Utilizing a Kidney-Targeting Peptide to Improve Renal Deposition of a Pro-Angiogenic Protein Biopolymer


Abstract
:
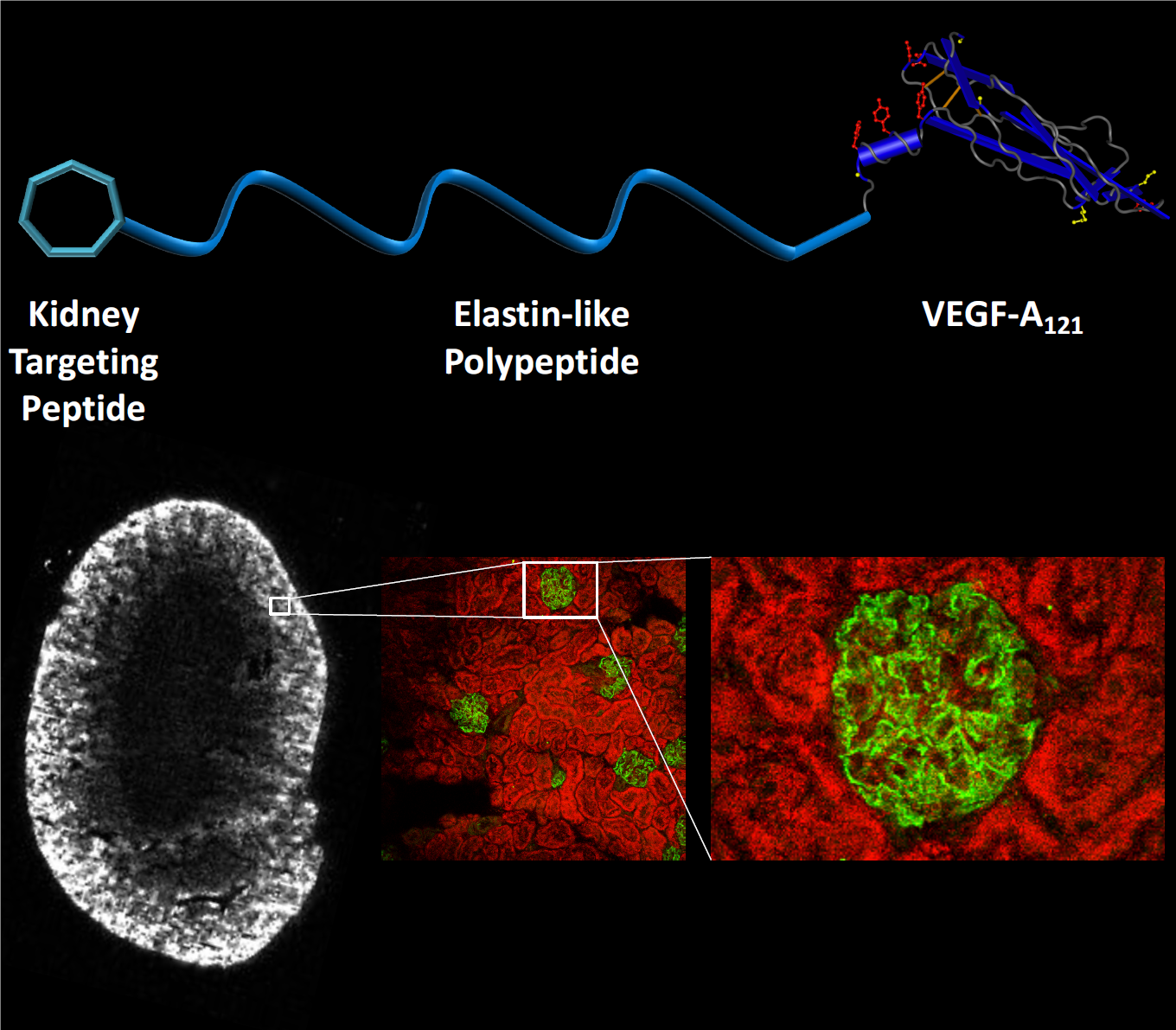{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Protein Expression and Purification
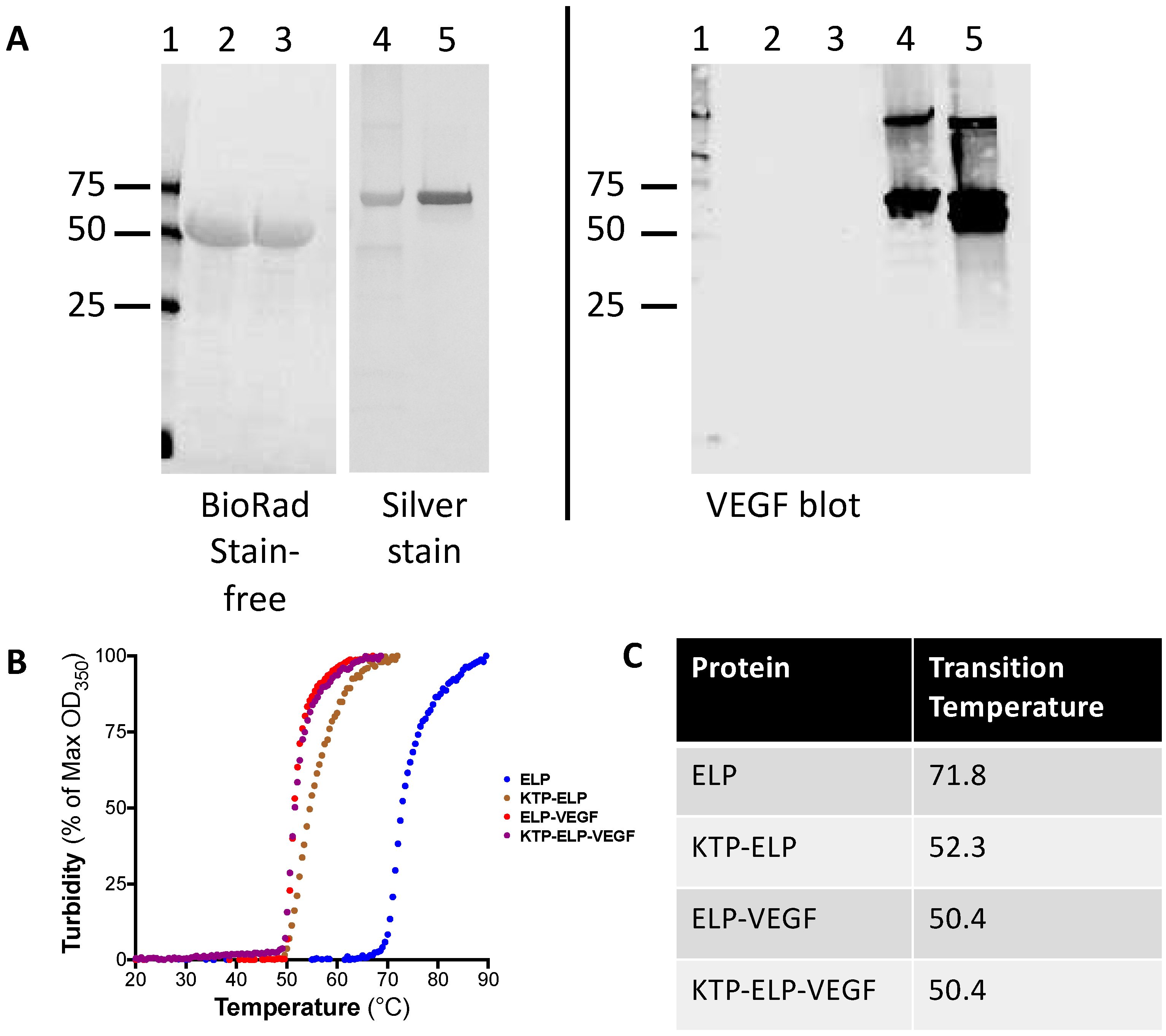
2.2. Determining the Transition Temperature of ELP Fusion Proteins
2.3. Cell Culture
2.4. Labeling Polypeptides with Fluorescent Probes
2.5. Western Blotting and Silver Staining
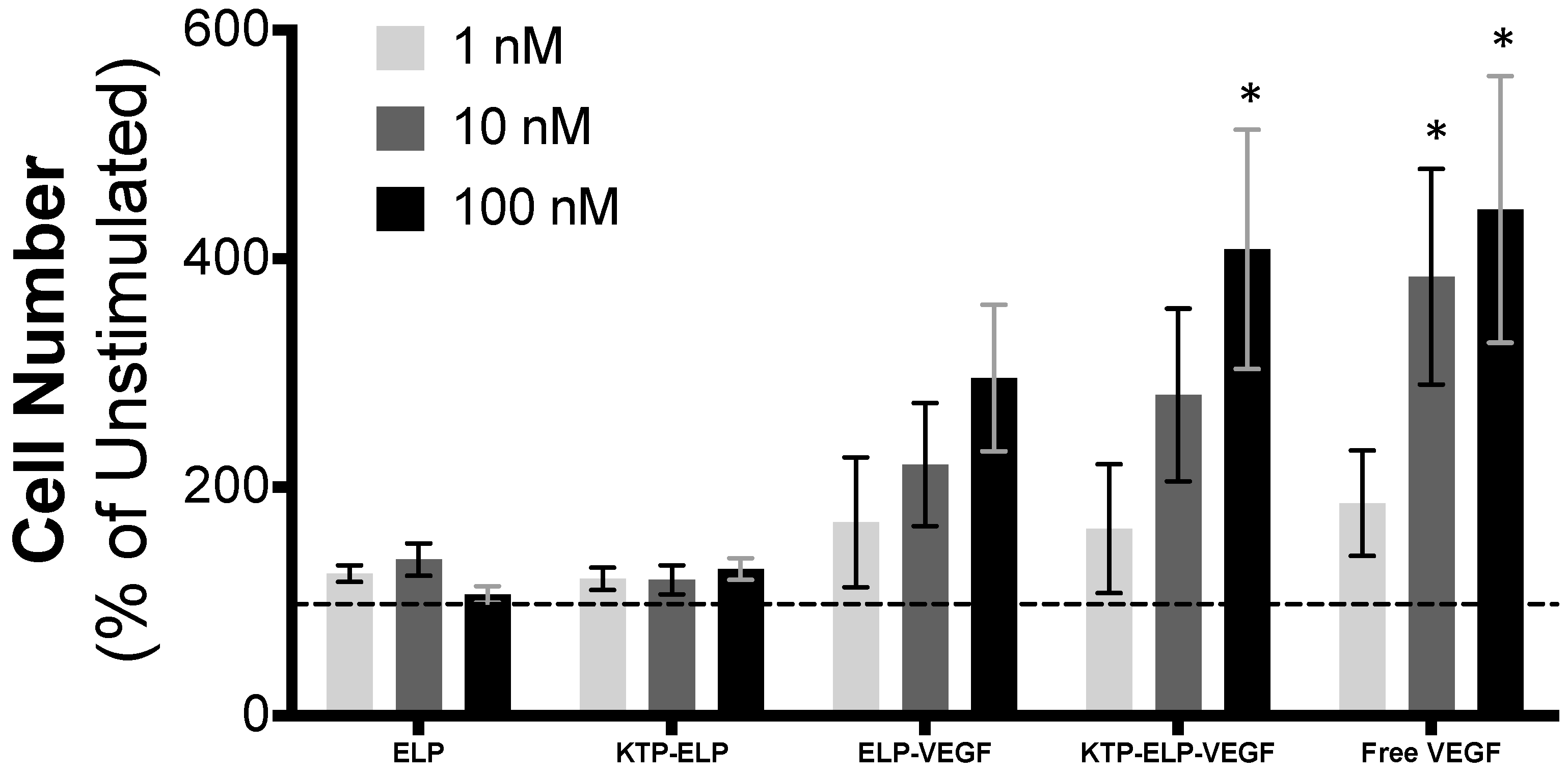
2.6. Proliferation Assay
2.7. Tube Formation Assay
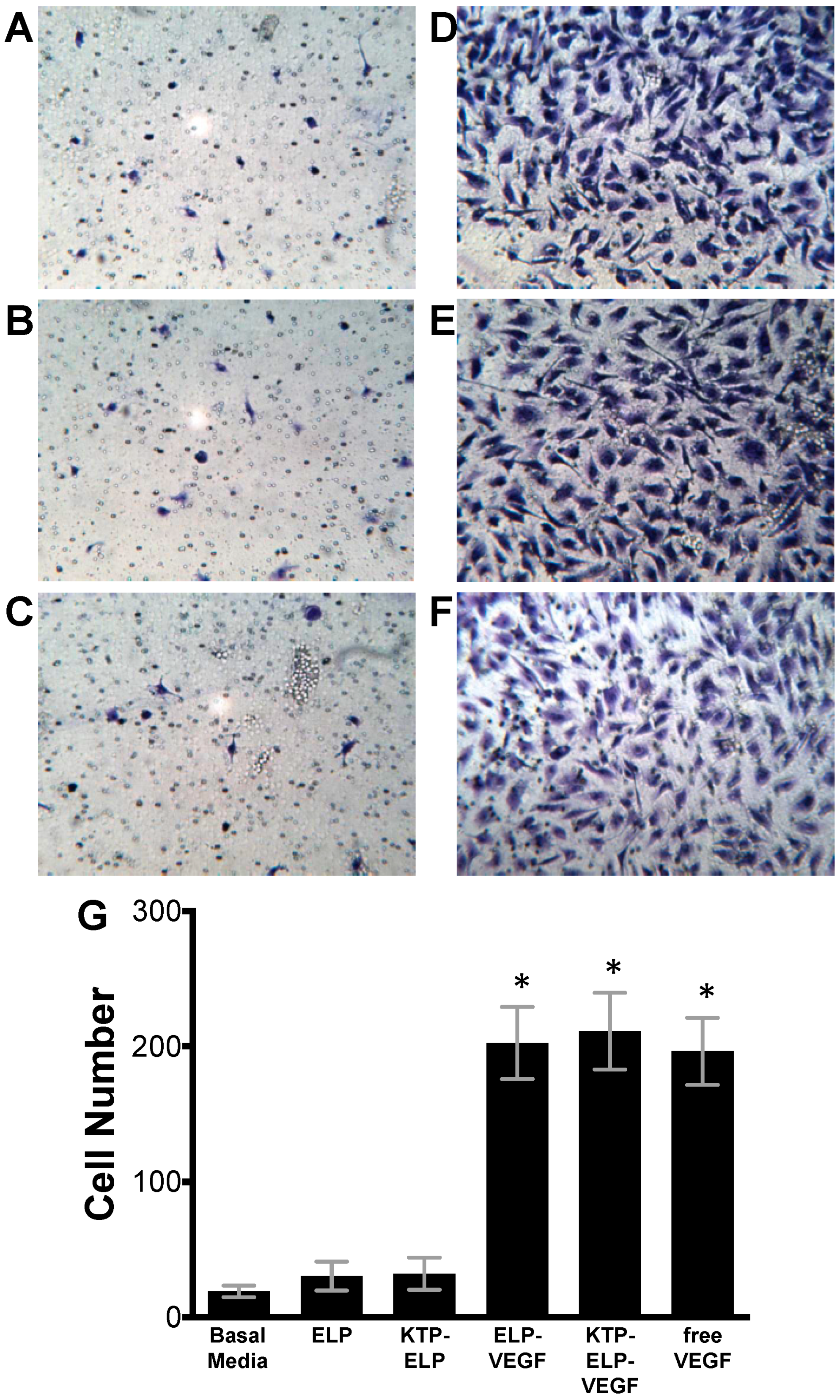
2.8. Migration Assay
2.9. Flow Cytometry
2.10. In Vivo Biodistribution Studies
2.11. Statistical Analysis
3. Results
3.1. Production of ELP Fusion Proteins
3.2. ELP-Fused VEGF Constructs Stimulate Angiogenic-Like Activity in Human Glomerular Microvascular Endothelial Cells
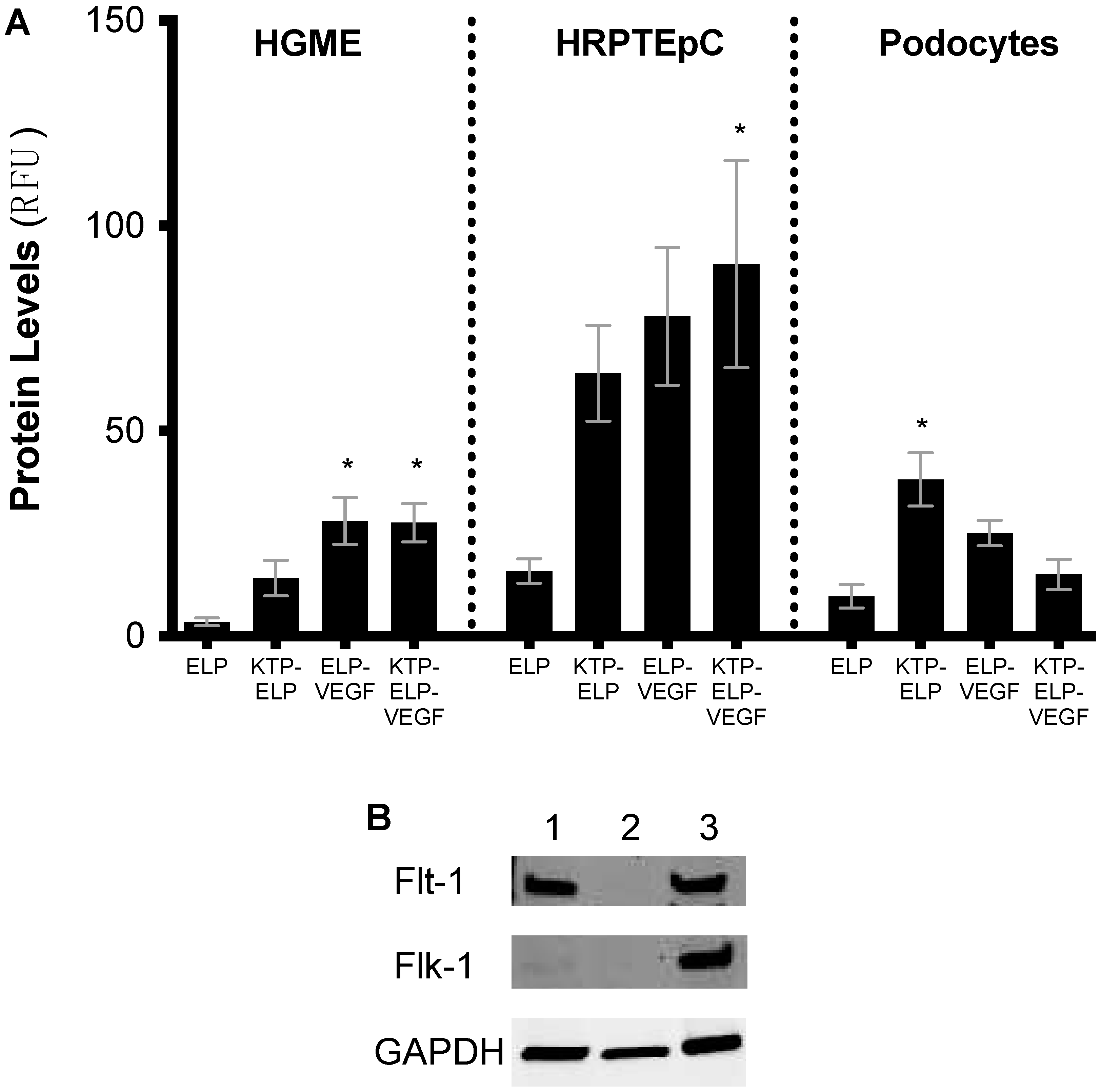
3.3. Cell Binding/Uptake of Polypeptides in Primary Human Renal Cells
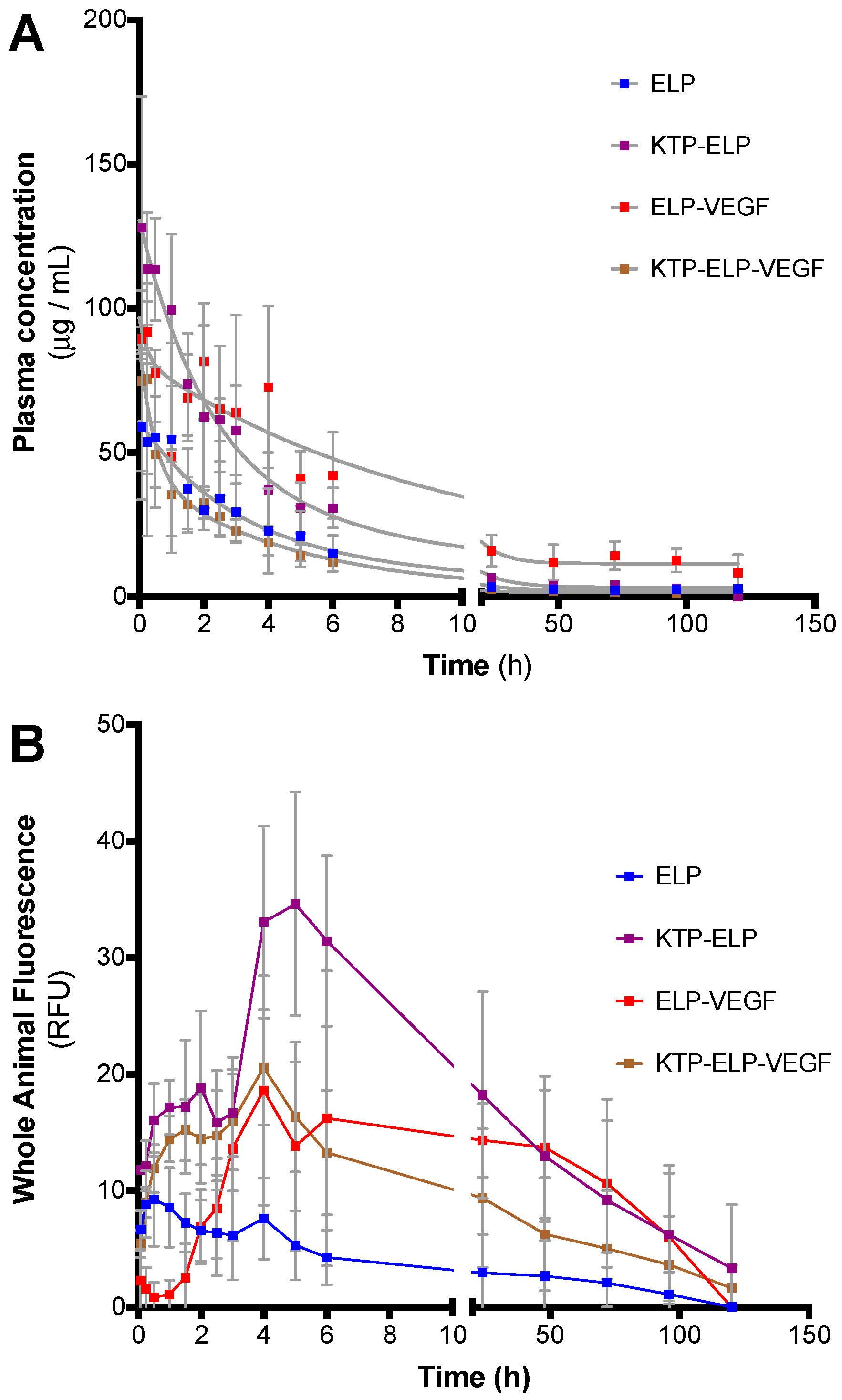
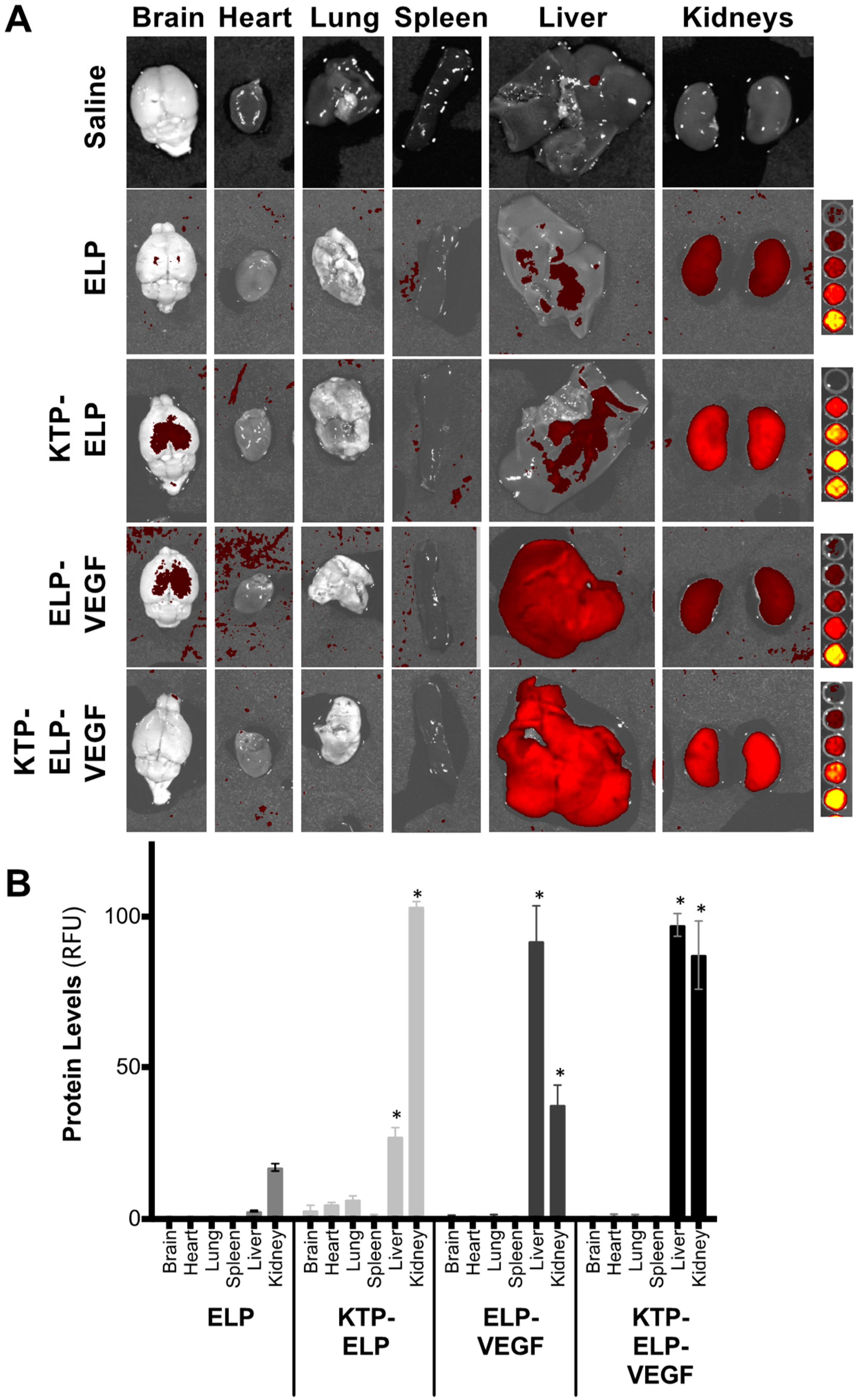
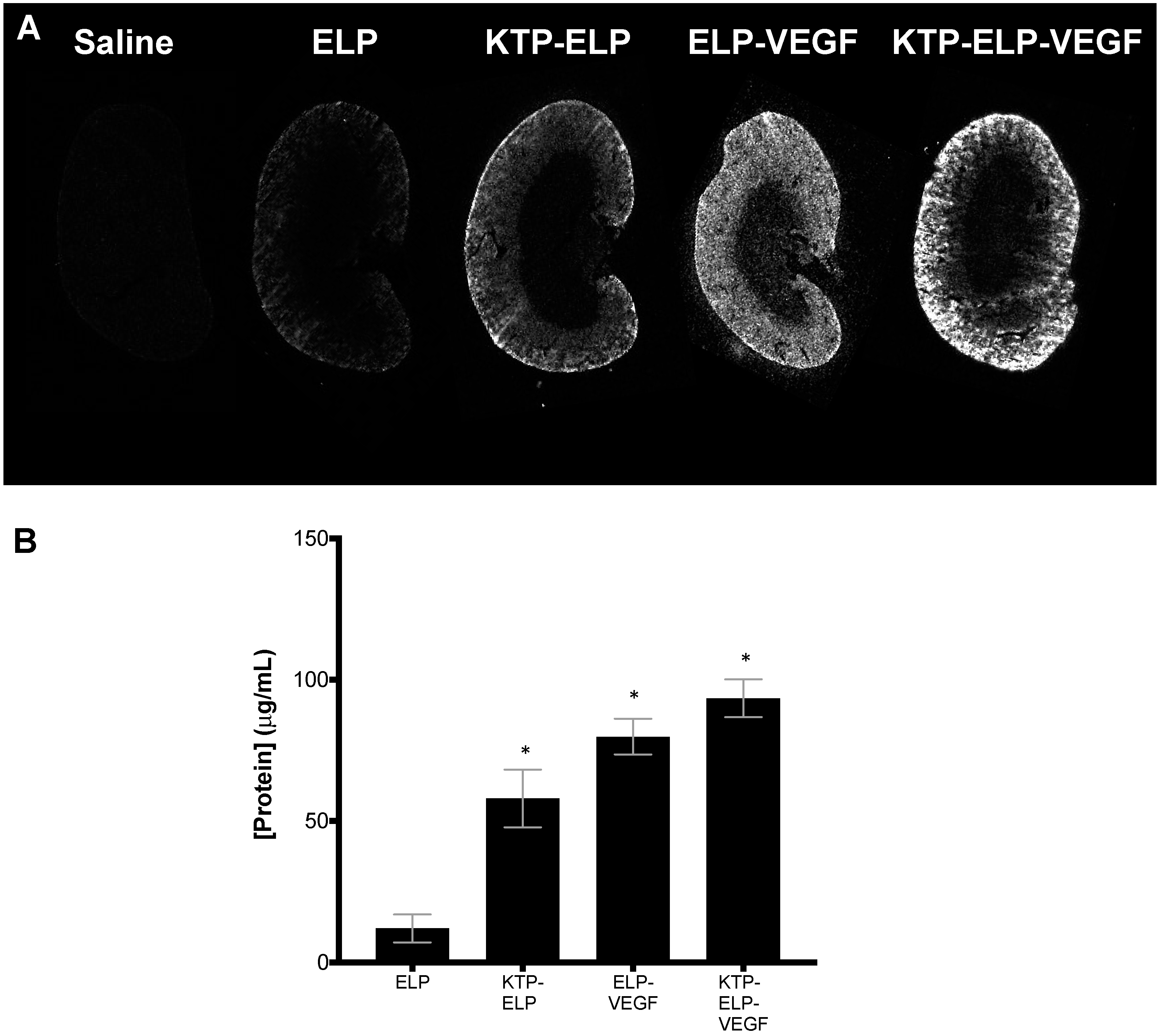
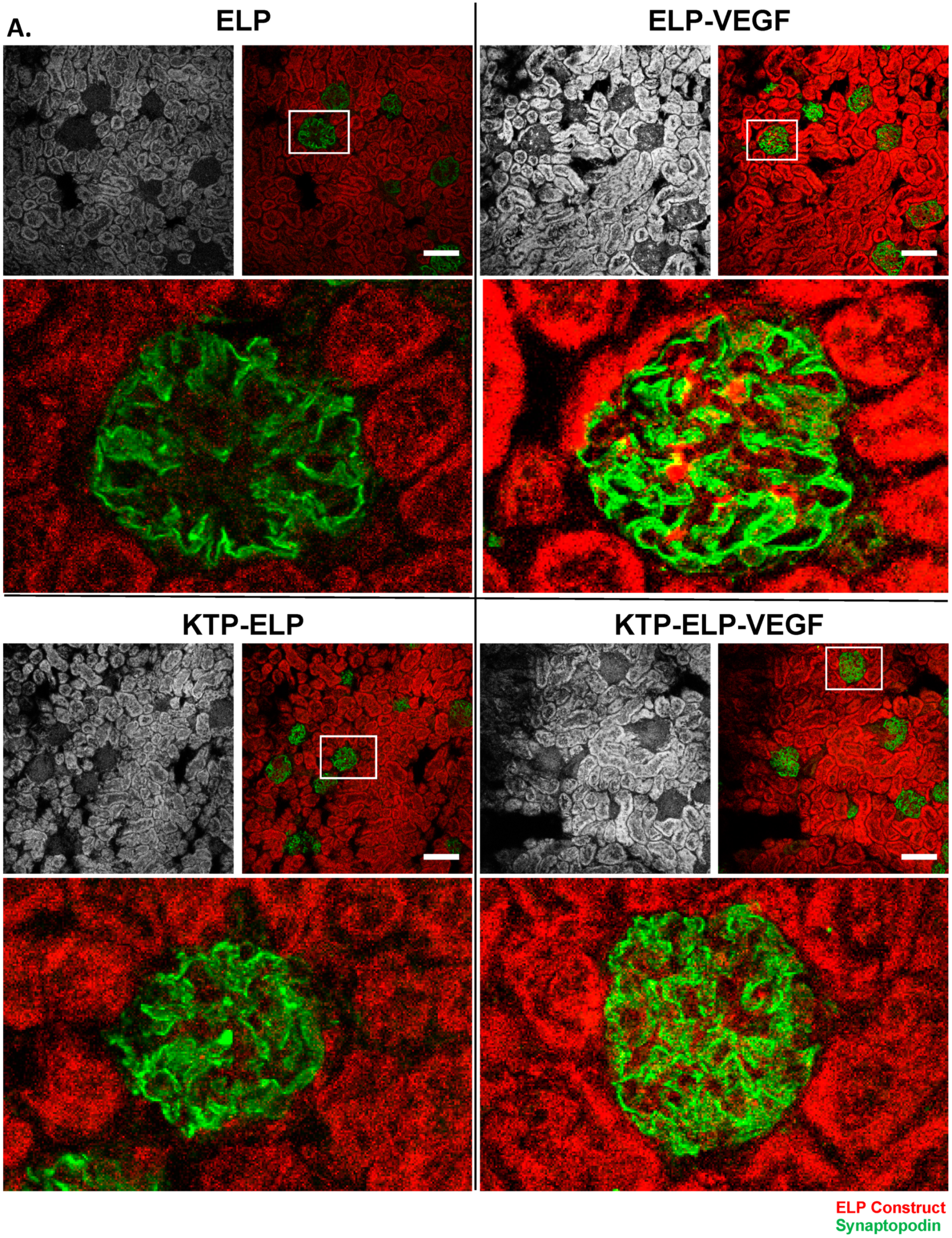
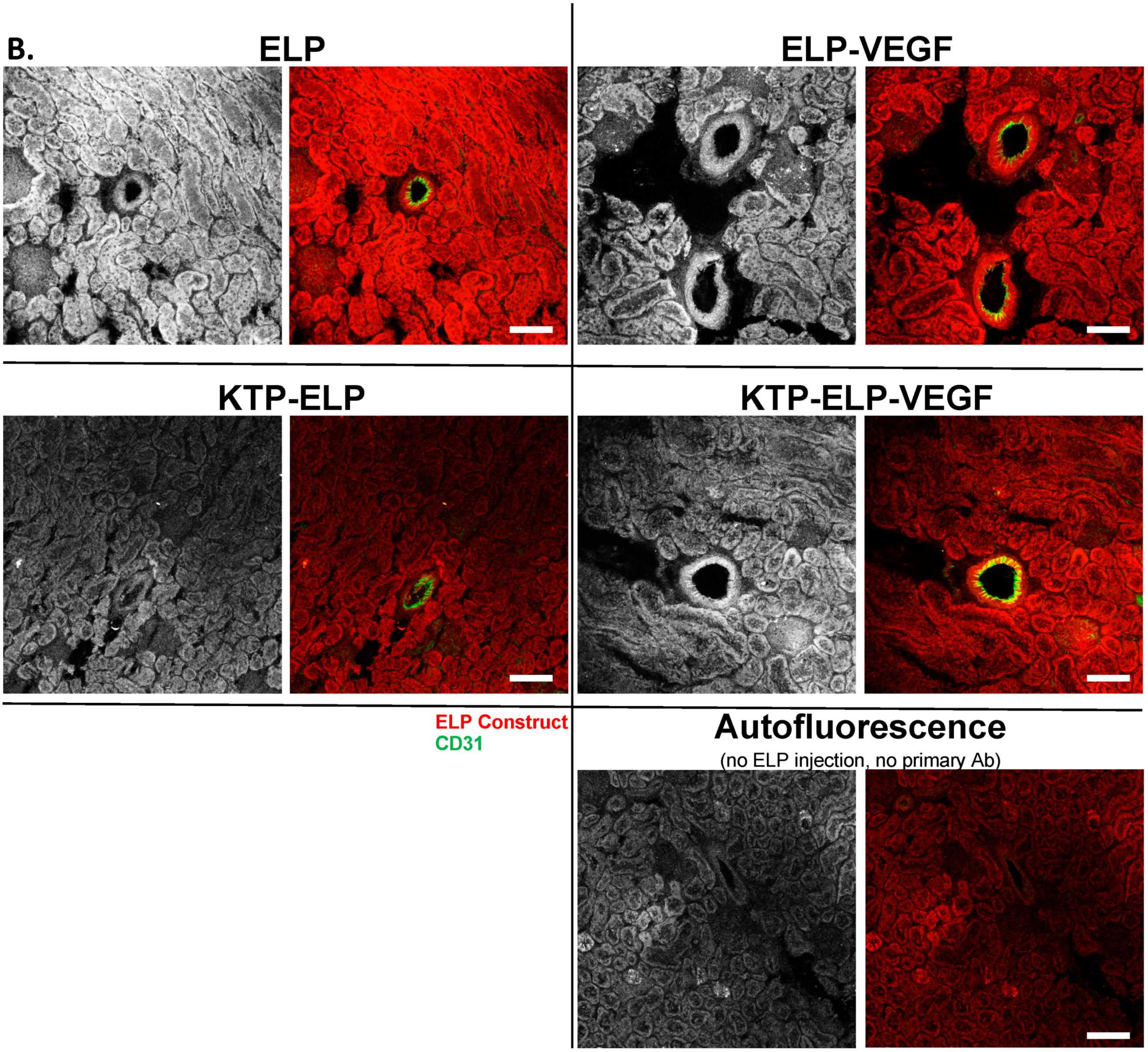
3.4. Pharmacokinetics and Biodistribution of ELP, KTP–ELP, ELP–VEGF, and KTP–ELP–VEGF
4. Discussion
5. Conclusions
6. Patents
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Urry, D.W.; Trapane, T.L.; Prasad, K.U. Phase-structure transitions of the elastin polypentapeptide-water system within the framework of composition-temperature studies. Biopolymers 1985, 24, 2345–2356. [Google Scholar] [CrossRef] [PubMed]
- Despanie, J.; Dhandhukia, J.P.; Hamm-Alvarez, S.F.; MacKay, J.A. Elastin-like polypeptides: Therapeutic applications for an emerging class of nanomedicines. J. Control. Release 2016, 240, 93–108. [Google Scholar] [CrossRef] [PubMed]
- Massodi, I.; Raucher, D. A thermally responsive Tat-elastin-like polypeptide fusion protein induces membrane leakage, apoptosis, and cell death in human breast cancer cells. J. Drug Target. 2007, 15, 611–622. [Google Scholar] [CrossRef] [PubMed]
- Bidwell, G.L.; Mahdi, F.; Shao, Q.; Logue, O.C.; Waller, J.P.; Reese, C.; Chade, A.R. A kidney-selective biopolymer for targeted drug delivery. Am. J. Physiol. Renal Physiol. 2017, 312, F54–F64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massodi, I.; Bidwell, G.L.; Raucher, D. Evaluation of cell penetrating peptides fused to elastin-like polypeptide for drug delivery. J. Control. Release 2005, 108, 396–408. [Google Scholar] [CrossRef]
- Nouri, F.S.; Wang, X.; Chen, X.; Hatefi, A. Reducing the Visibility of the Vector/DNA Nanocomplexes to the Immune System by Elastin-Like Peptides. Pharm. Res. 2015, 32, 3018–3028. [Google Scholar] [CrossRef] [PubMed]
- Changi, K.; Bosnjak, B.; Gonzalez-Obeso, C.; Kluger, R.; Rodríguez-Cabello, J.C.; Hoffmann, O.; Epstein, M.M. Biocompatibility and immunogenicity of elastin-like recombinamer biomaterials in mouse models. J. Biomed. Mater. Res. A 2018, 106, 924–934. [Google Scholar] [CrossRef]
- Shah, M.; Hsueh, P.-Y.; Sun, G.; Chang, H.Y.; Janib, S.M.; MacKay, J.A. Biodegradation of elastin-like polypeptide nanoparticles. Protein Sci. 2012, 21, 743–750. [Google Scholar] [CrossRef] [Green Version]
- Urry, D.W.; Luan, C.-H.; Parker, T.M.; Gowda, D.C.; Prasad, K.U.; Reid, M.C.; Safavy, A. Temperature of Polypeptide Inverse Temperature Transition Depends on Mean Residue Hydrophobicity. J. Am. Chem. Soc. 1991, 113, 4346–4348. [Google Scholar] [CrossRef]
- Amiram, M.; Luginbuhl, K.M.; Li, X.; Feinglos, M.N.; Chilkoti, A. Injectable protease-operated depots of glucagon-like peptide-1 provide extended and tunable glucose control. Proc. Natl. Acad. Sci. USA 2013, 110, 2792–2797. [Google Scholar] [CrossRef] [Green Version]
- Shamji, M.F.; Whitlatch, L.; Friedman, A.H.; Richardson, W.J.; Chilkoti, A.; Setton, L.A. An injectable and in situ-gelling biopolymer for sustained drug release following perineural administration. Spine 2008, 33, 748–754. [Google Scholar] [CrossRef] [PubMed]
- Luginbuhl, K.M.; Schaal, J.L.; Umstead, B.; Mastria, E.M.; Li, X.; Banskota, S.; Arnold, S.; Feinglos, M.; D’Alessio, D.; Chilkoti, A. One-week glucose control via zero-order release kinetics from an injectable depot of glucagon-like peptide-1 fused to a thermosensitive biopolymer. Nat. Biomed. Eng. 2017, 1, 0078. [Google Scholar] [CrossRef] [PubMed]
- Amiram, M.; Luginbuhl, K.M.; Li, X.; Feinglos, M.N.; Chilkoti, A. A depot-forming glucagon-like peptide-1 fusion protein reduces blood glucose for five days with a single injection. J. Control. Release 2013, 172, 144–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Jashnani, A.; Aluri, S.R.; Gustafson, J.A.; Hsueh, P.-Y.; Yarber, F.; McKown, R.L.; Laurie, G.W.; Hamm-Alvarez, S.F.; MacKay, J.A. A thermo-responsive protein treatment for dry eyes. J. Control. Release 2015, 199, 156–167. [Google Scholar] [CrossRef]
- Janib, S.M.; Pastuszka, M.; Aluri, S.; Folchman-Wagner, Z.; Hsueh, P.-Y.; Shi, P.; Lin, Y.A.; Cui, H.; Mackay, J.A. A quantitative recipe for engineering protein polymer nanoparticles. Polym. Chem. 2014, 5, 1614–1625. [Google Scholar] [CrossRef]
- MacKay, J.A.; Chen, M.; McDaniel, J.R.; Liu, W.; Simnick, A.J.; Chilkoti, A. Self-assembling chimeric polypeptide-doxorubicin conjugate nanoparticles that abolish tumours after a single injection. Nat. Mater. 2009, 8, 993–999. [Google Scholar] [CrossRef]
- Gonzalez-Valdivieso, J.; Girotti, A.; Muñoz, R.; Rodriguez-Cabello, J.C.; Arias, F.J. Self-Assembling ELR-Based Nanoparticles as Smart Drug-Delivery Systems Modulating Cellular Growth via Akt. Biomacromolecules 2019, 20, 1996–2007. [Google Scholar] [CrossRef]
- Wang, W.; Despanie, J.; Shi, P.; Edman-Woolcott, M.C.; Lin, Y.-A.; Cui, H.; Heur, J.M.; Fini, M.E.; Hamm-Alvarez, S.F.; MacKay, J.A. Lacritin-mediated regeneration of the corneal epithelia by protein polymer nanoparticles. J. Mater. Chem. B 2014, 2, 8131–8141. [Google Scholar] [CrossRef] [Green Version]
- Zhao, P.; Atanackovic, D.; Dong, S.; Yagita, H.; He, X.; Chen, M. An Anti-Programmed Death-1 Antibody (αPD-1) Fusion Protein That Self-Assembles into a Multivalent and Functional αPD-1 Nanoparticle. Mol. Pharm. 2017, 14, 1494–1500. [Google Scholar] [CrossRef]
- Meyer, D.E.; Kong, G.A.; Dewhirst, M.W.; Zalutsky, M.R.; Chilkoti, A. Targeting a Genetically Engineered Elastin-like Polypeptide to Solid Tumors by Local Hyperthermia. Cancer Res. 2001, 61, 1548–1554. [Google Scholar]
- Dreher, M.R.; Liu, W.; Michelich, C.R.; Dewhirst, M.W.; Chilkoti, A. Thermal cycling enhances the accumulation of a temperature-sensitive biopolymer in solid tumors. Cancer Res. 2007, 67, 4418–4424. [Google Scholar] [CrossRef] [PubMed]
- Bidwell, G.L.; Perkins, E.; Raucher, D. A thermally targeted c-Myc inhibitory polypeptide inhibits breast tumor growth. Cancer Lett. 2012, 319, 136–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bidwell, G.L.; Perkins, E.; Hughes, J.; Khan, M.; James, J.R.; Raucher, D. Thermally targeted delivery of a c-Myc inhibitory polypeptide inhibits tumor progression and extends survival in a rat glioma model. PLoS ONE 2013, 8, e55104. [Google Scholar] [CrossRef] [PubMed]
- Furgeson, D.Y.; Dreher, M.R.; Chilkoti, A. Structural optimization of a “smart” doxorubicin-polypeptide conjugate for thermally targeted delivery to solid tumors. J. Control. Release 2006, 110, 362–369. [Google Scholar] [CrossRef]
- Dhandhukia, J.P.; Shi, P.; Peddi, S.; Li, Z.; Aluri, S.; Ju, Y.; Brill, D.; Wang, W.; Janib, S.M.; Lin, Y.-A.; et al. Bifunctional Elastin-like Polypeptide Nanoparticles Bind Rapamycin and Integrins and Suppress Tumor Growth in Vivo. Bioconjug. Chem. 2017, 28, 2715–2728. [Google Scholar] [CrossRef]
- Du, K.; Sun, J.; Song, X.; Song, C.; Feng, W. Enhancement of the solubility and stability of D-amino acid oxidase by fusion to an elastin like polypeptide. J. Biotechnol. 2015, 212, 50–55. [Google Scholar] [CrossRef]
- Zhou, Y.; Li, X.; Yan, D.; Peprah, F.A.; Ji, X.; Fletcher, E.E.; Wang, Y.; Wang, Y.; Gu, J.; Lin, F.; et al. Multifunctional elastin-like polypeptide renders β-glucosidase enzyme phase transition and high stability. Biotechnol. Biofuels 2019, 12, 157. [Google Scholar] [CrossRef]
- Wang, Y.; Tan, X.; Zong, Y.; Lu, H.; Zhang, X.; Xia, X.; Sun, H. Enhancing purification and plasma stability of porcine interferon-α/γ by fusion to elastin-like polypeptide. Vet. Immunol. Immunopathol. 2018, 203, 60–64. [Google Scholar] [CrossRef]
- George, E.M.; Liu, H.; Robinson, G.G.; Mahdi, F.; Perkins, E.; Bidwell, G.L. Growth factor purification and delivery systems (PADS) for therapeutic angiogenesis. Vasc. Cell 2015, 7, 1. [Google Scholar] [CrossRef]
- Costa, S.A.; Mozhdehi, D.; Dzuricky, M.J.; Isaacs, F.J.; Brustad, E.M.; Chilkoti, A. Active Targeting of Cancer Cells by Nanobody Decorated Polypeptide Micelle with Bio-orthogonally Conjugated Drug. Nano Lett. 2019, 19, 247–254. [Google Scholar] [CrossRef]
- Mie, M.; Matsumoto, R.; Mashimo, Y.; Cass, A.E.G.; Kobatake, E. Development of drug-loaded protein nanoparticles displaying enzymatically-conjugated DNA aptamers for cancer cell targeting. Mol. Biol. Rep. 2019, 46, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Bidwell, G.L.; Davis, A.N.; Raucher, D. Targeting a c-Myc inhibitory polypeptide to specific intracellular compartments using cell penetrating peptides. J. Control. Release 2009, 135, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Kuna, M.; Mahdi, F.; Chade, A.R.; Bidwell, G.L. Molecular Size Modulates Pharmacokinetics, Biodistribution, and Renal Deposition of the Drug Delivery Biopolymer Elastin-like Polypeptide. Sci. Rep. 2018, 8, 7923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuna, M.; Waller, J.P.; Logue, O.C.; Bidwell, G.L. Polymer size affects biodistribution and placental accumulation of the drug delivery biopolymer elastin-like polypeptide in a rodent pregnancy model. Placenta 2018, 72–73, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Logue, O.C.; Mahdi, F.; Chapman, H.; George, E.M.; Bidwell, G.L. A Maternally Sequestered, Biopolymer-Stabilized Vascular Endothelial Growth Factor (VEGF) Chimera for Treatment of Preeclampsia. J. Am. Heart Assoc. 2017, 6, e007216. [Google Scholar] [CrossRef]
- Chade, A.R.; Tullos, N.A.; Harvey, T.W.; Mahdi, F.; Bidwell, G.L. Renal Therapeutic Angiogenesis Using a Bioengineered Polymer-Stabilized Vascular Endothelial Growth Factor Construct. J. Am. Soc. Nephrol. 2016, 27, 1741–1752. [Google Scholar] [CrossRef]
- Olsson, A.-K.; Dimberg, A.; Kreuger, J.; Claesson-Welsh, L. VEGF receptor signaling—In control of vascular function. Nat. Rev. Mol. Cell Biol. 2006, 7, 359–371. [Google Scholar] [CrossRef]
- Chade, A.R.; Kelsen, S. Reversal of renal dysfunction by targeted administration of VEGF into the stenotic kidney: A novel potential therapeutic approach. Am. J. Physiol. Ren. Physiol. 2012, 302, F1342–F1350. [Google Scholar] [CrossRef]
- Chade, A.R.; Rodriguez-Porcel, M.; Grande, J.P.; Zhu, X.; Sica, V.; Napoli, C.; Sawamura, T.; Textor, S.C.; Lerman, A.; Lerman, L.O. Mechanisms of renal structural alterations in combined hypercholesterolemia and renal artery stenosis. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 1295–1301. [Google Scholar] [CrossRef]
- Maynard, S.E.; Min, J.-Y.; Merchan, J.; Lim, K.-H.; Li, J.; Mondal, S.; Libermann, T.A.; Morgan, J.P.; Sellke, F.W.; Stillman, I.E.; et al. Excess placental soluble fms-like tyrosine kinase 1 (sFlt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in preeclampsia. J. Clin. Investig. 2003, 111, 649–658. [Google Scholar] [CrossRef] [Green Version]
- Eppler, S.M.; Combs, D.L.; Henry, T.D.; Lopez, J.J.; Ellis, S.G.; Yi, J.H.; Annex, B.H.; McCluskey, E.R.; Zioncheck, T.F. A target-mediated model to describe the pharmacokinetics and hemodynamic effects of recombinant human vascular endothelial growth factor in humans. Clin. Pharmacol. Ther. 2002, 72, 20–32. [Google Scholar] [CrossRef] [PubMed]
- Henry, T.D.; Rocha-Singh, K.; Isner, J.M.; Kereiakes, D.J.; Giordano, F.J.; Simons, M.; Losordo, D.W.; Hendel, R.C.; Bonow, R.O.; Eppler, S.M.; et al. Intracoronary administration of recombinant human vascular endothelial growth factor to patients with coronary artery disease. Am. Heart J. 2001, 142, 872–880. [Google Scholar] [CrossRef] [PubMed]
- Takeshita, S.; Zheng, L.P.; Brogi, E.; Kearney, M.; Pu, L.Q.; Bunting, S.; Ferrara, N.; Symes, J.F.; Isner, J.M. Therapeutic angiogenesis. A single intraarterial bolus of vascular endothelial growth factor augments revascularization in a rabbit ischemic hind limb model. J. Clin. Investig. 1994, 93, 662–670. [Google Scholar] [CrossRef] [PubMed]
- Houck, K.A.; Ferrara, N.; Winer, J.; Cachianes, G.; Li, B.; Leung, D.W. The vascular endothelial growth factor family: Identification of a fourth molecular species and characterization of alternative splicing of RNA. Mol. Endocrinol. 1991, 5, 1806–1814. [Google Scholar] [CrossRef]
- Houck, K.A.; Leung, D.W.; Rowland, A.M.; Winer, J.; Ferrara, N. Dual regulation of vascular endothelial growth factor bioavailability by genetic and proteolytic mechanisms. J. Biol. Chem. 1992, 267, 26031–26037. [Google Scholar]
- Chade, A.R.; Williams, M.L.; Guise, E.; Vincent, L.J.; Harvey, T.W.; Kuna, M.; Mahdi, F.; Bidwell, G.L. Systemic biopolymer-delivered vascular endothelial growth factor promotes therapeutic angiogenesis in experimental renovascular disease. Kidney Int. 2017, 93, 842–854. [Google Scholar] [CrossRef]
- Guise, E.; Engel, J.E.; Williams, M.L.; Mahdi, F.; Bidwell, G.L.; Chade, A.R. Biopolymer-delivered vascular endothelial growth factor improves renal outcomes following revascularization. Am. J. Physiol. Ren. Physiol. 2019, 316, F1016–F1025. [Google Scholar] [CrossRef]
- Engel, J.E.; Williams, E.; Williams, M.L.; Bidwell, G.L.; Chade, A.R. Targeted VEGF (Vascular Endothelial Growth Factor) Therapy Induces Long-Term Renal Recovery in Chronic Kidney Disease via Macrophage Polarization. Hypertension 2019, 74, 1113–1123. [Google Scholar] [CrossRef]
- Pasqualini, R.; Ruoslahti, E. Organ targeting in vivo using phage display peptide libraries. Nature 1996, 380, 364–366. [Google Scholar] [CrossRef]
- Bidwell, G.L.; Raucher, D. Application of thermally responsive polypeptides directed against c-Myc transcriptional function for cancer therapy. Mol. Cancer Ther. 2005, 4, 1076–1085. [Google Scholar] [CrossRef]
- National Research Council (US) Committee for the Update of the Guide for the Care and Use of Laboratory Animals. Guide for the Care and Use of Laboratory Animals; The National Academies Collection: Reports funded by National Institutes of Health, 8th ed.; National Academies Press: Washington, DC, USA, 2011; ISBN 978-0-309-15400-0.
- McGowan, J.W.D.; Bidwell, G.L. The Use of Ex Vivo Whole-organ Imaging and Quantitative Tissue Histology to Determine the Bio-distribution of Fluorescently Labeled Molecules. J. Vis. Exp. 2016, 118, e54987. [Google Scholar] [CrossRef] [PubMed]
- George, E.M.; Liu, H.; Robinson, G.G.; Bidwell, G.L. A polypeptide drug carrier for maternal delivery and prevention of fetal exposure. J. Drug Target. 2014, 22, 935–947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Logue, O.C.; McGowan, J.W.D.; George, E.M.; Bidwell, G.L. Therapeutic angiogenesis by vascular endothelial growth factor supplementation for treatment of renal disease. Curr. Opin. Nephrol. Hypertens. 2016, 25, 404–409. [Google Scholar] [CrossRef] [PubMed]
- Bidwell, G.L.; George, E.M. Maternally sequestered therapeutic polypeptides—A new approach for the management of preeclampsia. Front. Pharmacol. 2014, 5, 201. [Google Scholar] [CrossRef]
- Eddy, A.C.; Bidwell, G.L.; George, E.M. Pro-angiogenic therapeutics for preeclampsia. Biol. Sex Differ. 2018, 9, 36. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, R.; Christensen, E.I.; Birn, H. Megalin and cubilin in proximal tubule protein reabsorption: From experimental models to human disease. Kidney Int. 2016, 89, 58–67. [Google Scholar] [CrossRef] [PubMed]










© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mahdi, F.; Chade, A.R.; Bidwell, G.L., III. Utilizing a Kidney-Targeting Peptide to Improve Renal Deposition of a Pro-Angiogenic Protein Biopolymer. Pharmaceutics 2019, 11, 542. https://doi.org/10.3390/pharmaceutics11100542
Mahdi F, Chade AR, Bidwell GL III. Utilizing a Kidney-Targeting Peptide to Improve Renal Deposition of a Pro-Angiogenic Protein Biopolymer. Pharmaceutics. 2019; 11(10):542. https://doi.org/10.3390/pharmaceutics11100542
Chicago/Turabian StyleMahdi, Fakhri, Alejandro R. Chade, and Gene L. Bidwell, III. 2019. "Utilizing a Kidney-Targeting Peptide to Improve Renal Deposition of a Pro-Angiogenic Protein Biopolymer" Pharmaceutics 11, no. 10: 542. https://doi.org/10.3390/pharmaceutics11100542