Modulation of Hypoxia-Induced Chemoresistance to Polymeric Micellar Cisplatin: The Effect of Ligand Modification of Micellar Carrier Versus Inhibition of the Mediators of Drug Resistance

 ,
,
Abstract
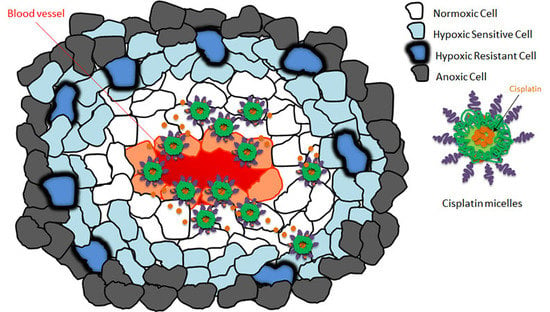
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.1.1. Synthesis of Block Copolymers with Functionalized Poly(ethylene oxide) (PEO)
2.1.2. Synthesis of GE11 Peptide and GE11 Conjugation to Poly(ethylene oxide)-poly(α-carboxyl-ε-caprolactone) (PEO-PCCL) Block Copolymers
2.1.3. Preparation of Plain and GE11 Cisplatin Micelles
2.1.4. Measurement of the Size and Zeta Potential of Plain and GE11 Cisplatin Micelles
2.1.5. Measurement of the Critical Micellar Concentration (CMC) of Plain and GE11 Cisplatin Micelles
2.1.6. Measurement of Cisplatin Encapsulation
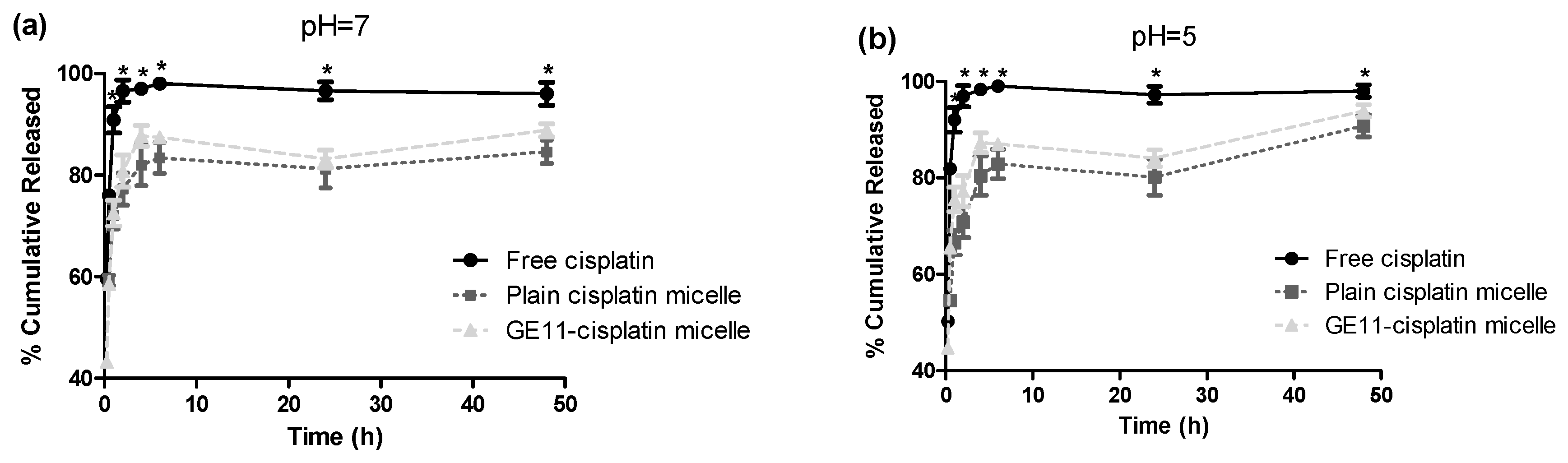
2.1.7. In Vitro Release Studies
2.1.8. Cell Culture
2.1.9. Flow Cytometric Detection of Apoptosis using Annexin V-FITC and Propidium Iodide
2.1.10. MTT Assay
2.1.11. Western Blot
2.1.12. Cisplatin Cellular Uptake
2.1.13. Statistical Analysis
3. Results
3.1. Successful Synthesis of GE11 Conjugated Poly(ethylene oxide)-poly(α-carboxyl-ε-caprolactone)(PEO-PCCL) Block Copolymer and Its Self-assembly
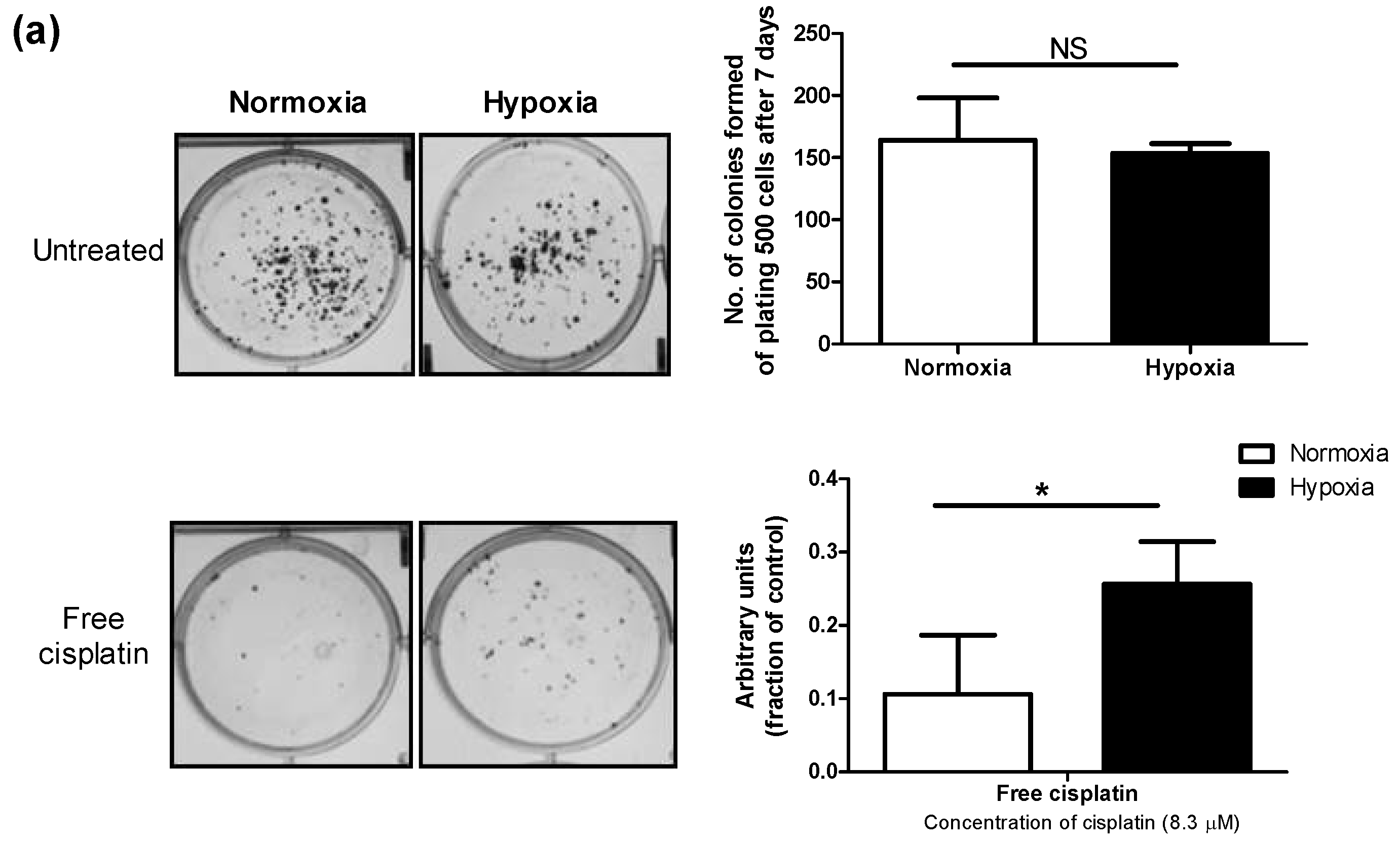
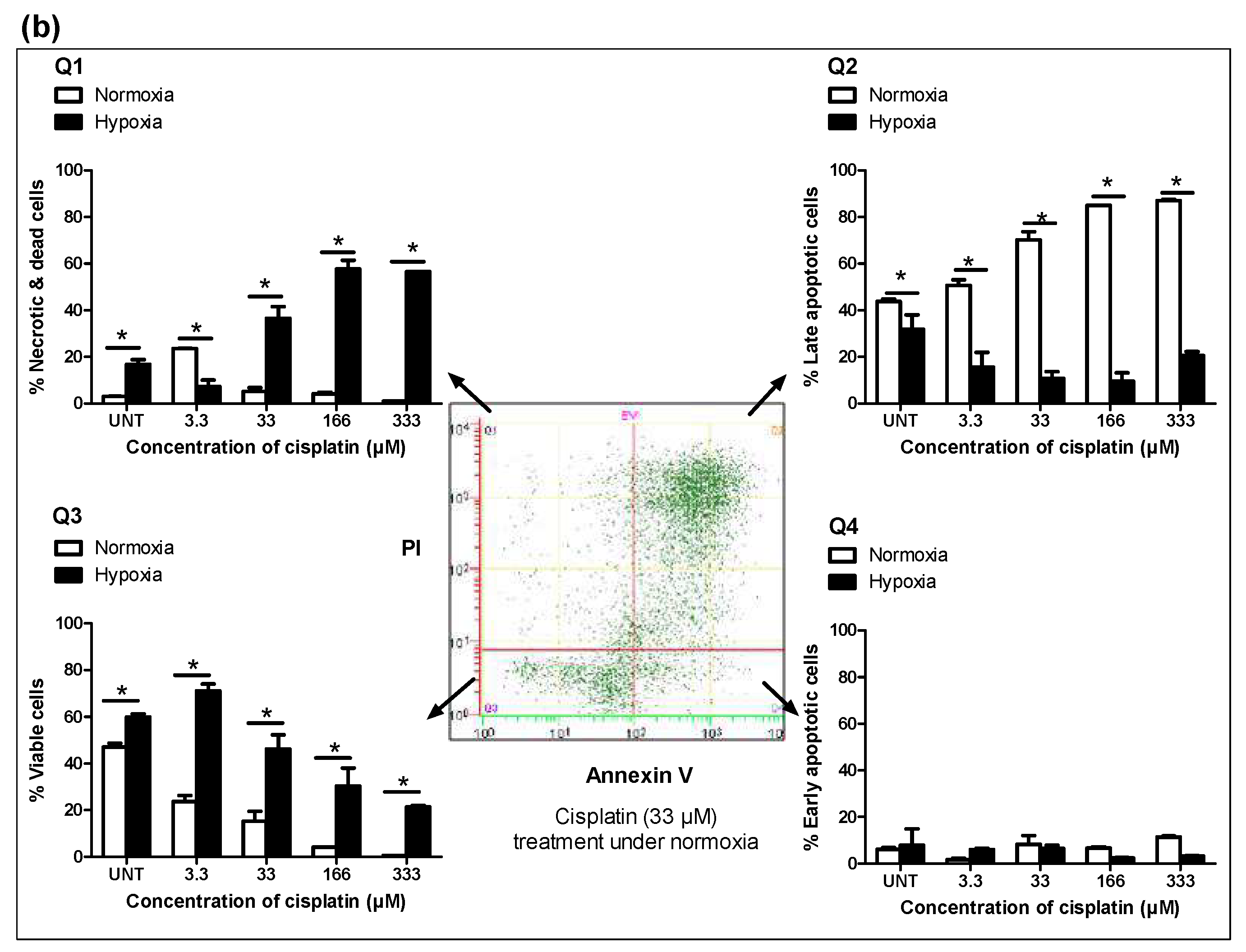
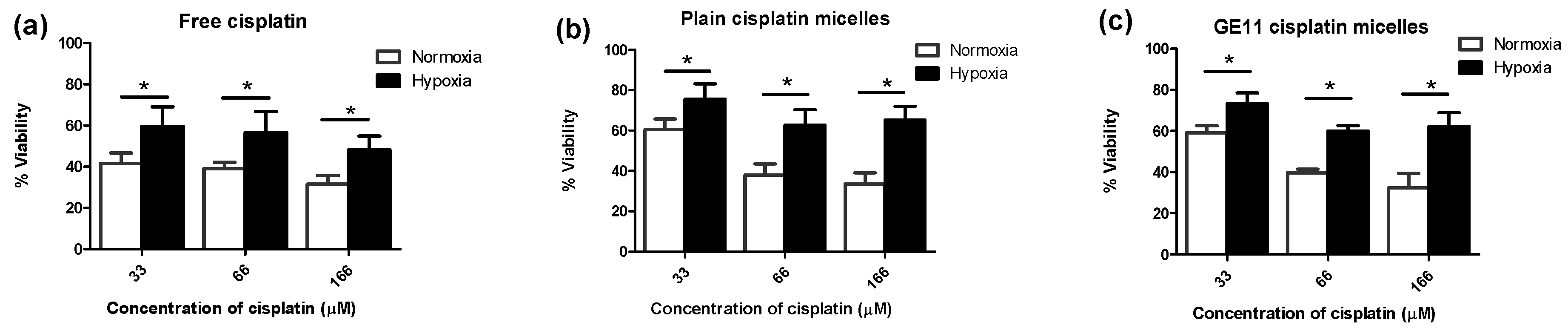
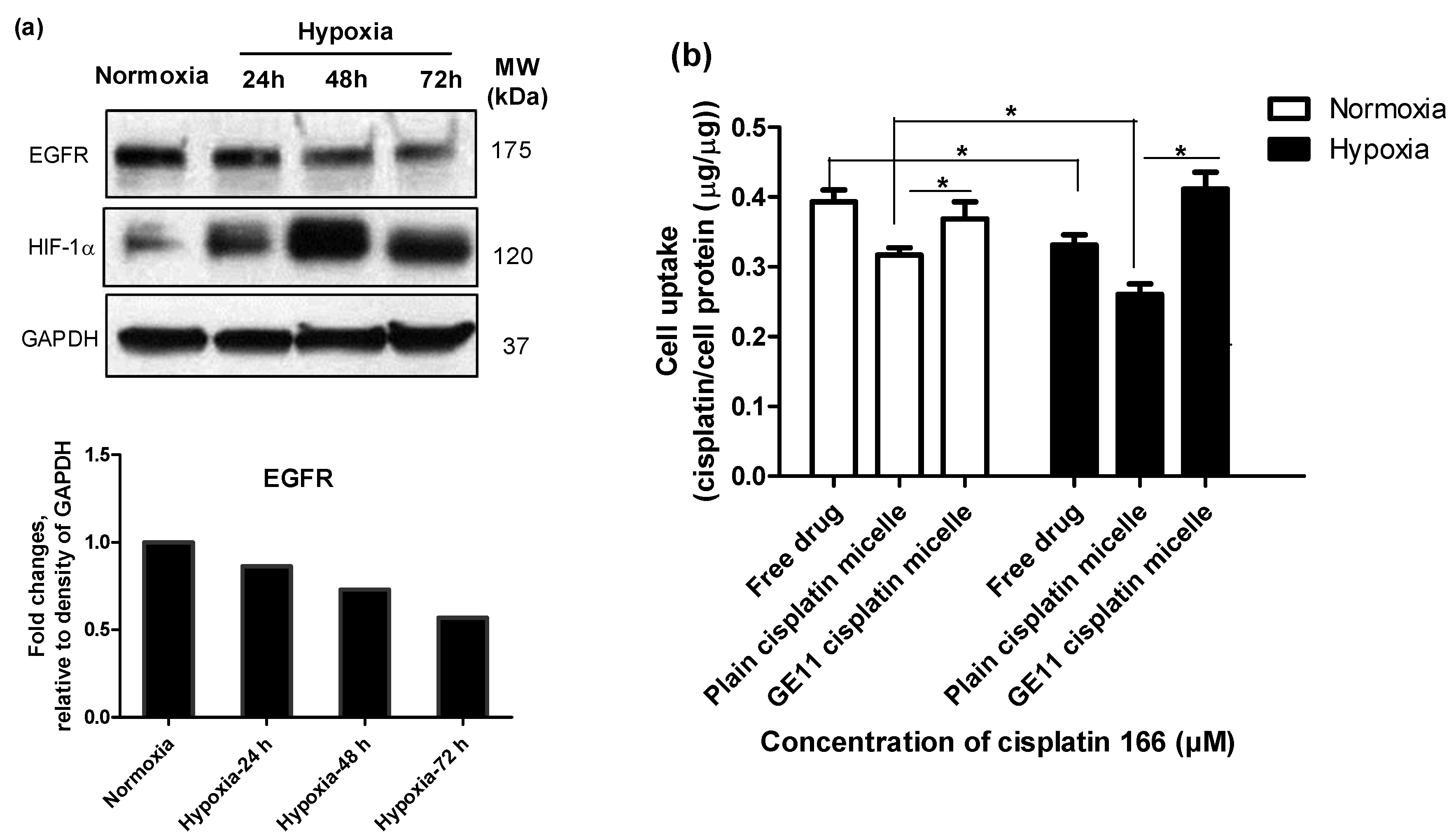
3.2. Hypoxia Induces Chemoresistance to Free Cisplatin in MDA-MB-231 Cells
3.3. Modification of Cisplatin Micelles with GE11 Peptide Enhances the Cellular Uptake of Cisplatin, but Does Not Affect its Cytotoxicity in MDA-MB-231 Cells
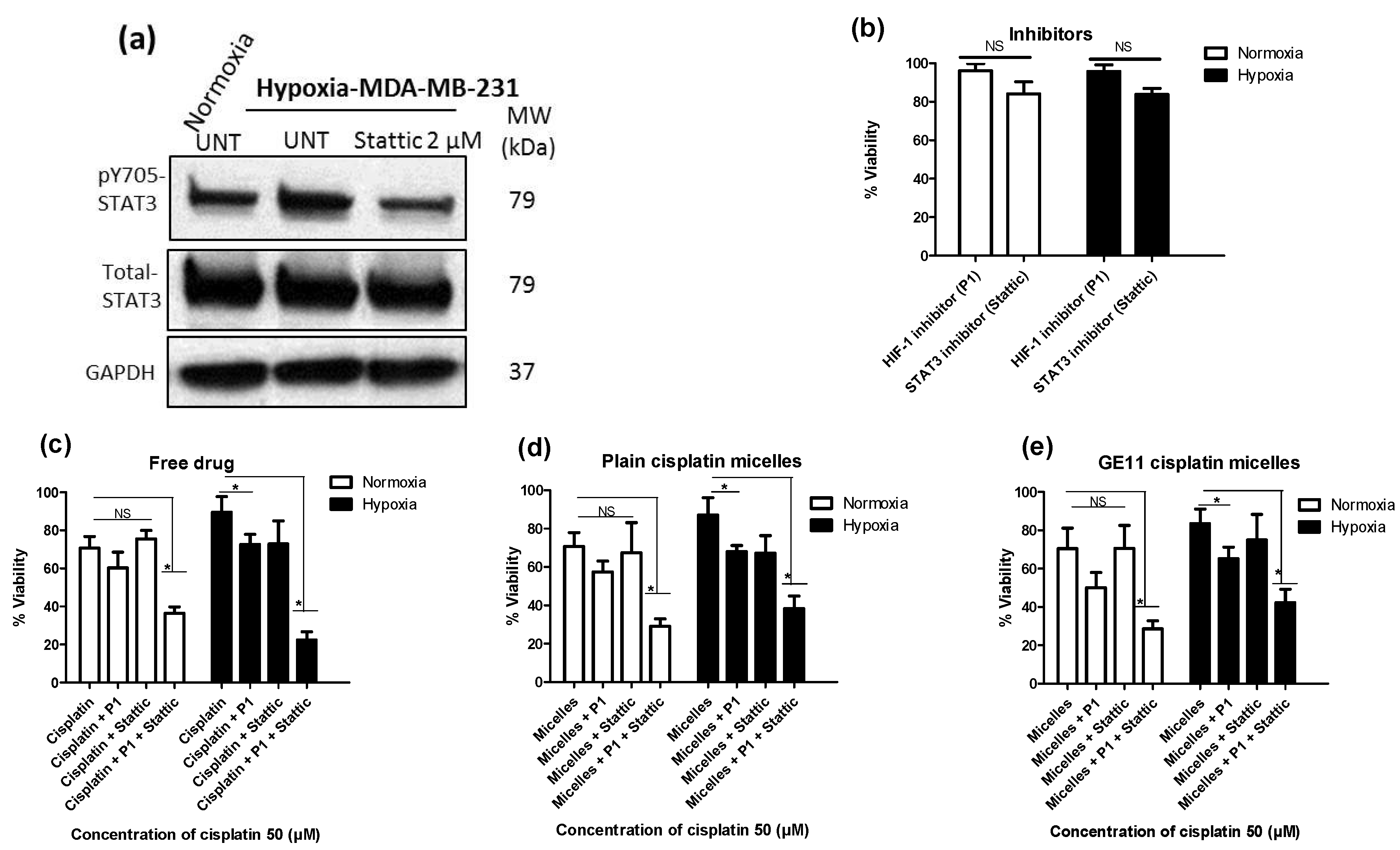
3.4. Co-Treatment with Pharmacological Inhibitors of HIF-1 and STAT3 Potentiates the Anticancer Activity of Free Cisplatin, as well as Its Micellar Formulations in Hypoxic MDA-MB-231 Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tredan, O.; Galmarini, C.M.; Patel, K.; Tannock, I.F. Drug resistance and the solid tumor microenvironment. J. Natl. Cancer Inst. 2007, 99, 1441–1454. [Google Scholar] [CrossRef] [PubMed]
- Mamede, A.C.; Abrantes, A.M.; Pedrosa, L.; Casalta-Lopes, J.E.; Pires, A.S.; Teixo, R.J.; Goncalves, A.C.; Sarmento-Ribeiro, A.B.; Maia, C.J.; Botelho, M.F. Beyond the limits of oxygen: Effects of hypoxia in a hormone-independent prostate cancer cell line. ISRN Oncol. 2013, 2013, 918207. [Google Scholar] [CrossRef] [PubMed]
- Selvendiran, K.; Bratasz, A.; Kuppusamy, M.L.; Tazi, M.F.; Rivera, B.K.; Kuppusamy, P. Hypoxia induces chemoresistance in ovarian cancer cells by activation of signal transducer and activator of transcription 3. Int. J. Cancer 2009, 125, 2198–2204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sullivan, R.; Pare, G.C.; Frederiksen, L.J.; Semenza, G.L.; Graham, C.H. Hypoxia-induced resistance to anticancer drugs is associated with decreased senescence and requires hypoxia-inducible factor-1 activity. Mol. Cancer Ther. 2008, 7, 1961–1973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Notte, A.; Ninane, N.; Arnould, T.; Michiels, C. Hypoxia counteracts taxol-induced apoptosis in MDA-MB-231 breast cancer cells: Role of autophagy and JNK activation. Cell Death Dis. 2013, 4, e638. [Google Scholar] [CrossRef] [PubMed]
- Foulkes, W.D.; Smith, I.E.; Reis-Filho, J.S. Triple-negative breast cancer. N. Engl. J. Med. 2010, 363, 1938–1948. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, E.A.; Gubbins, L.; Sharma, S.; Tully, R.; Guang, M.H.; Weiner-Gorzel, K.; McCaffrey, J.; Harrison, M.; Furlong, F.; Kell, M.; et al. The fate of chemoresistance in triple negative breast cancer (TNBC). BBA Clin. 2015, 3, 257–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.R.; Chen, A. Poly(Adenosine diphosphate-ribose) polymerase inhibitors in cancer treatment. Hematol. Oncol. Clin. N. Am. 2012, 26, 649–670. [Google Scholar] [CrossRef] [PubMed]
- Hang, Z.; Cooper, M.A.; Ziora, Z.M. Platinum-based anticancer drugs encapsulated liposome and polymeric micelle formulation in clinical trials. Biochem. Compd. 2016, 4, 2. [Google Scholar] [CrossRef]
- Matsumura, Y.; Kataoka, K. Preclinical and clinical studies of anticancer agent-incorporating polymer micelles. Cancer Sci. 2009, 100, 572–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zamboni, W.C.; Gervais, A.C.; Egorin, M.J.; Schellens, J.H.; Zuhowski, E.G.; Pluim, D.; Joseph, E.; Hamburger, D.R.; Working, P.K.; Colbern, G.; et al. Systemic and tumor disposition of platinum after administration of cisplatin or STEALTH liposomal-cisplatin formulations (SPI-077 and SPI-077 B103) in a preclinical tumor model of melanoma. Cancer Chemother. Pharmacol. 2004, 53, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.-R.; Stanton-Maxey, K.J.; Stanley, J.K.; Levin, C.S.; Bardhan, R.; Akin, D.; Badve, S.; Sturgis, J.; Robinson, J.P.; Bashir, R.; et al. A cellular Trojan horse for delivery of therapeutic nanoparticles into tumors. Nano Lett. 2007, 7, 3759–3765. [Google Scholar] [CrossRef] [PubMed]
- Mooney, R.; Weng, Y.; Garcia, E.; Bhojane, S.; Smith-Powell, L.; Kim, S.U.; Annala, A.J.; Aboody, K.S.; Berlin, J.M. Conjugation of pH-responsive nanoparticles to neural stem cells improves intratumoral therapy. J. Controll. Release 2014, 191, 82–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, W.; Dong, Z.; Fu, T.; Liu, J.; Chen, Q.; Li, Y.; Zhu, R.; Xu, L.; Liu, Z. Modulation of hypoxia in solid tumor microenvironment with MnO2 nanoparticles to enhance photodynamic therapy. Adv. Funct. Mater. 2016, 26, 5490–5498. [Google Scholar] [CrossRef]
- Shahin, M.; Safaei-Nikouei, N.; Lavasanifar, A. Polymeric micelles for pH-responsive delivery of cisplatin. J. Drug Target. 2014, 22, 629–637. [Google Scholar] [CrossRef] [PubMed]
- Franovic, A.; Gunaratnam, L.; Smith, K.; Robert, I.; Patten, D.; Lee, S. Translational up-regulation of the EGFR by tumor hypoxia provides a nonmutational explanation for its overexpression in human cancer. Proc. Natl. Acad. Sci. USA 2007, 104, 13092–13097. [Google Scholar] [CrossRef] [PubMed]
- Milane, L.; Duan, Z.; Amiji, M. Development of EGFR-targeted polymer blend nanocarriers for combination paclitaxel/lonidamine delivery to treat multi-drug resistance in human breast and ovarian tumor cells. Mol. Pharm. 2011, 8, 185–203. [Google Scholar] [CrossRef] [PubMed]
- Rojo, F.; Albanell, J.; Rovira, A.; Corominas, J.M.; Manzarbeitia, F. Targeted therapies in breast cancer. Semin. Diagn. Pathol. 2008, 25, 245–261. [Google Scholar] [CrossRef] [PubMed]
- Nie, S.; Xing, Y.; Kim, G.J.; Simons, J.W. Nanotechnology applications in cancer. Annu. Rev. Biomed. Eng. 2007, 9, 257–288. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.E.; Chen, Z.G.; Shin, D.M. Nanoparticle therapeutics: An emerging treatment modality for cancer. Nat. Rev. Drug Discov. 2008, 7, 771–782. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.D.; Shin, D.M.; Simons, J.W.; Nie, S. Nanotechnology for targeted cancer therapy. Expert Rev. Anticancer Ther. 2007, 7, 833–837. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, N.; Okazaki, S.; Cabral, H.; Miyamoto, M.; Kato, Y.; Sugiyama, Y.; Nishio, K.; Matsumura, Y.; Kataoka, K. Novel cisplatin-incorporated polymeric micelles can eradicate solid tumors in mice. Cancer Res. 2003, 63, 8977–8983. [Google Scholar] [PubMed]
- Nida, D.L.; Rahman, M.S.; Carlson, K.D.; Richards-Kortum, R.; Follen, M. Fluorescent nanocrystals for use in early cervical cancer detection. Gynecol. Oncol. 2005, 99, S89–S94. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Eom, K.; Lim, E.K.; Park, J.; Kang, Y.; Yoon, D.S.; Na, S.; Koh, E.K.; Suh, J.S.; Huh, Y.M.; et al. In situ detection of live cancer cells by using bioprobes based on Au nanoparticles. Langmuir 2008, 24, 12112–12115. [Google Scholar] [CrossRef] [PubMed]
- Melancon, M.P.; Lu, W.; Yang, Z.; Zhang, R.; Cheng, Z.; Elliot, A.M.; Stafford, J.; Olson, T.; Zhang, J.Z.; Li, C. In vitro and in vivo targeting of hollow gold nanoshells directed at epidermal growth factor receptor for photothermal ablation therapy. Mol. Cancer Ther. 2008, 7, 1730–1739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patra, C.R.; Bhattacharya, R.; Wang, E.; Katarya, A.; Lau, J.S.; Dutta, S.; Muders, M.; Wang, S.; Buhrow, S.A.; Safgren, S.L.; et al. Targeted delivery of gemcitabine to pancreatic adenocarcinoma using cetuximab as a targeting agent. Cancer Res. 2008, 68, 1970–1978. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Barth, R.F.; Yang, W.; Kawabata, S.; Zhang, L.; Green-Church, K. Targeted delivery of methotrexate to epidermal growth factor receptor-positive brain tumors by means of cetuximab (IMC-C225) dendrimer bioconjugates. Mol. Cancer Ther. 2006, 5, 52–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Master, A.M.; Sen Gupta, A. EGF receptor-targeted nanocarriers for enhanced cancer treatment. Nanomedicine 2012, 7, 1895–1906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Zhao, R.; Wu, X.; Sun, Y.; Yao, M.; Li, J.; Xu, Y.; Gu, J. Identification and characterization of a novel peptide ligand of epidermal growth factor receptor for targeted delivery of therapeutics. FASEB J. 2005, 19, 1978–1985. [Google Scholar] [CrossRef] [PubMed]
- Soleymani Abyaneh, H.; Gupta, N.; Radziwon-Balicka, A.; Jurasz, P.; Seubert, J.; Lai, R.; Lavasanifar, A. STAT3 but not HIF-1alpha is important in mediating hypoxia-induced chemoresistance in MDA-MB-231, a triple negative breast cancer cell line. Cancers 2017, 9, 137. [Google Scholar] [CrossRef] [PubMed]
- Storey, R.F.; Sherman, J.W. Kinetics and mechanism of the stannous octoate-catalyzed bulk polymerization of ε-Caprolactone. Macromolecules 2002, 35, 1504–1512. [Google Scholar] [CrossRef]
- Mahmud, A.; Xiong, X.-B.; Lavasanifar, A. Novel self-associating poly(ethylene oxide)-block-poly(ε-caprolactone) block copolymers with functional side groups on the polyester block for drug delivery. Macromolecules 2006, 39, 9419–9428. [Google Scholar] [CrossRef]
- Nagasaki, Y.; Kutsuna, T.; Iijima, M.; Kato, M.; Kataoka, K.; Kitano, S.; Kadoma, Y. Formyl-ended heterobifunctional poly(ethylene oxide): Synthesis of poly(ethylene oxide) with a formyl group at one end and a hydroxyl group at the other end. Bioconjugate Chem. 1995, 6, 231–233. [Google Scholar] [CrossRef]
- Xiong, X.B.; Mahmud, A.; Uludag, H.; Lavasanifar, A. Conjugation of arginine-glycine-aspartic acid peptides to poly(ethylene oxide)-b-poly(epsilon-caprolactone) micelles for enhanced intracellular drug delivery to metastatic tumor cells. Biomacromolecules 2007, 8, 874–884. [Google Scholar] [CrossRef] [PubMed]
- Soudy, R.; Gill, A.; Sprules, T.; Lavasanifar, A.; Kaur, K. Proteolytically stable cancer targeting peptides with high affinity for breast cancer cells. J. Med. Chem. 2011, 54, 7523–7534. [Google Scholar] [CrossRef] [PubMed]
- Topel, Ö.; Çakır, B.A.; Budama, L.; Hoda, N. Determination of critical micelle concentration of polybutadiene-block-poly(ethyleneoxide) diblock copolymer by fluorescence spectroscopy and dynamic light scattering. J. Mol. Liq. 2013, 177, 40–43. [Google Scholar] [CrossRef]
- Shahin, M.; Lavasanifar, A. Novel self-associating poly(ethylene oxide)-b-poly(epsilon-caprolactone) based drug conjugates and nano-containers for paclitaxel delivery. Int. J. Pharm. 2010, 389, 213–222. [Google Scholar] [CrossRef] [PubMed]
- Costa, P.; Sousa Lobo, J.M. Modeling and comparison of dissolution profiles. Eur. J. Pharm. Sci. 2001, 13, 123–133. [Google Scholar] [CrossRef]
- Miranda, E.; Nordgren, I.K.; Male, A.L.; Lawrence, C.E.; Hoakwie, F.; Cuda, F.; Court, W.; Fox, K.R.; Townsend, P.A.; Packham, G.K.; et al. A cyclic peptide inhibitor of HIF-1 heterodimerization that inhibits hypoxia signaling in cancer cells. J. Am. Chem. Soc. 2013, 135, 10418–10425. [Google Scholar] [CrossRef] [PubMed]
- Thews, O.; Gassner, B.; Kelleher, D.K.; Schwerdt, G.; Gekle, M. Impact of hypoxic and acidic extracellular conditions on cytotoxicity of chemotherapeutic drugs. Adv. Exp. Med. Biol. 2007, 599, 155–161. [Google Scholar] [PubMed]
- Soleymani Abyaneh, H.; Gupta, N.; Alshareef, A.; Gopal, K.; Lavasanifar, A.; Lai, R. Hypoxia Induces the Acquisition of Cancer Stem-like Phenotype Via Upregulation and Activation of Signal Transducer and Activator of Transcription-3 (STAT3) in MDA-MB-231, a Triple Negative Breast Cancer Cell Line. Cancer Microenviron. 2018. [Google Scholar] [CrossRef] [PubMed]
- Schust, J.; Sperl, B.; Hollis, A.; Mayer, T.U.; Berg, T. Stattic: A small-molecule inhibitor of STAT3 activation and dimerization. Chem. Biol. 2006, 13, 1235–1242. [Google Scholar] [CrossRef] [PubMed]
- Mistry, I.N.; Tavassoli, A. Reprogramming the transcriptional response to hypoxia with a chromosomally encoded cyclic peptide HIF-1 inhibitor. ACS Synth. Biol. 2017, 6, 518–527. [Google Scholar] [CrossRef] [PubMed]
- Hoogsteen, I.J.; Marres, H.A.; van den Hoogen, F.J.; Rijken, P.F.; Lok, J.; Bussink, J.; Kaanders, J.H. Expression of EGFR under tumor hypoxia: Identification of a subpopulation of tumor cells responsible for aggressiveness and treatment resistance. Int. J. Radiat. Oncol. Biol. Phys. 2012, 84, 807–814. [Google Scholar] [CrossRef] [PubMed]
- Anselmo, A.C.; Mitragotri, S. Nanoparticles in the clinic. Bioeng. Transl. Med. 2016, 1, 10–29. [Google Scholar] [CrossRef] [PubMed]
- Aldea, M.; Florian, I.A.; Kacso, G.; Craciun, L.; Boca, S.; Soritau, O.; Florian, I.S. Nanoparticles for targeting intratumoral hypoxia: exploiting a potential weakness of glioblastoma. Pharm. Res. 2016, 33, 2059–2077. [Google Scholar] [CrossRef] [PubMed]
- Brahimi-Horn, C.; Pouyssegur, J. The role of the hypoxia-inducible factor in tumor metabolism growth and invasion. Bull. Cancer 2006, 93, 10073–10080. [Google Scholar]
- Ono, M.; Kuwano, M. Molecular mechanisms of epidermal growth factor receptor (EGFR) activation and response to gefitinib and other EGFR-targeting drugs. Clin. Cancer Res. 2006, 12, 7242–7251. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Defining the role of hypoxia-inducible factor 1 in cancer biology and therapeutics. Oncogene 2010, 29, 625–634. [Google Scholar] [CrossRef] [PubMed]
- Rohwer, N.; Cramer, T. Hypoxia-mediated drug resistance: Novel insights on the functional interaction of HIFs and cell death pathways. Drug Resist. Update 2011, 14, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Klauber-DeMore, N.; Schulte, B.A.; Wang, G.Y. Targeting MYC for triple-negative breast cancer treatment. Oncoscience 2018, 5, 120–121. [Google Scholar] [CrossRef] [PubMed]
- Robey, I.F.; Lien, A.D.; Welsh, S.J.; Baggett, B.K.; Gillies, R.J. Hypoxia-inducible factor-1alpha and the glycolytic phenotype in tumors. Neoplasia 2005, 7, 324–330. [Google Scholar] [CrossRef] [PubMed]
- Berishaj, M.; Gao, S.P.; Ahmed, S.; Leslie, K.; Al-Ahmadie, H.; Gerald, W.L.; Bornmann, W.; Bromberg, J.F. Stat3 is tyrosine-phosphorylated through the interleukin-6/glycoprotein 130/Janus kinase pathway in breast cancer. Breast Cancer Res. 2007, 9, R32. [Google Scholar] [CrossRef] [PubMed]
- Godse, P.; Kumar, P.; Yewalkar, N.; Deore, V.; Lohar, M.; Mundada, R.; Padgaonkar, A.; Manohar, S.; Joshi, A.; Bhatia, D.; et al. Discovery of P3971 an orally efficacious novel anticancer agent targeting HIF-1alpha and STAT3 pathways. Anticancer Agents Med. Chem. 2013, 13, 1460–1466. [Google Scholar] [CrossRef] [PubMed]
- Reddy, K.R.; Guan, Y.; Qin, G.; Zhou, Z.; Jing, N. Combined treatment targeting HIF-1alpha and Stat3 is a potent strategy for prostate cancer therapy. Prostate 2011, 71, 1796–1809. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Micelle a | Average Diameter ± SD (nm) b | PDI ± SD c | Zeta potential ± SD (mV) | CMC ± SD (μg/mL) d | EE ± SD (%) e | DL ± SD (%) f | Drug/polymer ± SD (mol/mol) |
|---|---|---|---|---|---|---|---|
| Cisplatin plain micelle | 84.4 ± 2.6 | 0.263 ± 0.11 | −13.3 ± 1.2 | 65.1 ± 5.5 | 12.4 ± 0.99 | 12.0 ± 1.41 | 3.93 ± 0.31 |
| GE11 cisplatin micelle | 84.1 ± 3.2 | 0.235 ± 0.18 | −13.6 ± 0.95 | 70.5 ± 7.2 | 13.0 ± 2.95 | 15.5 ± 3.53 | 4.01 ± 0.93 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Soleymani Abyaneh, H.; Soleimani, A.H.; Vakili, M.R.; Soudy, R.; Kaur, K.; Cuda, F.; Tavassoli, A.; Lavasanifar, A. Modulation of Hypoxia-Induced Chemoresistance to Polymeric Micellar Cisplatin: The Effect of Ligand Modification of Micellar Carrier Versus Inhibition of the Mediators of Drug Resistance. Pharmaceutics 2018, 10, 196. https://doi.org/10.3390/pharmaceutics10040196
Soleymani Abyaneh H, Soleimani AH, Vakili MR, Soudy R, Kaur K, Cuda F, Tavassoli A, Lavasanifar A. Modulation of Hypoxia-Induced Chemoresistance to Polymeric Micellar Cisplatin: The Effect of Ligand Modification of Micellar Carrier Versus Inhibition of the Mediators of Drug Resistance. Pharmaceutics. 2018; 10(4):196. https://doi.org/10.3390/pharmaceutics10040196
Chicago/Turabian StyleSoleymani Abyaneh, Hoda, Amir Hassan Soleimani, Mohammad Reza Vakili, Rania Soudy, Kamaljit Kaur, Francesco Cuda, Ali Tavassoli, and Afsaneh Lavasanifar. 2018. "Modulation of Hypoxia-Induced Chemoresistance to Polymeric Micellar Cisplatin: The Effect of Ligand Modification of Micellar Carrier Versus Inhibition of the Mediators of Drug Resistance" Pharmaceutics 10, no. 4: 196. https://doi.org/10.3390/pharmaceutics10040196