Brain Distribution and Metabolism of Flupirtine, a Nonopioid Analgesic Drug with Antiseizure Effects, in Neonatal Rats

Abstract
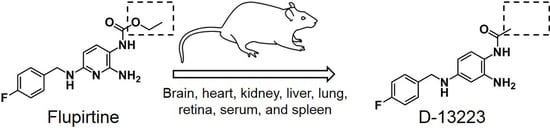
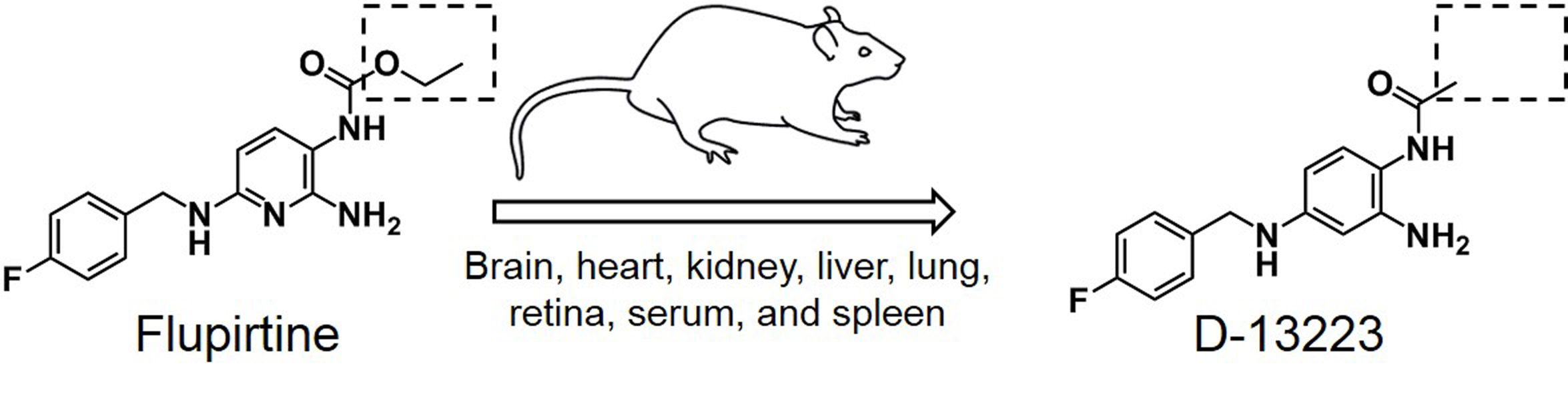
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
2.2.1. Animals
2.2.2. Tissue Distribution Studies
2.2.3. Sample Preparation for In Vivo Tissue Distribution
2.2.4. Flupirtine Metabolism in Isolated Tissues
2.2.5. Sample Preparation for Ex Vivo Metabolism
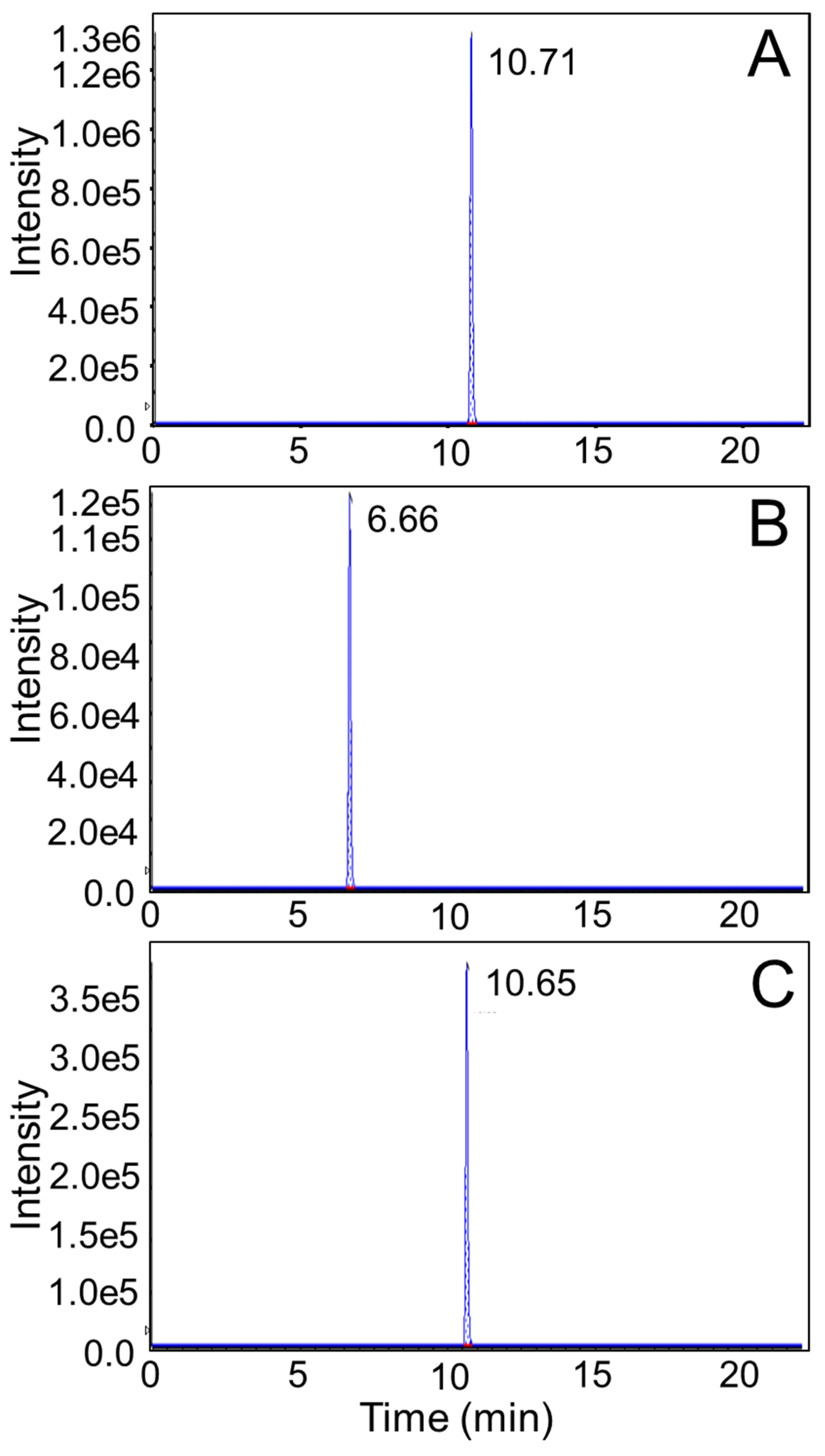
2.2.6. LC-MS/MS Analysis
3. Data Analysis
4. Results
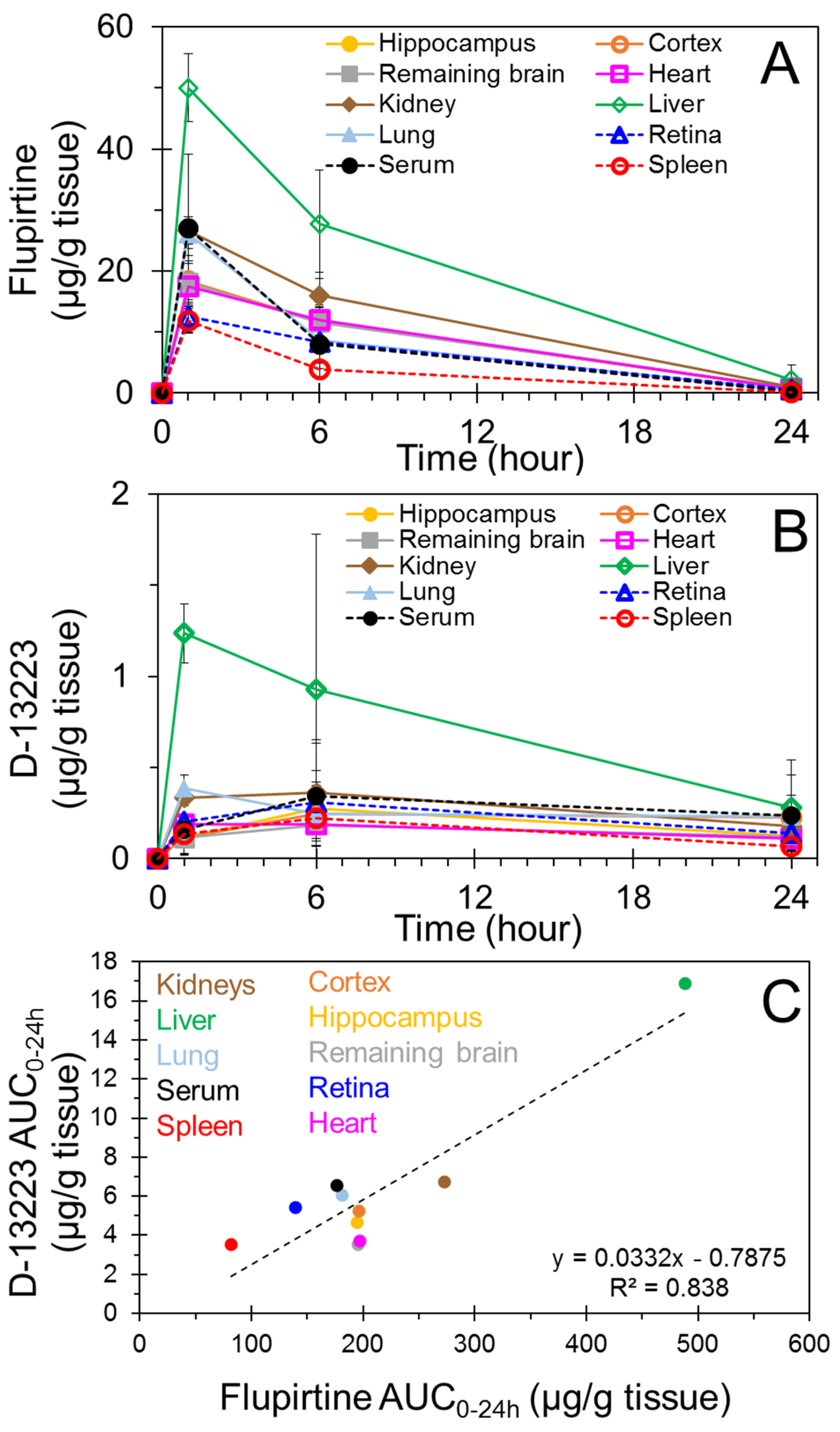
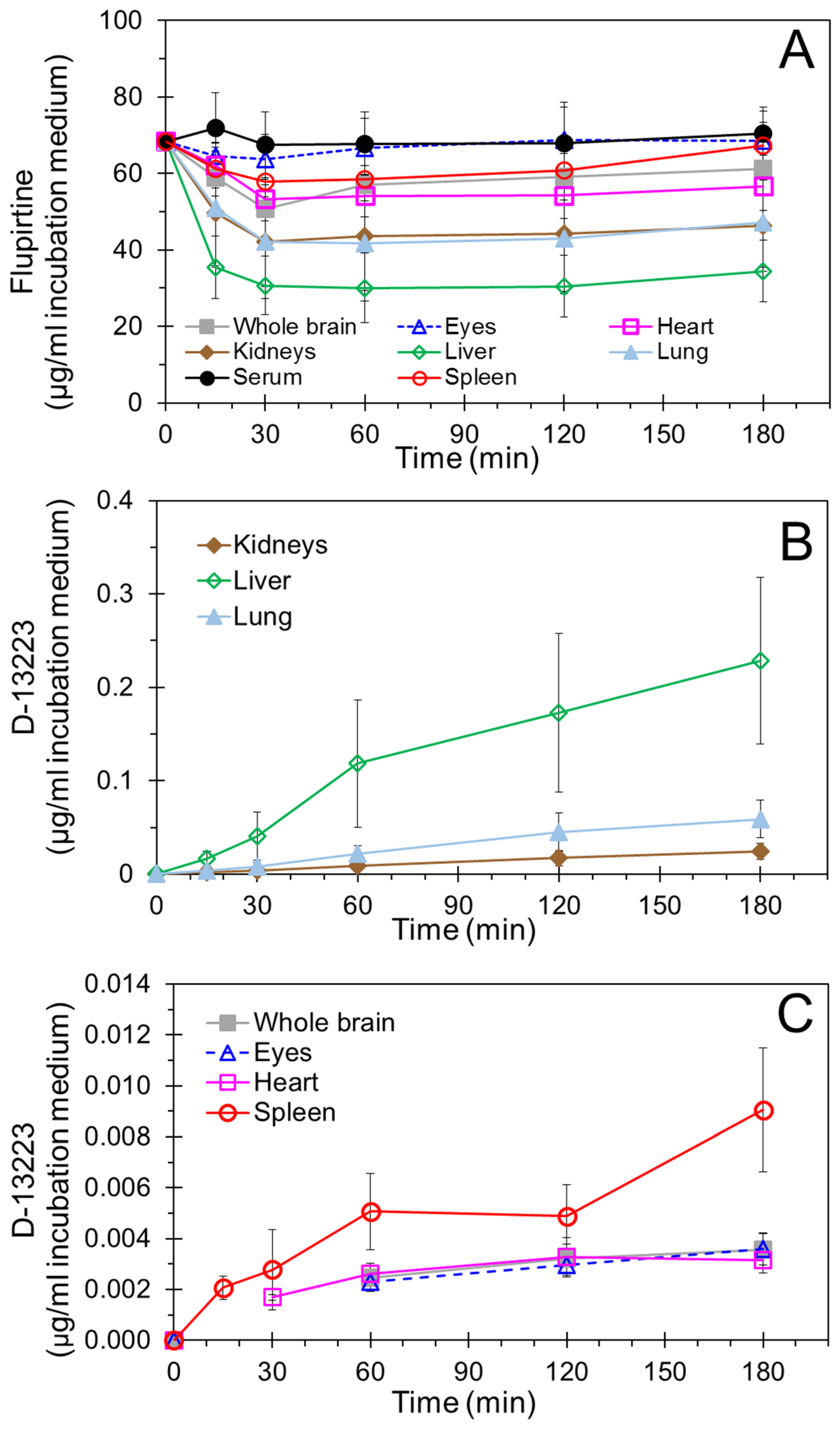
4.1. Biodistribution and Pharmacokinetics of Flupirtine
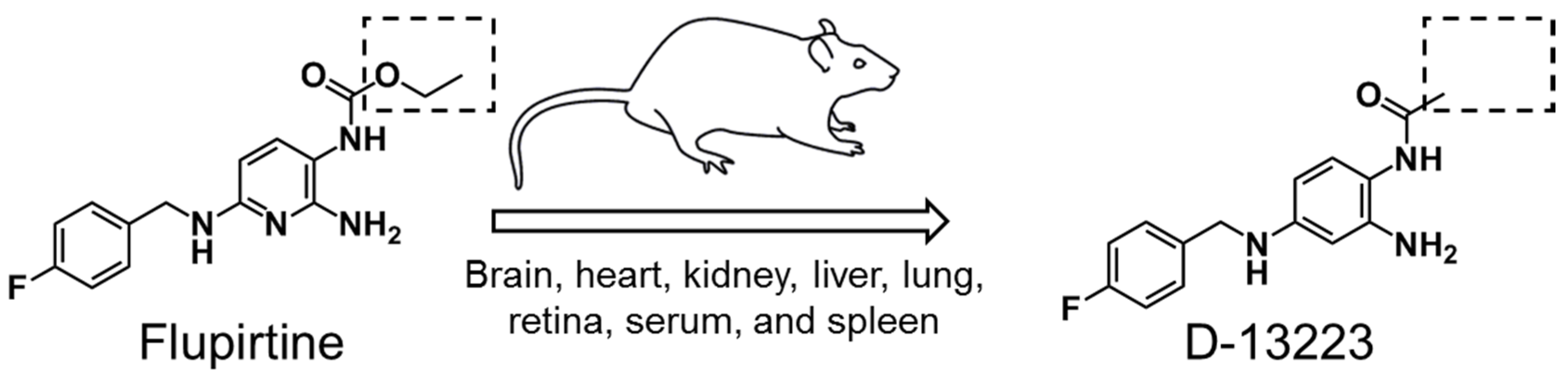
4.2. Conversion of Flupirtine to D-13223 in Isolated Rat Tissues
4.3. Tissue Partitioning In Ex Vivo Vs In Vivo Studies
5. Discussion
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Szelenyi, I. Flupirtine, a re-discovered drug, revisited. Inflamm. Res. 2013, 62, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Harish, S.; Bhuvana, K.; Bengalorkar, G.M.; Kumar, T. Flupirtine: Clinical pharmacology. J. Anaesthesiol. Clin. Pharmacol. 2012, 28, 172–177. [Google Scholar] [CrossRef] [PubMed]
- Douros, A.; Bronder, E.; Andersohn, F.; Klimpel, A.; Thomae, M.; Orzechowski, H.D.; Kreutz, R.; Garbe, E. Flupirtine-induced liver injury—Seven cases from the Berlin Case-control Surveillance Study and review of the German spontaneous adverse drug reaction reporting database. Eur. J. Clin. Pharmacol. 2014, 70, 453–459. [Google Scholar] [CrossRef] [PubMed]
- Klinger, F.; Geier, P.; Dorostkar, M.M.; Chandaka, G.K.; Yousuf, A.; Salzer, I.; Kubista, H.; Boehm, S. Concomitant facilitation of GABAA receptors and KV7 channels by the non-opioid analgesic flupirtine. Br. J. Pharmacol. 2012, 166, 1631–1642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Millichap, J.J.; Cooper, E.C. KCNQ2 Potassium Channel Epileptic Encephalopathy Syndrome: Divorce of an Electro-Mechanical Couple? Epilepsy Curr. 2012, 12, 150–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jakob, R.; Krieglstein, J. Influence of flupirtine on a G-protein coupled inwardly rectifying potassium current in hippocampal neurones. Br. J. Pharmacol. 1997, 122, 1333–1338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kornhuber, J.; Bleich, S.; Wiltfang, J.; Maler, M.; Parsons, C.G. Flupirtine shows functional NMDA receptor antagonism by enhancing Mg2+ block via activation of voltage independent potassium channels. Rapid communication. J. Neural. Transm. 1999, 106, 857–867. [Google Scholar] [CrossRef]
- Raffa, R.B.; Pergolizzi, J.V. The evolving understanding of the analgesic mechanism of action of flupirtine. J. Clin. Pharm. Ther. 2012, 37, 4–6. [Google Scholar] [CrossRef]
- Montandon, G.; Ren, J.; Victoria, N.C.; Liu, H.; Wickman, K.; Greer, J.J.; Horner, R.L. G-protein-gated Inwardly Rectifying Potassium Channels Modulate Respiratory Depression by Opioids. Anesthesiology 2016, 124, 641–650. [Google Scholar] [CrossRef]
- Martire, M.; Castaldo, P.; D’Amico, M.; Preziosi, P.; Annunziato, L.; Taglialatela, M. M channels containing KCNQ2 subunits modulate norepinephrine, aspartate, and GABA release from hippocampal nerve terminals. J. Neurosci. 2004, 24, 592–597. [Google Scholar] [CrossRef]
- Rupalla, K.; Cao, W.; Krieglstein, J. Flupirtine protects neurons against excitotoxic or ischemic damage and inhibits the increase in cytosolic Ca2+ concentration. Eur. J. Pharmacol. 1995, 294, 469–473. [Google Scholar] [CrossRef]
- Klepacki, A.; Zalewski, J.; Zaremba, L. Inhibition of fibrinolysis in acute necrotic pancreatitis. Pol. Przegl. Chir. 1974, 46, 985–989. [Google Scholar]
- Block, F.; Pergande, G.; Schwarz, M. Flupirtine reduces functional deficits and neuronal damage after global ischemia in rats. Brain Res. 1997, 754, 279–284. [Google Scholar] [CrossRef]
- Raol, Y.H.; Lapides, D.A.; Keating, J.G.; Brooks-Kayal, A.R.; Cooper, E.C. A KCNQ channel opener for experimental neonatal seizures and status epilepticus. Ann. Neurol. 2009, 65, 326–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sampath, D.; Shmueli, D.; White, A.M.; Raol, Y.H. Flupirtine effectively prevents development of acute neonatal seizures in an animal model of global hypoxia. Neurosci. Lett. 2015, 607, 46–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sampath, D.; Valdez, R.; White, A.M.; Raol, Y.H. Anticonvulsant effect of flupirtine in an animal model of neonatal hypoxic-ischemic encephalopathy. Neuropharmacology 2017, 123, 126–135. [Google Scholar] [CrossRef] [PubMed]
- Hlavica, P.; Niebch, G. Pharmacokinetics and biotransformation of the analgesic flupirtine in humans. Arzneimittelforschung 1985, 35, 67–74. [Google Scholar] [PubMed]
- Abrams, S.M.; Baker, L.R.; Crome, P.; White, A.S.; Johnston, A.; Ankier, S.I.; Warrington, S.J.; Turner, P.; Niebch, G. Pharmacokinetics of flupirtine in elderly volunteers and in patients with moderate renal impairment. Postgrad. Med. J. 1988, 64, 361–363. [Google Scholar] [CrossRef]
- Chen, X.; Zhong, D.; Xu, H.; Schug, B.; Blume, H. Simultaneous determination of flupirtine and its major active metabolite in human plasma by liquid chromatography-tandem mass spectrometry. J. Chromatogr. B Biomed. Sci. Appl. 2001, 755, 195–202. [Google Scholar] [CrossRef]
- Narang, P.K.; Tourville, J.F.; Chatterji, D.C.; Gallelli, J.F. Quantitation of flupirtine and its active acetylated metabolite by reversed-phase high-performance liquid chromatography using fluorometric detection. J. Chromatogr. 1984, 305, 135–143. [Google Scholar] [CrossRef]
- Obermeier, K.; Niebch, G.; Thiemer, K. Pharmacokinetics and biotransformation of the analgesic flupirtine in the rat and dog. Arzneimittelforschung 1985, 35, 60–67. [Google Scholar] [PubMed]
- De Vito, V.; Lebkowska-Wieruszewska, B.; Owen, H.; Kowalski, C.J.; Giorgi, M. Pharmacokinetic profiles of the analgesic drug flupirtine in cats. Vet. J. 2014, 202, 309–313. [Google Scholar] [CrossRef] [Green Version]
- Giorgi, M.; De Vito, V.; Poapolathep, A.; Rychshanova, R.; Sgorbini, M.; Owen, H. Pharmacokinetics and disposition of flupirtine in the horse. Vet. J. 2016, 208, 76–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, A.L.; Xiang, X.; Ong, P.S.; Mitchell, E.Q.Y.; Syn, N.; Wee, I.; Kumar, A.P.; Yong, W.P.; Sethi, G.; Goh, B.C.; et al. A Review on Liquid Chromatography-Tandem Mass Spectrometry Methods for Rapid Quantification of Oncology Drugs. Pharmaceutics 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Kadam, S.D.; Dudek, F.E. Neuropathogical features of a rat model for perinatal hypoxic-ischemic encephalopathy with associated epilepsy. J. Comp. Neurol. 2007, 505, 716–737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devulder, J. Flupirtine in pain management: Pharmacological properties and clinical use. CNS Drugs 2010, 24, 867–881. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency (EMA). Assessment Report for Flupirtine Containing Medicinal Products; Volume: EMEA/H/A-107i/1363; European Medicines Agency (EMA): London, UK, 2013. [Google Scholar]
- Food and Drug Administration. Guidance for Industry—Estimating the Maximum Safe Starting Dose in Initial Clinical Trials for Therapeutics in Adult Healthy Volunteers; Food and Drug Administration: Silver Spring, MD, USA, 2015. [Google Scholar]
- Methling, K.; Reszka, P.; Lalk, M.; Vrana, O.; Scheuch, E.; Siegmund, W.; Terhaag, B.; Bednarski, P.J. Investigation of the in vitro metabolism of the analgesic flupirtine. Drug Metab. Dispos. 2009, 37, 479–493. [Google Scholar] [CrossRef]
- Barker, D.F.; Walraven, J.M.; Ristagno, E.H.; Doll, M.A.; States, J.C.; Hein, D.W. Quantitative tissue and gene-specific differences and developmental changes in Nat1, Nat2, and Nat3 mRNA expression in the rat. Drug Metab. Dispos. 2008, 36, 2445–2451. [Google Scholar] [CrossRef]
- Puls, F.; Agne, C.; Klein, F.; Koch, M.; Rifai, K.; Manns, M.P.; Borlak, J.; Kreipe, H.H. Pathology of flupirtine-induced liver injury: A histological and clinical study of six cases. Virchows Arch. 2011, 458, 709–716. [Google Scholar] [CrossRef]
- Michel, M.C.; Radziszewski, P.; Falconer, C.; Marschall-Kehrel, D.; Blot, K. Unexpected frequent hepatotoxicity of a prescription drug, flupirtine, marketed for about 30 years. Br. J. Clin. Pharmacol. 2012, 73, 821–825. [Google Scholar] [CrossRef] [Green Version]
- Doerge, D.R.; Twaddle, N.C.; Vanlandingham, M.; Fisher, J.W. Pharmacokinetics of bisphenol A in neonatal and adult Sprague-Dawley rats. Toxicol. Appl. Pharmacol. 2010, 247, 158–165. [Google Scholar] [CrossRef] [PubMed]
- Doerge, D.R.; Twaddle, N.C.; Vanlandingham, M.; Fisher, J.W. Pharmacokinetics of bisphenol A in neonatal and adult CD-1 mice: Inter-species comparisons with Sprague-Dawley rats and rhesus monkeys. Toxicol. Lett. 2011, 207, 298–305. [Google Scholar] [CrossRef] [PubMed]
- Statler, P.A.; McPherson, R.J.; Bauer, L.A.; Kellert, B.A.; Juul, S.E. Pharmacokinetics of high-dose recombinant erythropoietin in plasma and brain of neonatal rats. Pediatr. Res. 2007, 61, 671–675. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tissue | Flupirtine | D-13223 | |||
|---|---|---|---|---|---|
| AUC0–24h (µg·h/g Tissue) | Elimination Rate Constant (ke, h−1) | Half-Life (t1/2, h) | AUC0–24h (µg·h/g Tissue) | Half-Life (t1/2, h) | |
| Hippocampus | 195 | 0.140 | 4.9 | 4.70 | NE |
| Cortex | 196 | 0.142 | 4.9 | 5.25 | NE |
| Remaining brain | 195 | 0.135 | 5.1 | 3.56 | NE |
| Heart | 197 | 0.145 | 4.8 | 3.75 | NE |
| Kidney | 273 | 0.144 | 4.8 | 6.74 | NE |
| Liver | 488 | 0.137 | 5.1 | 16.9 | 10.7 |
| Lung | 181 | 0.179 | 3.9 | 6.09 | NE |
| Retina | 139 | 0.134 | 5.2 | 5.43 | NE |
| Serum | 176 | 0.192 | 3.6 | 6.56 | NE |
| Spleen | 82 | 0.222 | 3.1 | 3.54 | NE |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Patil, M.A.; Matter, B.A.; Raol, Y.H.; Bourne, D.W.A.; Kelley, R.A.; Kompella, U.B. Brain Distribution and Metabolism of Flupirtine, a Nonopioid Analgesic Drug with Antiseizure Effects, in Neonatal Rats. Pharmaceutics 2018, 10, 281. https://doi.org/10.3390/pharmaceutics10040281
Patil MA, Matter BA, Raol YH, Bourne DWA, Kelley RA, Kompella UB. Brain Distribution and Metabolism of Flupirtine, a Nonopioid Analgesic Drug with Antiseizure Effects, in Neonatal Rats. Pharmaceutics. 2018; 10(4):281. https://doi.org/10.3390/pharmaceutics10040281
Chicago/Turabian StylePatil, Madhoosudan A., Brock A. Matter, Yogendra H. Raol, David W. A. Bourne, Ryan A. Kelley, and Uday B. Kompella. 2018. "Brain Distribution and Metabolism of Flupirtine, a Nonopioid Analgesic Drug with Antiseizure Effects, in Neonatal Rats" Pharmaceutics 10, no. 4: 281. https://doi.org/10.3390/pharmaceutics10040281