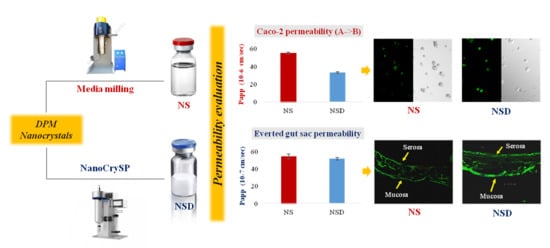
Permeability Behavior of Nanocrystalline Solid Dispersion of Dipyridamole Generated Using NanoCrySP Technology


Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
2.2.1. Generation of Nanocrystals of Dipyridamole (DPM)
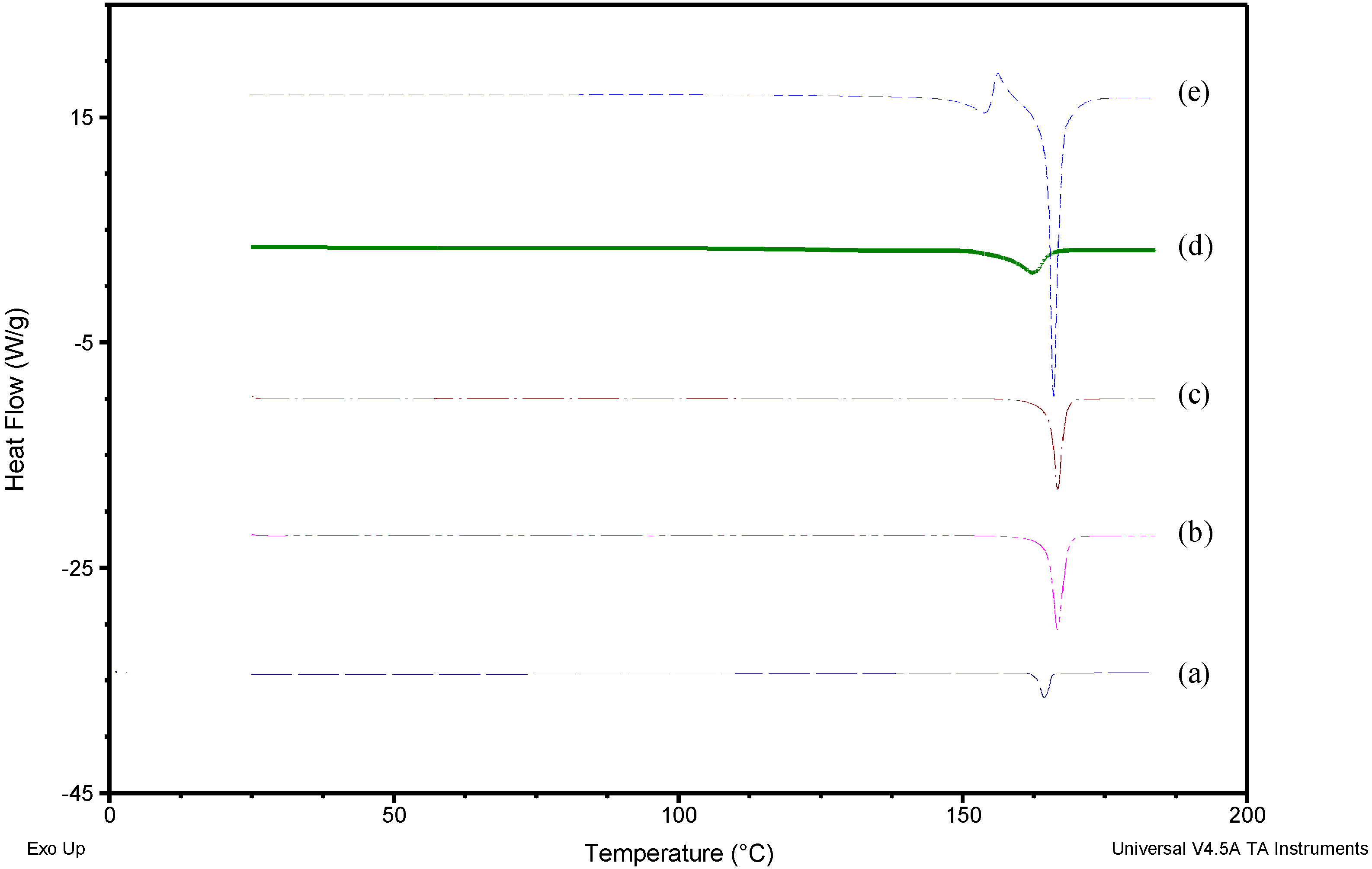
2.2.2. Differential Scanning Calorimetry (DSC)
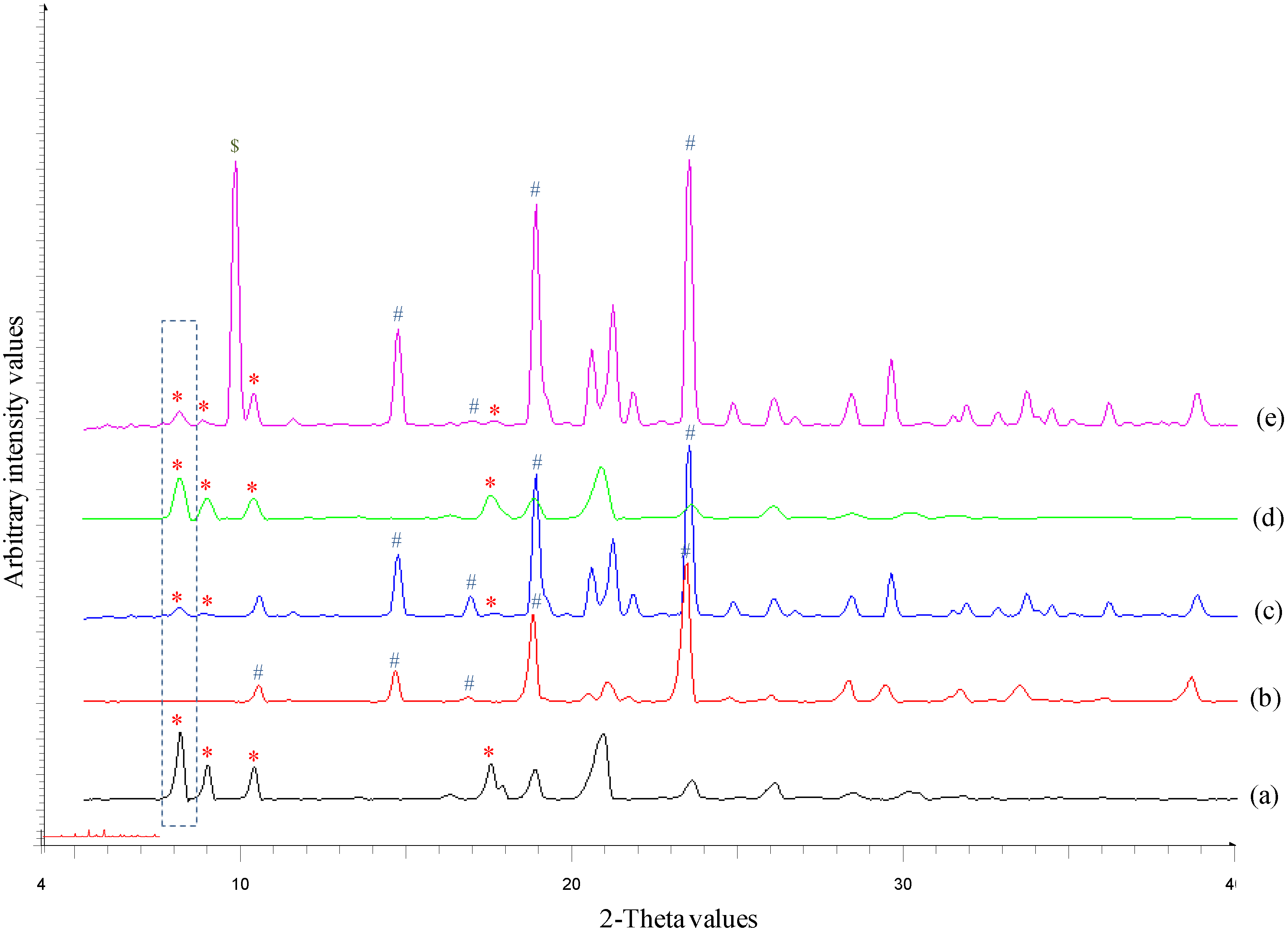
2.2.3. Powder X-ray Diffraction (PXRD)
2.2.4. Scanning Electron Microscopy (SEM)
2.2.5. Dynamic Light Scattering (DLS)
2.2.6. Drug Content Determination
2.2.7. Solubility Study
2.2.8. HPLC Analysis of DPM
2.2.9. Cell Culture
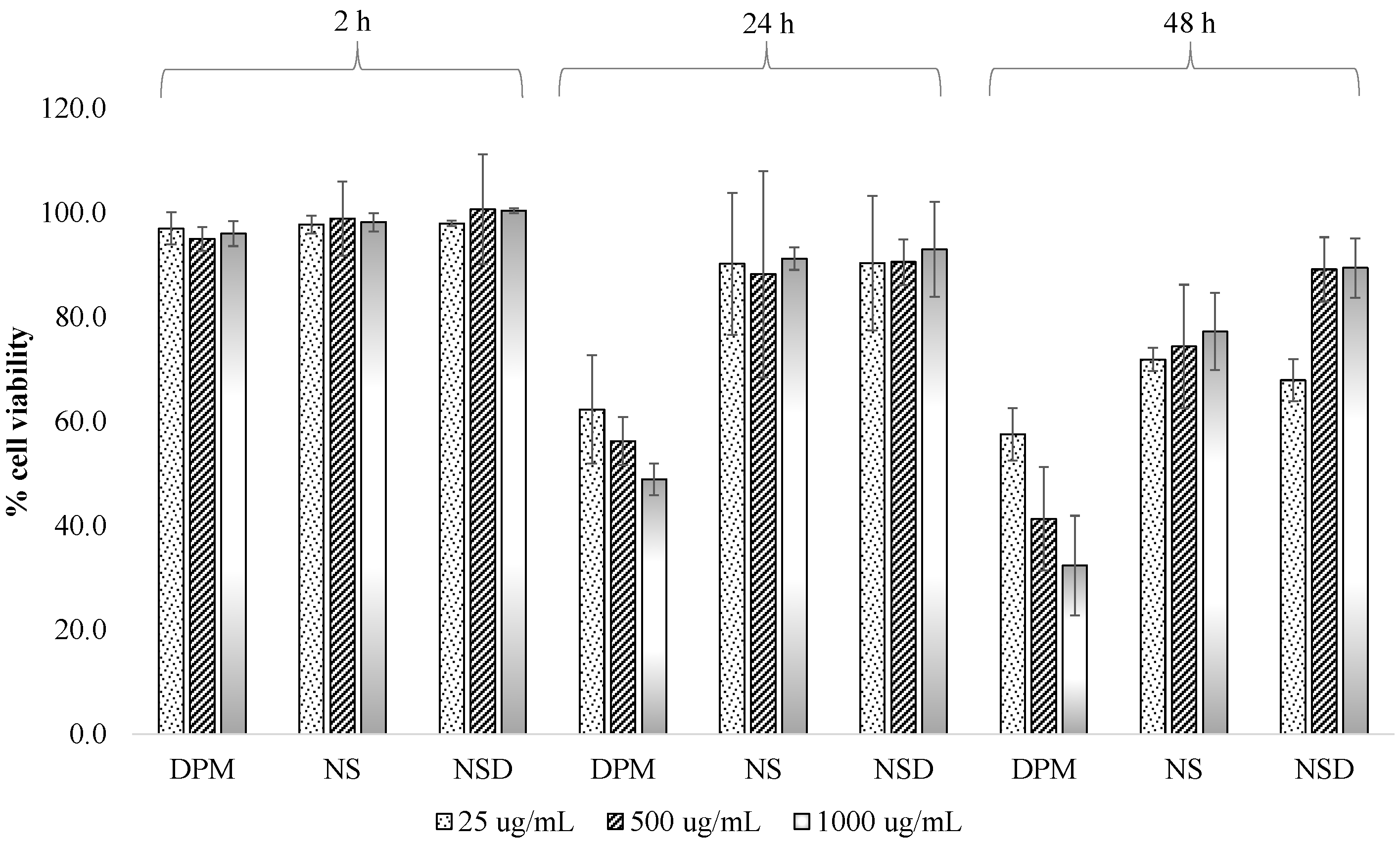
2.2.10. Cell Viability Studies
2.2.11. In Vitro Transport Studies
Monolayer Integrity Tests
Permeability Study
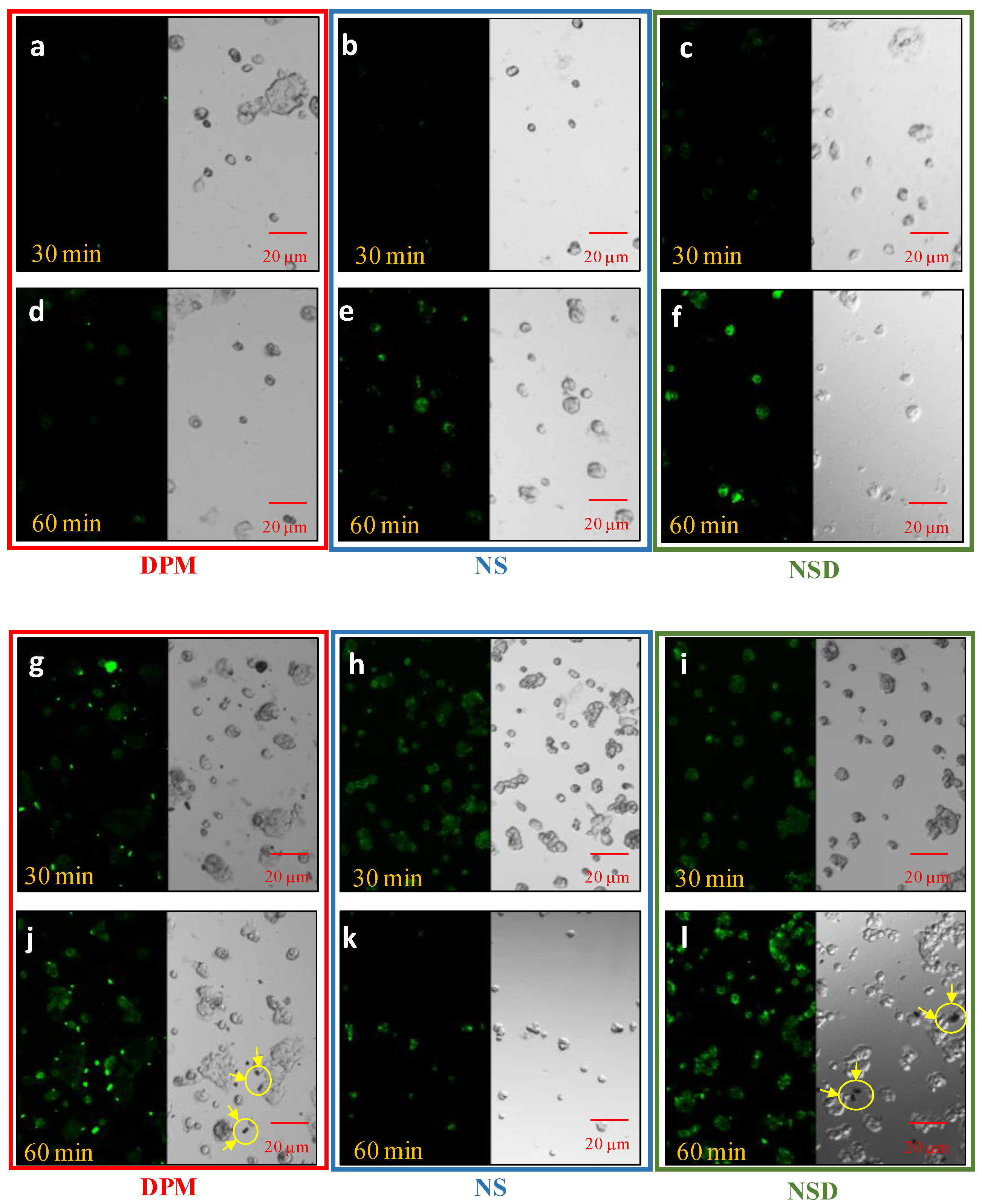
2.2.12. Cellular Uptake Studies
2.2.13. Absorption and Desorption Studies
2.2.14. Ex Vivo Study using Everted Gut Sac
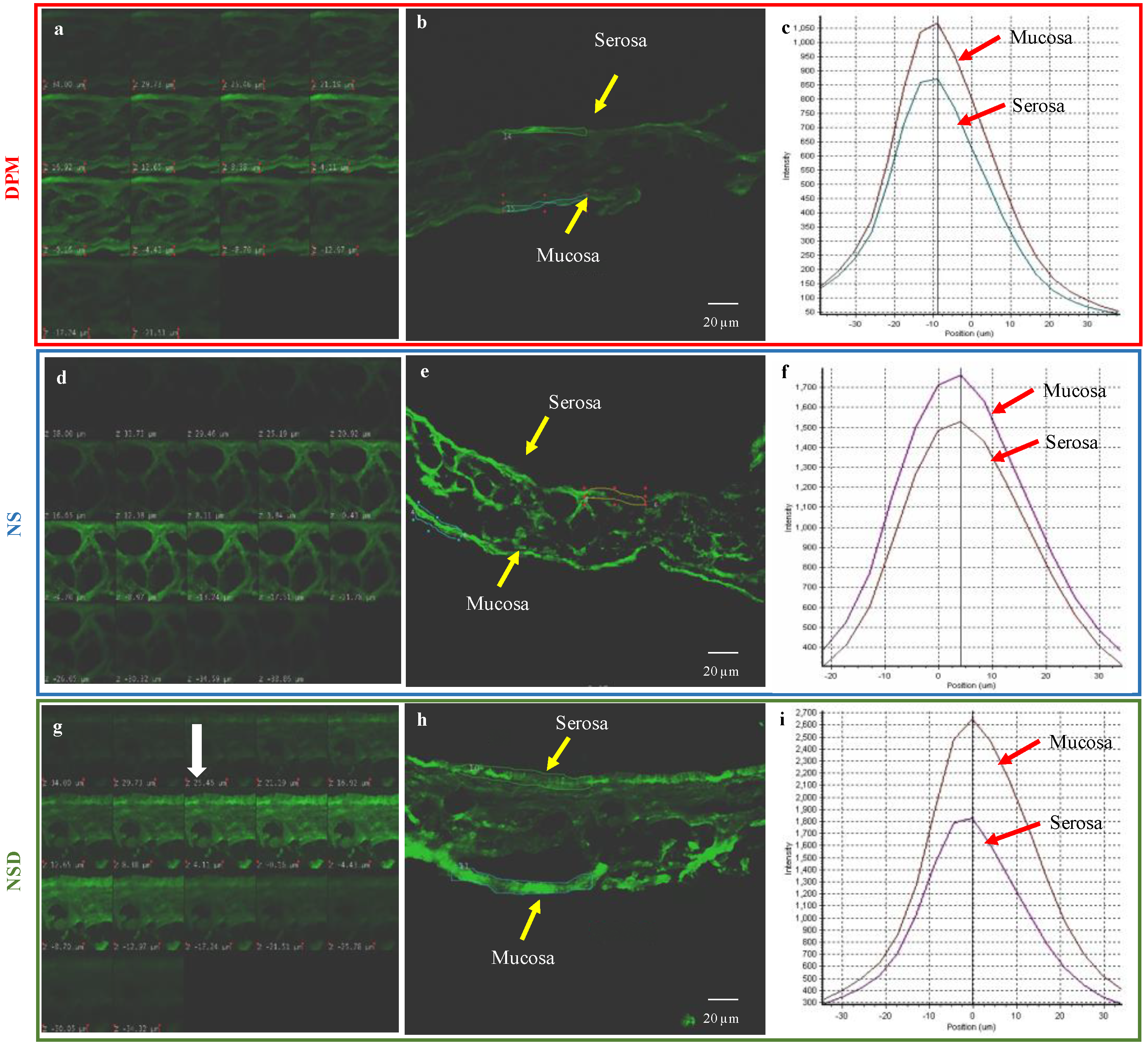
Permeability Study
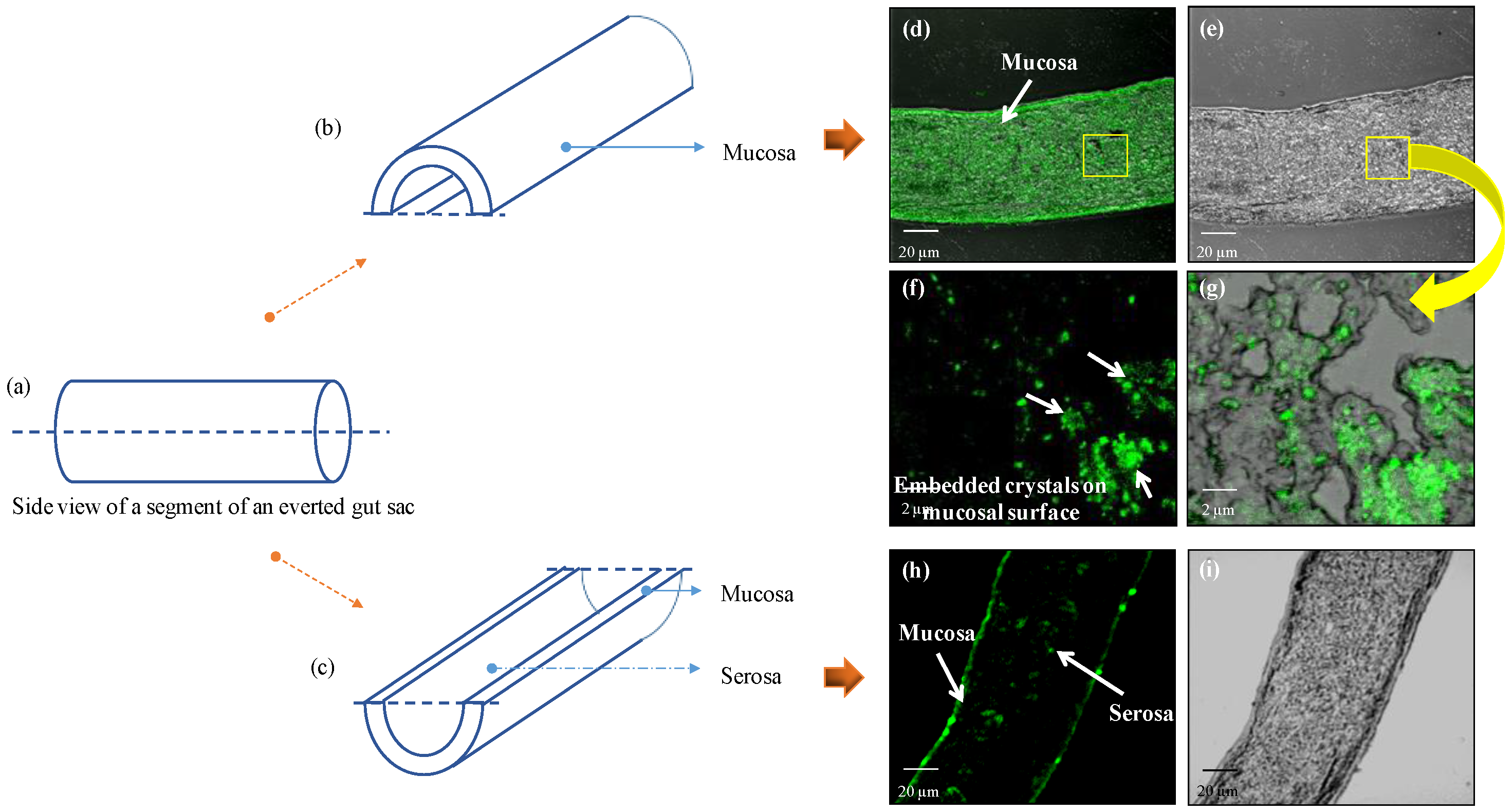
Mucoadhesion Study
2.2.15. Statistical Analysis
3. Results
3.1. Solid-State Characterization
3.2. Solubility Study
3.3. Cell Viability Studies
3.4. In Vitro Transport Studies
3.4.1. Cellular Uptake Studies
3.4.2. Absorption and Desorption Studies
3.5. Ex Vivo Study
3.5.1. Permeability Study
3.5.2. Mucoadhesion Study
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kaushal, A.M.; Gupta, P.; Bansal, A.K. Amorphous drug delivery systems: Molecular aspects, design, and performance. Crit. Rev. Ther. Drug Carrier Syst. 2004, 21, 133–193. [Google Scholar] [CrossRef] [PubMed]
- Yu, L. An integrated absorption model for determining dissolution, permeability, and solubility limited absorption. Pharm. Res. 1999, 16, 1884–1888. [Google Scholar]
- Riehemann, K.; Schneider, S.W.; Luger, T.A.; Godin, B.; Ferrari, M.; Fuchs, H. Nanomedicine-challenge and perspectives. Angew. Chem. Int. Ed. 2009, 48, 872–897. [Google Scholar] [CrossRef] [PubMed]
- Bamrungsap, S.; Zhao, Z.; Chen, T.; Wang, L.; Li, C.; Fu, T.; Tan, W. Nanotechnology in therapeutics: A focus on nanoparticles as a drug delivery system. Nanomedicine 2012, 7, 1253–1271. [Google Scholar] [CrossRef] [PubMed]
- Junghanns, J.-U.A.; Müller, R.H. Nanocrystal technology, drug delivery and clinical applications. Int. J. Nanomed. 2008, 3, 295–310. [Google Scholar]
- George, M.; Ghosh, I. Identifying the correlation between drug/stabilizer properties and critical quality attributes (CQAs) of nanosuspension formulation prepared by wet media milling technology. Eur. J. Pharm. Sci. 2013, 48, 142–152. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Matas, M.D.; Zhang, J.; Anwar, J. Nanocrystal preparation: Low-energy precipitation method revisited. Cryst. Growth Des. 2013, 13, 2766–2777. [Google Scholar] [CrossRef]
- Shete, G.; Bansal, A.K. NanoCrySP technology for generation of drug nanocrystals: Translational aspects and business potential. Drug Deliv. Trans. Res. 2016, 6, 392–398. [Google Scholar] [CrossRef] [PubMed]
- Bansal, A.K.; Dantuluri, A.K.R.; Bhaskarao, S.G.; Bapurao, P.Y. Nanocrystalline Solid Dispersion Compositions and Process of Preparation Thereof. WO2013132457A2. 7 March 2012. [Google Scholar]
- Keck, C.M.; Müller, R.H. Nanotoxicological classification system (NCS)—A guide for the risk-benefit assessment of nanoparticulate drug delivery systems. Eur. J. Pharm. Biopharm. 2013, 84, 445–448. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Kadomura, S.; Takekuma, Y.; Sugawara, M.; Miyazaki, K. A new system for the prediction of drug absorption using a pH-controlled Caco-2 model: Evaluation of pH-dependent soluble drug absorption and pH-related changes in absorption. J. Pham. Sci. 2004, 93, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Kostewicz, E.S.; Brauns, U.; Becker, R.; Dressman, J.B. Forecasting the oral absorption behavior of poorly soluble weak bases using solubility and dissolution studies in biorelevant media. Pharm. Res. 2002, 19, 345–349. [Google Scholar] [CrossRef] [PubMed]
- Iuliano, L.; Piccheri, C.; Coppola, I.; Pratico, D.; Micheletta, F.; Violi, F. Fluorescence quenching of dipyridamole associated to peroxyl radical scavenging: A versatile probe to measure the chain breaking antioxidant activity of biomolecules. Biochim. Biophys. Acta 2000, 1474, 177–182. [Google Scholar] [CrossRef]
- Bagul, P.; Khomane, K.S.; Bansal, A.K. Investigating permeability related hurdles in oral delivery of 11-keto-β-boswellic acid. Int. J. Pharm. 2014, 464, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Wahlang, B.; Pawar, Y.B.; Bansal, A.K. Identification of permeability-related hurdles in oral delivery of curcumin using the Caco-2 cell model. Eur. J. Pharm. Biopharm. 2011, 77, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Cares-Pacheco, M.; Vaca-Medina, G.; Calvet, R.; Espitalier, F.; Letourneau, J.-J.; Rouilly, A.; Rodier, E. Physicochemical characterization of D-mannitol polymorphs: The challenging surface energy determination by inverse gas chromatography in the infinite dilution region. Int. J. Pharm. 2014, 475, 69–81. [Google Scholar] [CrossRef] [PubMed]
- Artursson, P.; Karlsson, J. Correlation between oral drug absorption in humans and apparent drug permeability coefficients in human intestinal epithelial (Caco-2) cells. Biochem. Biophys. Res. Commun. 1991, 175, 880–885. [Google Scholar] [CrossRef]
- Hidalgo, I.J.; Raub, T.J.; Borchardt, R.T. Characterization of the human colon carcinoma cell line (Caco-2) as a model system for intestinal epithelial permeability. Gastroenterology 1989, 96, 736–749. [Google Scholar] [CrossRef]
- Yee, S. In vitro permeability across Caco-2 cells (colonic) can predict in vivo (small intestinal) absorption in man—fact or myth. Pharm. Res. 1997, 14, 763–766. [Google Scholar] [CrossRef] [PubMed]
- Elsby, R.; Surry, D.; Smith, V.; Gray, A. Validation and application of Caco-2 assays for the in vitro evaluation of development candidate drugs as substrates or inhibitors of P-glycoprotein to support regulatory submissions. Xenobiotica 2008, 38, 1140–1164. [Google Scholar] [CrossRef] [PubMed]
- Dokoumetzidis, A.; Macheras, P. A century of dissolution research: From Noyes and Whitney to the biopharmaceutics classification system. Int. J. Pharm. 2006, 321, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Williams, H.D.; Trevaskis, N.L.; Charman, S.A.; Shanker, R.M.; Charman, W.N.; Pouton, C.W.; Porter, C.J. Strategies to address low drug solubility in discovery and development. Pharmacol. Rev. 2013, 65, 315–499. [Google Scholar] [CrossRef] [PubMed]
- Müller, R.H.; Gohla, S.; Keck, C.M. State of the art of nanocrystals–special features, production, nanotoxicology aspects and intracellular delivery. Eur. J. Pharm. Biopharm. 2011, 78, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Li, T. Cellular uptake mechanism of paclitaxel nanocrystals determined by confocal imaging and kinetic measurement. AAPS J. 2015, 17, 1126–1134. [Google Scholar] [CrossRef] [PubMed]
- Swenson, E.S.; Curatolo, W.J. (C) Means to enhance penetration: (2) Intestinal permeability enhancement for proteins, peptides and other polar drugs: Mechanisms and potential toxicity. Adv. Drug Deliv. Rev. 1992, 8, 39–92. [Google Scholar] [CrossRef]
- Dimitrijevic, D.; Shaw, A.J.; Florence, A.T. Effects of some non-ionic surfactants on transepithelial permeability in Caco-2 cells. J. Pharm. Pharmacol. 2000, 52, 157–162. [Google Scholar] [CrossRef] [PubMed]
- Boulenc, X.; Breul, T.; Gautier, J.-C.; Saudemon, P.; Joyeux, H.; Roques, C.; Berger, Y.; Fabre, G. Sodium lauryl sulphate increases tiludronate paracellular transport using human epithelial caco-2 monolayers. Int. J. Pharm. 1995, 123, 71–83. [Google Scholar] [CrossRef]
- Peltonen, L.; Strachan, C. Understanding critical quality attributes for nanocrystals from preparation to delivery. Molecules 2015, 20, 22286–22300. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Time (min) | HBSS pH 6.5 (Apical Medium) | HBSS pH 7.4 (Basolateral Medium) | Medium 199 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| DPM | NS | NSD | DPM | NS | NSD | DPM | NS | NSD | |
| 15 | 4.39 ± 0.06 | 10.32 ± 0.25 | 8.78 ± 0.33 | 7.77 ± 1.11 | 23.46 ± 1.52 | 13.24 ± 1.85 | 5.05 ± 0.11 | 10.32 ± 0.25 | 10.41 ± 0.74 |
| 30 | 8.95 ± 0.98 | 31.40 ± 0.92 | 20.83 ± 0.95 | 14.88 ± 0.48 | 46.62 ± 2.59 | 22.77 ± 1.37 | 16.62 ± 2.44 | 31.40 ± 0.92 | 24.79 ± 1.86 |
| 60 | 24.03 ± 1.38 | 47.46 ± 2.88 | 40.74 ± 4.03 | 16.99 ± 2.50 | 57.97 ± 0.11 | 41.93 ± 0.64 | 33.65 ± 0.71 | 47.76 ± 2.88 | 44.32 ± 2.17 |
| 90 | 36.97 ± 1.82 | 71.93 ± 2.43 | 48.50 ± 3.79 | 31.41 ± 1.56 | 70.50 ± 1.54 | 53.89 ± 0.29 | 35.71 ± 0.15 | 60.41 ± 0.10 | 50.09 ± 0.31 |
| 120 | 52.18 ± 0.74 | 85.61 ± 1.78 | 75.21 ± 1.40 | 47.06 ± 8.42 | 79.36 ± 2.21 | 63.76 ± 0.28 | 39.53 ± 2.57 | 71.93 ± 2.43 | 59.97 ± 0.31 |
| Sample | Papp (Apical-to-Basolateral) (10−6 cm/s) | Papp (Basolateral-to-Apical) (10−6 cm/s) | ER |
|---|---|---|---|
| DPM | 16.84 ± 0.37 | 61.01 ± 0.51 | 3.60 ± 0.28 |
| NS | 55.42 ± 1.08 | 25.87 ± 0.25 | 0.46 ± 0.23 |
| NSD | 33.58 ± 0.91 | 28.25 ± 0.48 | 0.84 ± 0.35 |
| Sample | % DPM Absorbed | % DPM Desorbed |
|---|---|---|
| DPM | 3.55 ± 1.56 | 25.58 ± 2.13 |
| NS | 12.86 ± 1.01 | 55.05 ± 3.20 |
| NSD | 9.91 ± 1.20 | 51.72 ± 2.30 |
| Sample | Papp (Jejunum) (10−7 cm/s) | Papp (Ileum) (10−7 cm/s) | Intestinal Tissue Uptake (ng/cm2) |
|---|---|---|---|
| DPM | 29.64 ± 1.42 | 8.36 ± 0.52 | 336 ± 29 |
| NS | 54.34 ± 2.91 | 35.34 ± 2.51 | 707 ± 31 |
| NSD | 51.68 ± 1.58 | 40.73 ± 2.70 | 975 ± 53 |
| Contributors | DPM * | NS | NSD |
|---|---|---|---|
| Solubility (in HBSS pH 6.5 after 120 min) | 1.00 | 1.64 | 1.44 |
| Solubility (in medium 199 after 120 min) | 1.00 | 1.82 | 1.52 |
| Papp (apical-to-basolateral) | 1.00 | 3.30 | 2.35 |
| Papp (basolateral-to-apical) | 1.00 | 0.54 | 0.87 |
| Papp (mucosa-to-serosa) | 1.00 | 1.83 | 1.74 |
| Internalization (absorption-desorption study) | 1.00 | 3.62 | 2.79 |
| Mucoadhesion | 1.00 | 1.42 | 2.47 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Girdhar, A.; Thakur, P.S.; Sheokand, S.; Bansal, A.K. Permeability Behavior of Nanocrystalline Solid Dispersion of Dipyridamole Generated Using NanoCrySP Technology. Pharmaceutics 2018, 10, 160. https://doi.org/10.3390/pharmaceutics10030160
Girdhar A, Thakur PS, Sheokand S, Bansal AK. Permeability Behavior of Nanocrystalline Solid Dispersion of Dipyridamole Generated Using NanoCrySP Technology. Pharmaceutics. 2018; 10(3):160. https://doi.org/10.3390/pharmaceutics10030160
Chicago/Turabian StyleGirdhar, Ashish, Poonam Singh Thakur, Sneha Sheokand, and Arvind K. Bansal. 2018. "Permeability Behavior of Nanocrystalline Solid Dispersion of Dipyridamole Generated Using NanoCrySP Technology" Pharmaceutics 10, no. 3: 160. https://doi.org/10.3390/pharmaceutics10030160