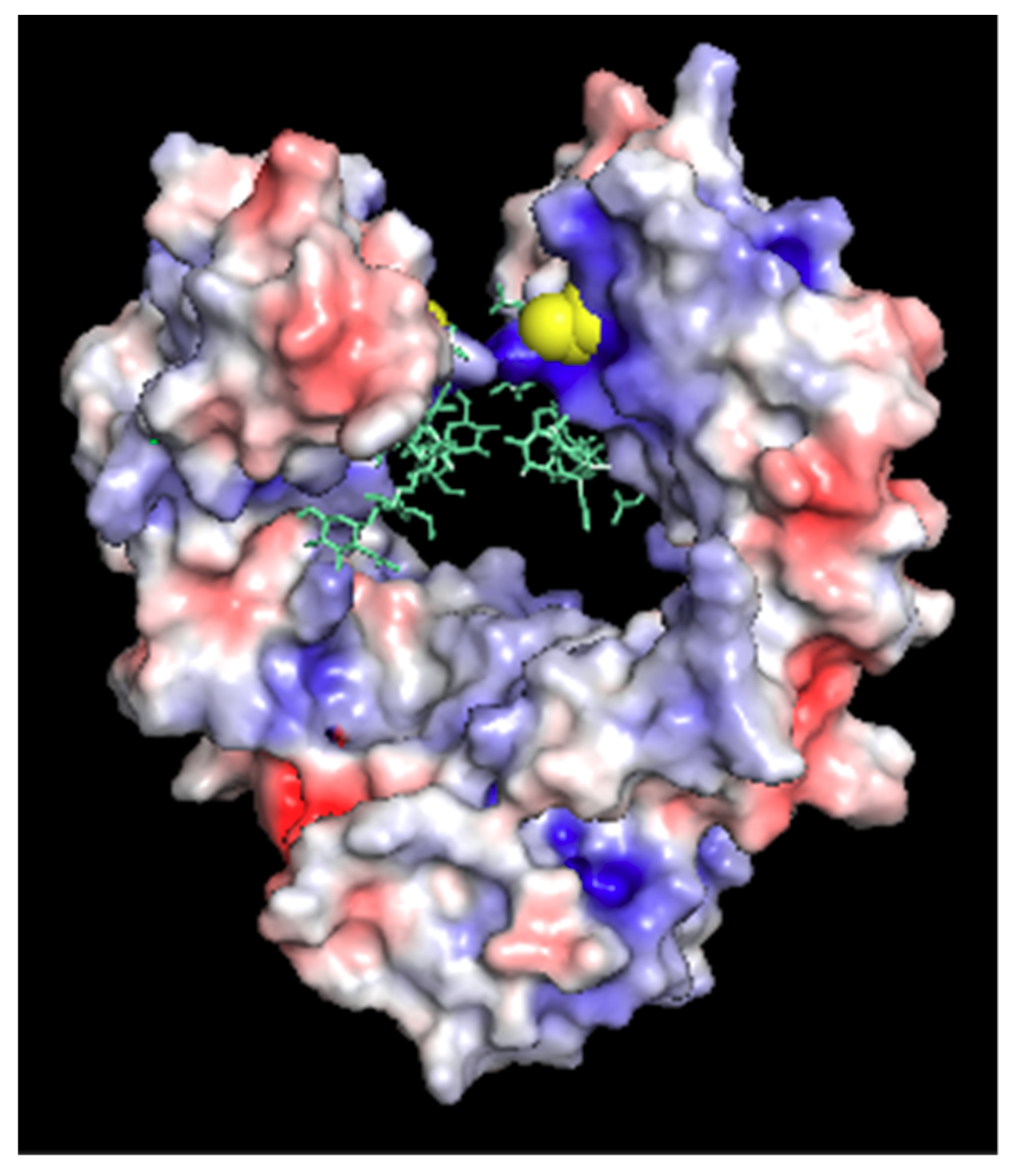
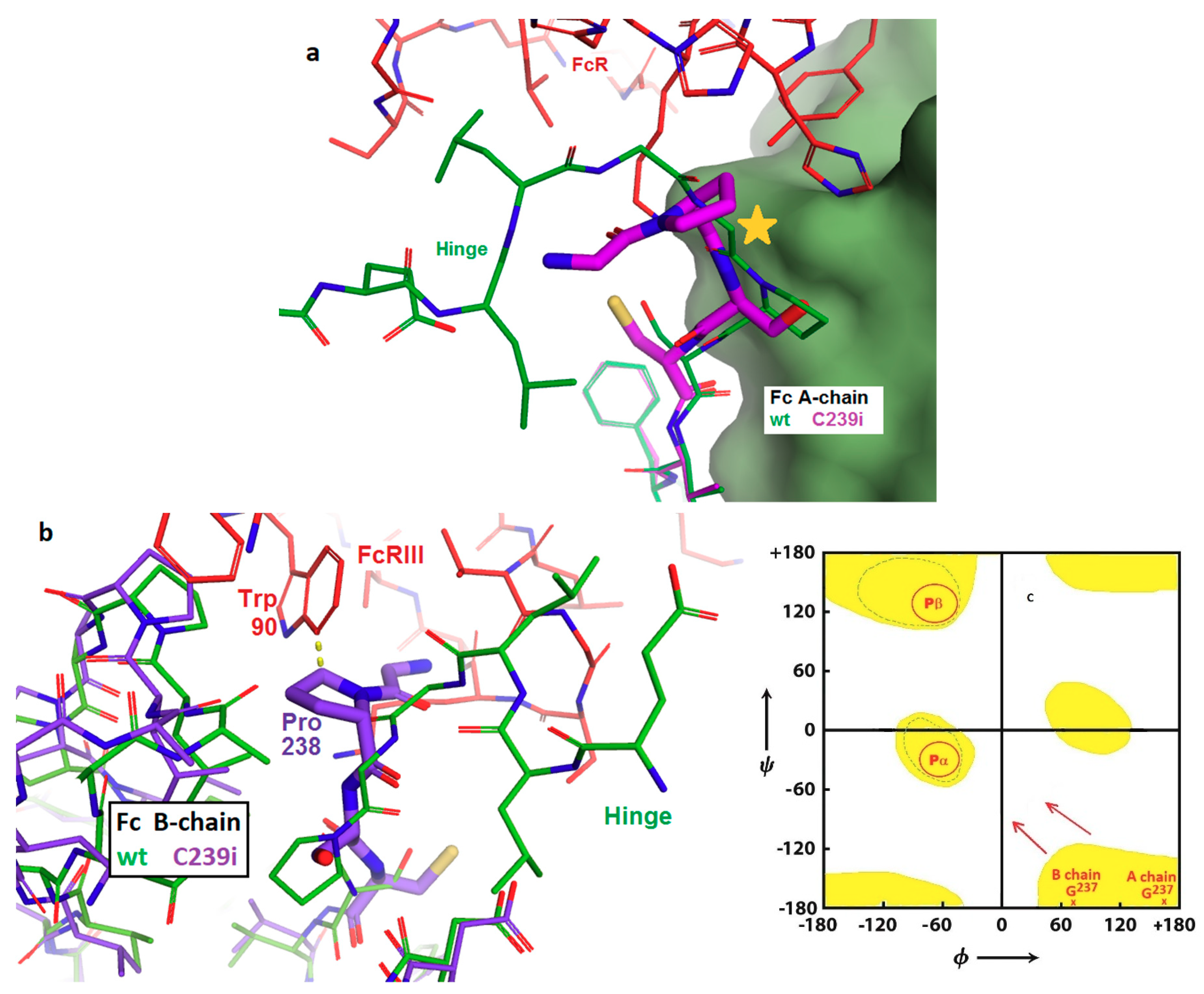
3.1. Structure of Fc-C239i
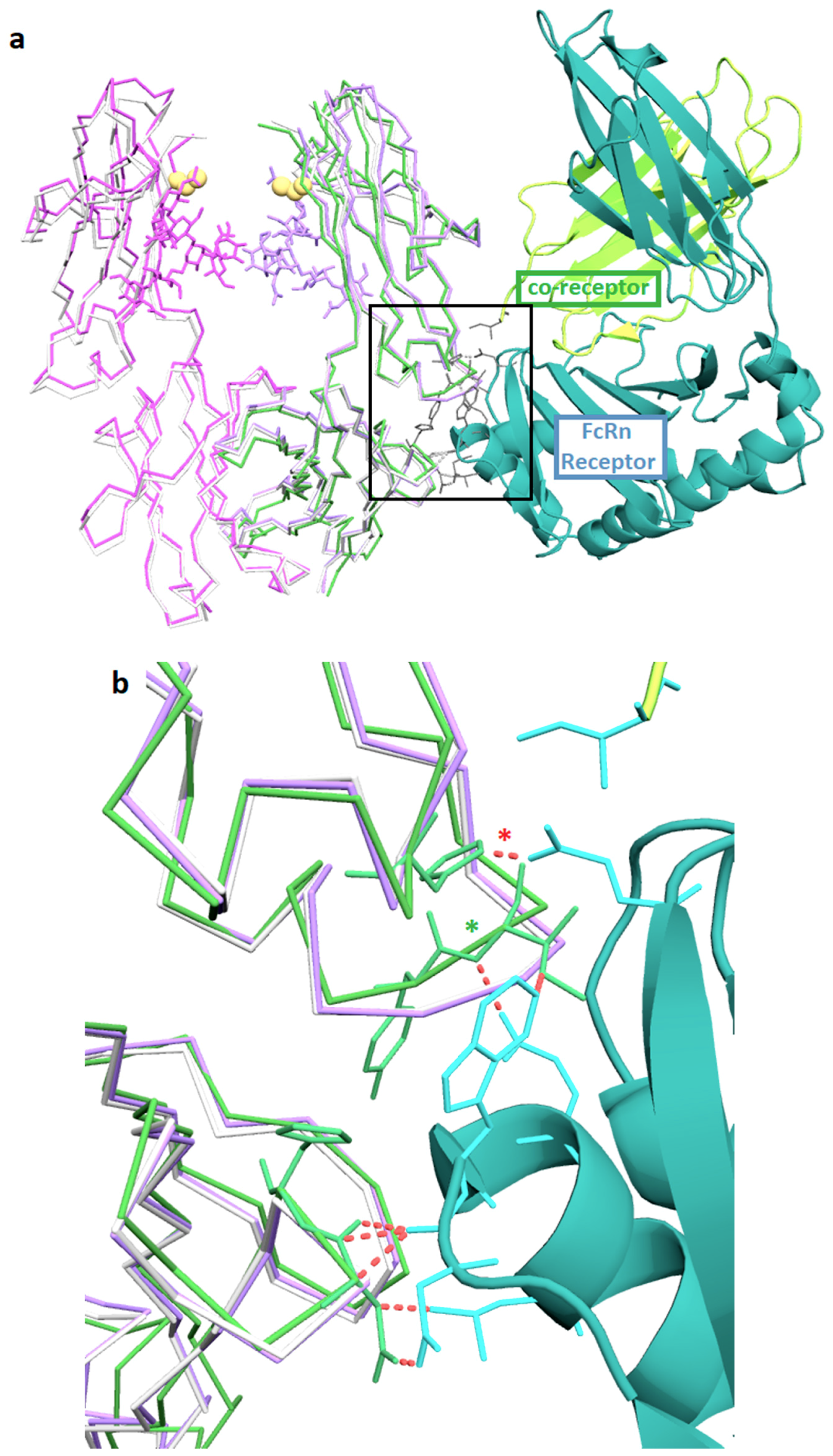
3.2. Structures of the Fc-C239i Adducts
3.3. Fc-C239i Dynamics
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Beck, A.; Goetsch, L.; Dumontet, C.; Corvaia, N. Strategies and challenges for the next generation of antibody-drug conjugates. Nat. Rev. Drug Discov. 2017, 16, 315–337. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, P.; Bertozzi, C.R. Site-specific antibody-drug conjugates: The nexus of bioorthogonal chemistry, protein engineering, and drug development. Bioconjugate Chem. 2015, 26, 176–192. [Google Scholar] [CrossRef] [PubMed]
- Dimasi, N.; Fleming, R.; Zhong, H.; Bezabeh, B.; Kinneer, K.; Christie, R.J.; Fazenbaker, C.; Wu, H.; Gao, C. Efficient preparation of site-specific antibody-drug conjugates using cysteine insertion. Mol. Pharm. 2017, 14, 1501–1516. [Google Scholar] [CrossRef] [PubMed]
- Dimasi, N.; Fleming, R.; Wu, H.; Gao, C. Molecular engineering strategies and methods for the expression and purification of IgG1-based bispecific bivalent antibodies. Methods 2019, 154, 77–86. [Google Scholar] [CrossRef]
- Thompson, P.; Fleming, R.; Bezabeh, B.; Huang, F.; Mao, S.; Chen, C.; Harper, J.; Zhong, H.; Gao, X.; Yu, X.Q.; et al. Rational design, biophysical and biological characterization of site-specific antibody-tubulysin conjugates with improved stability, efficacy and pharmacokinetics. J. Control. Release 2016, 236, 100–116. [Google Scholar] [CrossRef]
- Berman, H.M.; Henrick, K.; Nakamura, H. Announcing the worldwide Protein Data Bank. Nat. Struct. Biol. 2003, 10, 980. [Google Scholar] [CrossRef]
- Winn, M.D.; Ballard, C.C.; Cowtan, K.D.; Dodson, E.J.; Emsley, P.; Evans, P.R.; Keegan, R.M.; Krissinel, E.B.; Leslie, A.G.; McCoy, A.; et al. Overview of the CCP4 suite and current developments. Acta Cryst. Sect. D Biol. Crystallogr. 2011, D67, 235–242. [Google Scholar] [CrossRef]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Cryst. Sect. D Biol. Crystallogr. 2010, D66, 486–501. [Google Scholar] [CrossRef]
- DeLano, W.L. The PyMOL Molecular Graphics System; Version 2.0 Schrödinger; LLC: New York, NY, USA, 2002; Available online: http://www.pymol.org (accessed on 1 February 2019).
- Wales, T.E.; Fadgen, K.E.; Gerhardt, G.C.; Engen, J.R. High-speed and high-resolution UPLC separation at zero degrees Celsius. Anal. Chem. 2008, 80, 6815–6820. [Google Scholar] [CrossRef]
- Geromanos, S.J.; Vissers, J.P.; Silva, J.C.; Dorschel, C.A.; Li, G.Z.; Gorenstein, M.V.; Bateman, R.H.; Langridge, J.I. The detection, correlation, and comparison of peptide precursor and product ions from data independent LC-MS with data dependant LC-MS/MS. Proteomics 2009, 9, 1683–1695. [Google Scholar] [CrossRef]
- Silva, J.C.; Denny, R.; Dorschel, C.; Gorenstein, M.V.; Li, G.Z.; Richardson, K.; Wall, D.; Geromanos, S.J. Simultaneous qualitative and quantitative analysis of the Escherichia coli proteome: A sweet tale. Mol. Cell. Proteom. 2006, 5, 589–607. [Google Scholar] [CrossRef] [PubMed]
- Li, G.Z.; Vissers, J.P.; Silva, J.C.; Golick, D.; Gorenstein, M.V.; Geromanos, S.J. Database searching and accounting of multiplexed precursor and product ion spectra from the data independent analysis of simple and complex peptide mixtures. Proteomics 2009, 9, 1696–1719. [Google Scholar] [CrossRef] [PubMed]
- Iacob, R.E.; Bou-Assaf, G.M.; Makowski, L.; Engen, J.R.; Berkowitz, S.A.; Houde, D. Investigating monoclonal antibody aggregation using a combination of H/DX-MS and other biophysical measurements. J. Pharm. Sci. 2013, 102, 4315–4329. [Google Scholar] [CrossRef] [PubMed]
- Iacob, R.E.; Chen, G.; Ahn, J.; Houel, S.; Wei, H.; Mo, J.; Tao, L.; Cohen, D.; Xie, D.; Lin, Z.; et al. The influence of adnectin binding on the extracellular domain of epidermal growth factor receptor. J. Am. Soc. Mass Spectrom. 2014, 25, 2093–2102. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.; Jung, M.C.; Wyndham, K.; Yu, Y.Q.; Engen, J.R. Pepsin immobilized on high-strength hybrid particles for continuous flow online digestion at 10,000 psi. Anal. Chem. 2012, 84, 7256–7262. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, D.T.; Galvin, C.V.; Karageorgos, I. Structure of the Fc fragment of the NIST reference antibody RM 8671. Acta Cryst. Sect. F Struct. Boil. Commun. 2018, F74, 524–529. [Google Scholar] [CrossRef]
- Kinneer, K.; Flynn, M.; Thomas, S.B.; Meekin, J.; Varkey, R.; Xiao, X.; Zhong, H.; Breen, S.; Hynes, P.G.; Fleming, R.; et al. Preclinical assessment of an antibody-PBD conjugate that targets BCMA on multiple myeloma and myeloma progenitor cells. Leukemia 2019, 33, 766–771. [Google Scholar] [CrossRef]
- NCI Clinical Trials. Available online: https://www.cancer.gov/about-cancer/treatment/clinical-trials/intervention/anti-bcmapbd-adc-medi2228?redirect=true (accessed on 7 February 2019).
- Krissinel, E.; Henrick, K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 2007, 372, 774–797. [Google Scholar] [CrossRef]
- Roopenian, D.C.; Akilesh, S. FcRn: The neonatal Fc receptor comes of age. Nat. Rev. Immunol. 2007, 7, 715–725. [Google Scholar] [CrossRef]
- Oganesyan, V.; Damschroder, M.M.; Cook, K.E.; Li, Q.; Gao, C.; Wu, H.; Dall’acqua, W.F. Structural Insights into Neonatal Fc Receptor-based Recycling Mechanisms. J. Biol. Chem. 2014, 289, 7812–7824. [Google Scholar] [CrossRef] [Green Version]
- Junatala, R.J.; Raab, H.; Clark, S.; Bhakta, S.; Leipold, D.D.; Weir, S.; Chen, Y.; Simpson, M.; Tsai, S.P.; Dennis, M.S.; et al. Site-specific conjugation of a cytotoxic drug to an antibody improves the therapeutic index. Nat. Biotech. 2008, 26, 925–932. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, T.; Yagi, H.; Takemoto, E.; Shibata-Koyama, M.; Isoda, Y.; Iida, S.; Masuda, K.; Satoh, M.; Kato, K. Structural basis for improved efficacy of therapeutic antibodies on defucosylation of their Fc glycans. Genes Cells 2011, 16, 1071–1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, T.; DiLillo, D.J.; Bournazos, S.; Giddens, J.P.; Ravetch, J.V.; Wang, L.-X. Modulating IgG effector function by Fc glycan engineering. Proc. Natl. Acad. Sci. USA 2017, 114, 3485–3490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faller, C.E.; Reilly, K.A.; Hills, R.D., Jr.; Guvench, O. Peptide backbone sampling convergence with the adaptive biasing force algorithm. J. Phys. Chem. B 2013, 117, 518–526. [Google Scholar] [CrossRef]
- Uppal, H.; Doudement, E.; Mahapatra, K.; Darbonne, W.C.; Bumbaca, D.; Shen, B.Q.; Du, X.; Saad, O.; Bowles, K.; Olsen, S.; et al. Potential mechanisms for thrombocytopenia development with trastuzumab emtansine (T-DM1). Clin. Cancer Res. 2015, 21, 123–133. [Google Scholar] [CrossRef]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gallagher, D.T.; McCullough, C.; Brinson, R.G.; Ahn, J.; Marino, J.P.; Dimasi, N. Structure and Dynamics of a Site-Specific Labeled Fc Fragment with Altered Effector Functions. Pharmaceutics 2019, 11, 546. https://doi.org/10.3390/pharmaceutics11100546
Gallagher DT, McCullough C, Brinson RG, Ahn J, Marino JP, Dimasi N. Structure and Dynamics of a Site-Specific Labeled Fc Fragment with Altered Effector Functions. Pharmaceutics. 2019; 11(10):546. https://doi.org/10.3390/pharmaceutics11100546
Chicago/Turabian StyleGallagher, D. Travis, Chris McCullough, Robert G. Brinson, Joomi Ahn, John P. Marino, and Nazzareno Dimasi. 2019. "Structure and Dynamics of a Site-Specific Labeled Fc Fragment with Altered Effector Functions" Pharmaceutics 11, no. 10: 546. https://doi.org/10.3390/pharmaceutics11100546